
Single-Cell Gene Network Analysis and Transcriptional Landscape of MYCN-Amplified Neuroblastoma Cell Lines

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
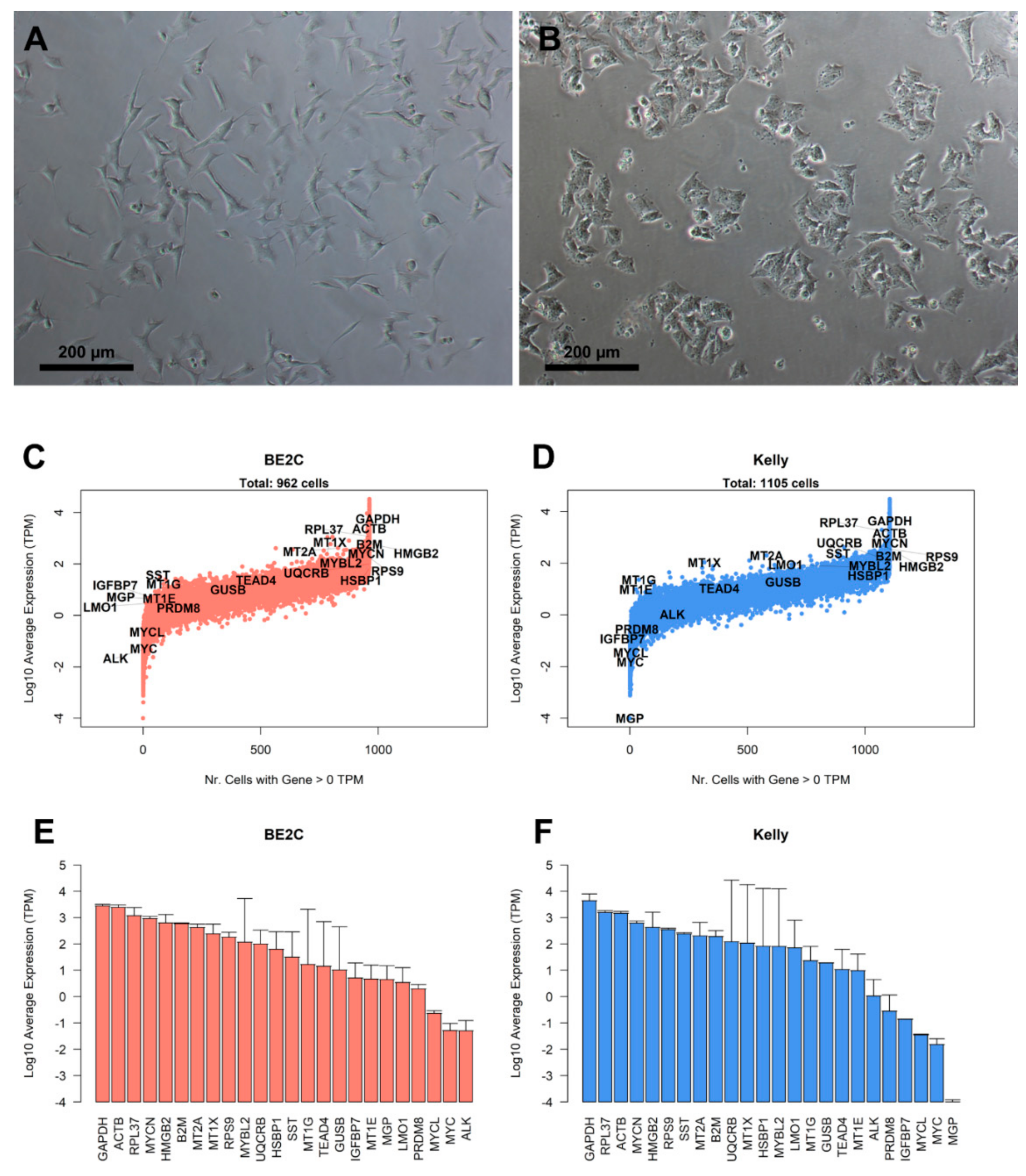
2.1. Cell Cultures
2.2. 10× Genomics Library Preparation and Sequencing
2.3. Data Processing
3. Results
3.1. Characterization of Landmark Gene Expression
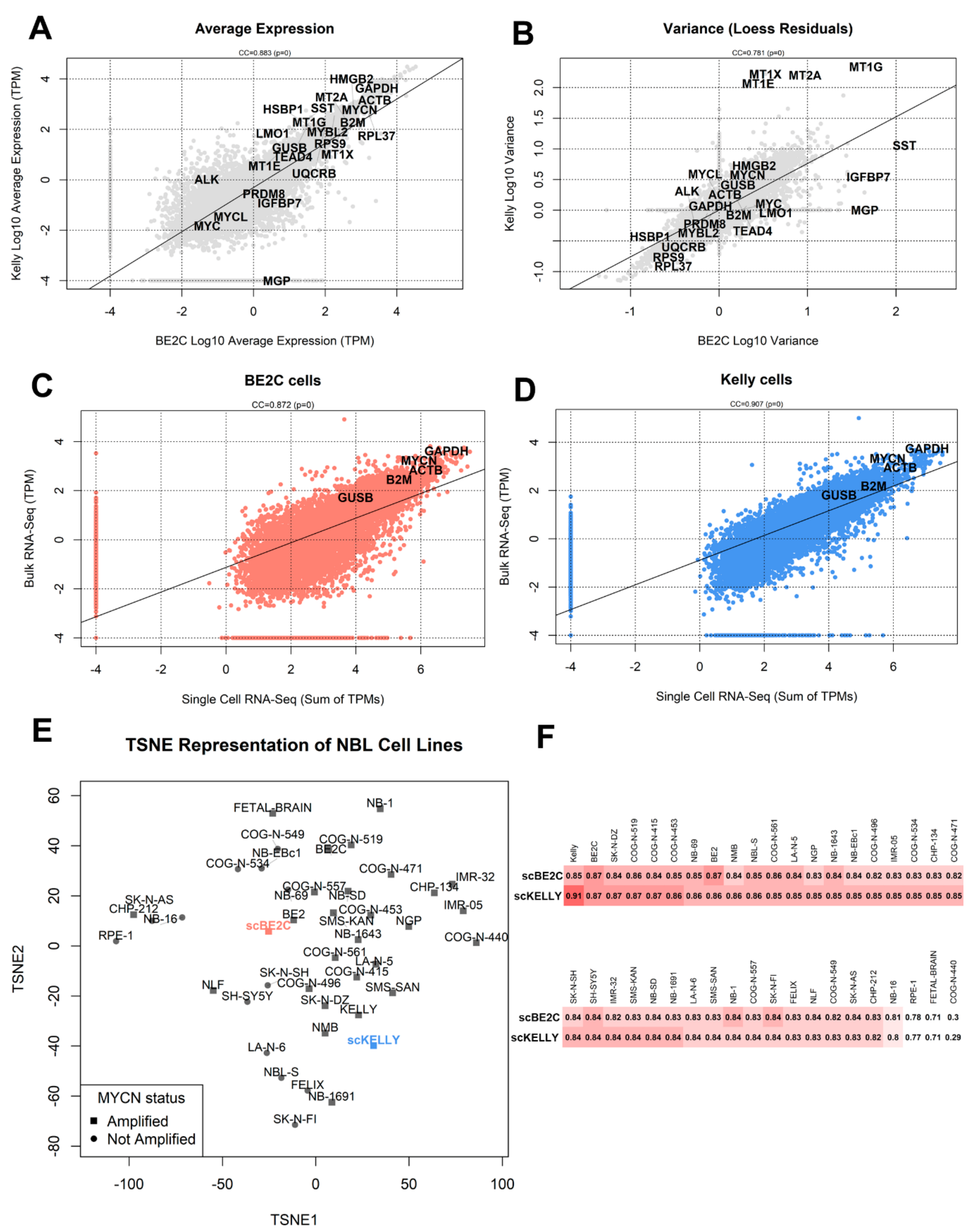
3.2. Comparison with Bulk RNA-Seq
3.3. Dimensionality Reduction and Clustering of Cells
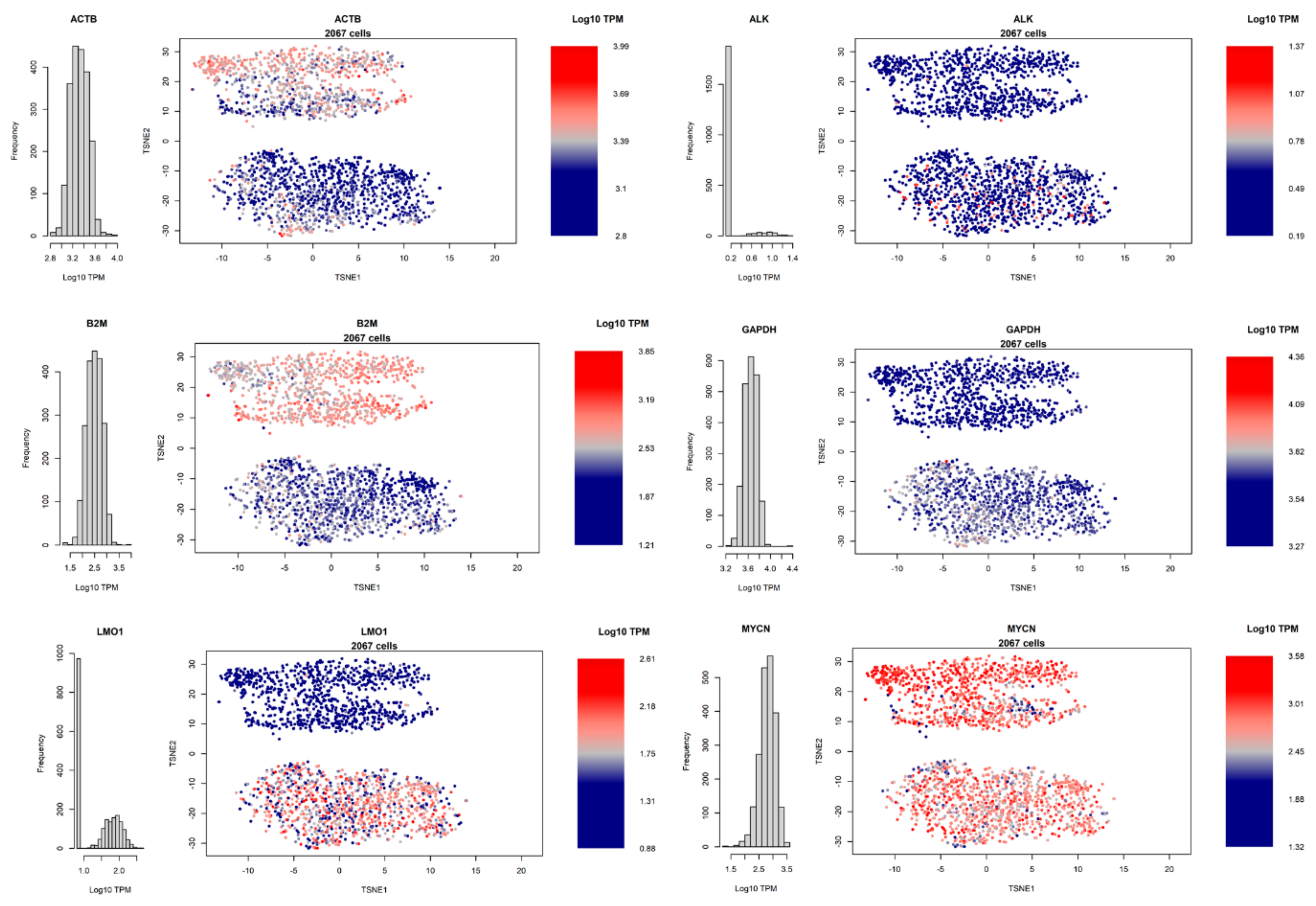
3.4. Heterogeneity of Gene Expression
3.5. Differential Gene Expression
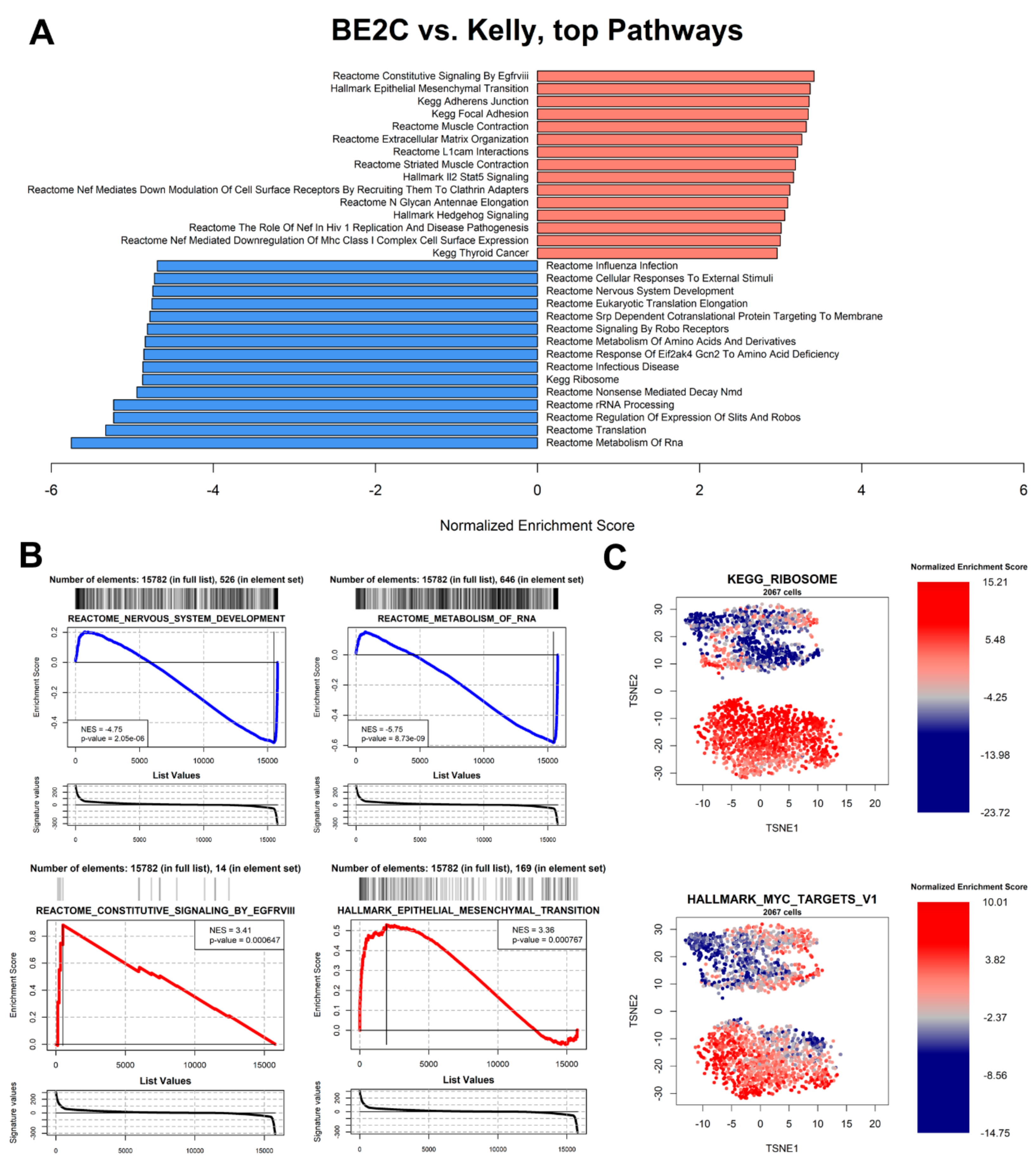
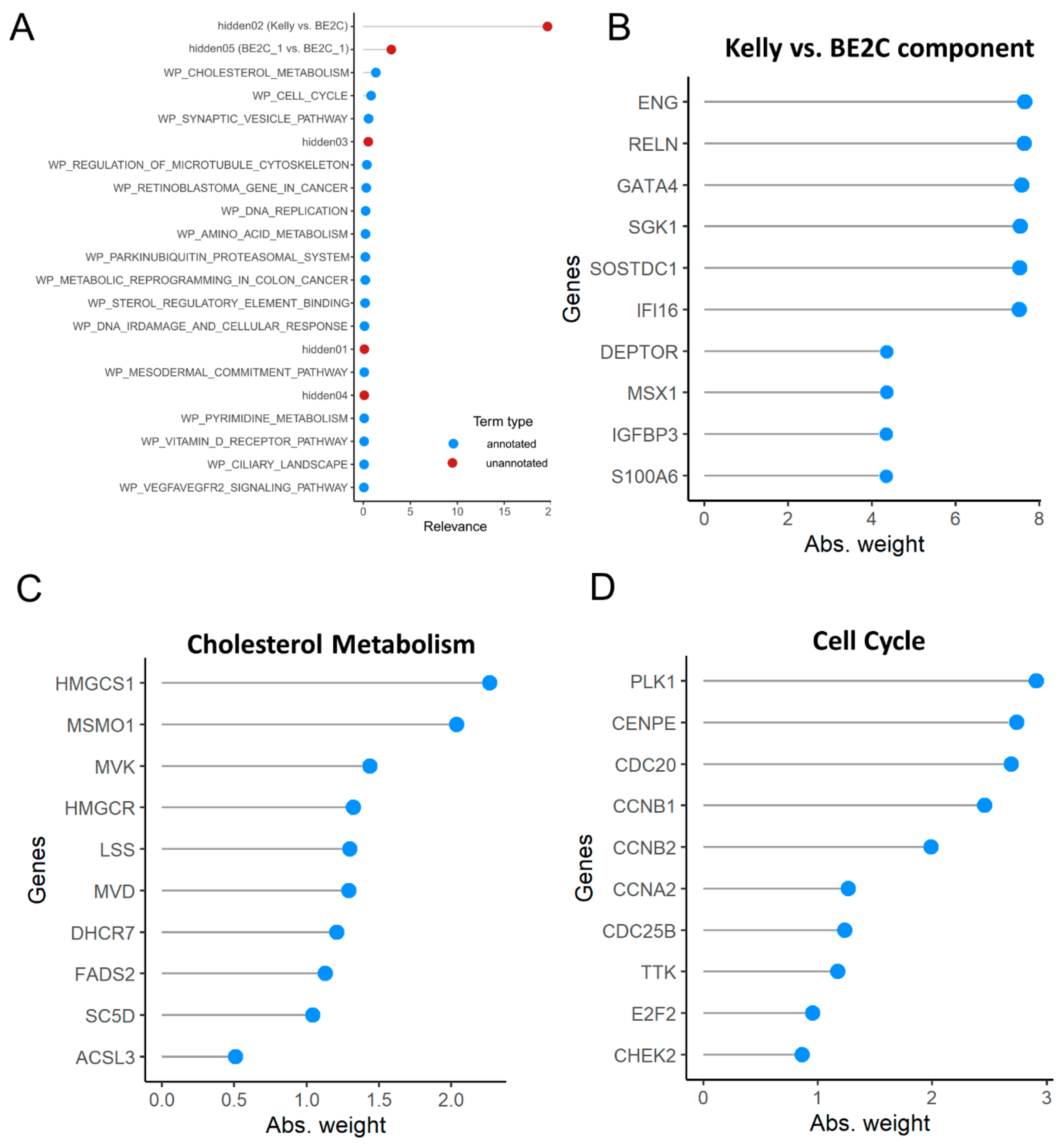
3.6. Pathway Analysis
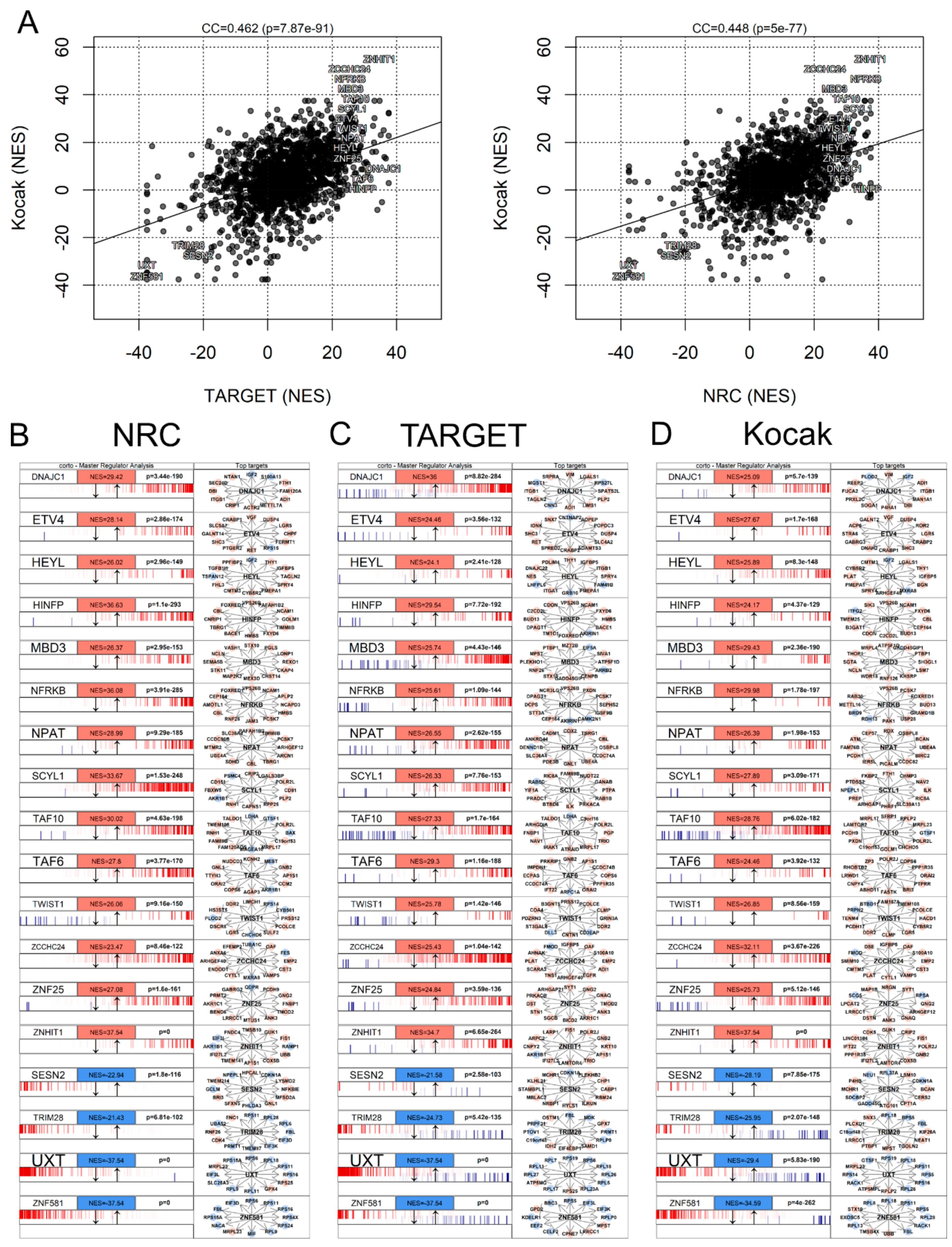
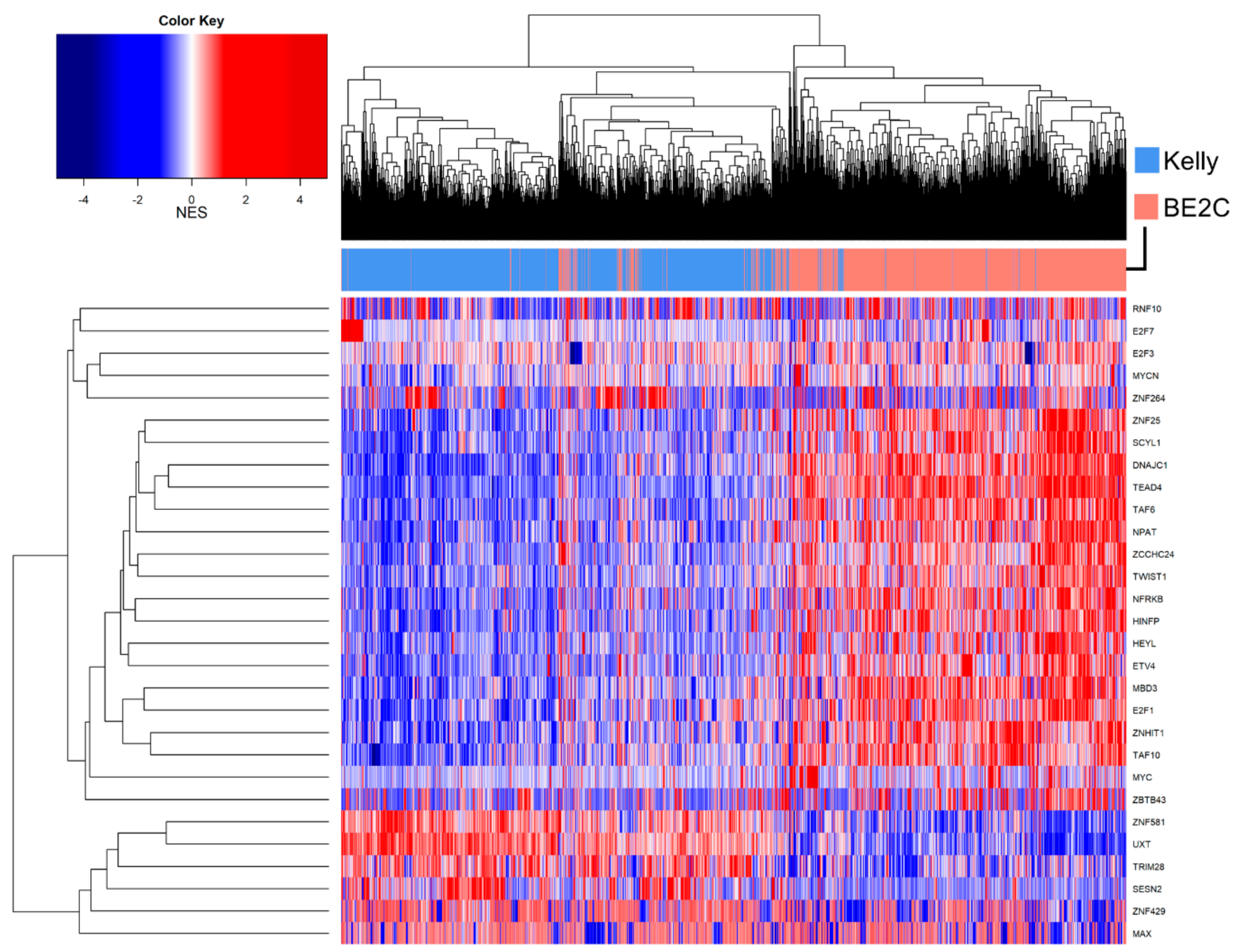
3.7. Master Regulator Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol. Cancer Res. 2016, 14, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Klinghammer, K.; Walther, W.; Hoffmann, J. Choosing wisely-Preclinical test models in the era of precision medicine. Cancer Treat. Rev. 2017, 55, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.; Schildknecht, S. In Vitro Research Reproducibility: Keeping Up High Standards. Front. Pharmacol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb. Perspect. Med. 2016, 6, a026583. [Google Scholar] [CrossRef]
- Hynds, R.E.; Vladimirou, E.; Janes, S.M. The secret lives of cancer cell lines. Dis. Models Mech. 2018, 11, dmm037366. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Lachmann, A.; Giorgi, F.M.; Alvarez, M.J.; Califano, A. Detection and removal of spatial bias in multiwell assays. Bioinformatics 2016, 32, 1959–1965. [Google Scholar] [CrossRef]
- He, S.; Wang, L.-H.; Liu, Y.; Li, Y.-Q.; Chen, H.-T.; Xu, J.-H.; Peng, W.; Lin, G.-W.; Wei, P.-P.; Li, B.; et al. Single-cell transcriptome profiling of an adult human cell atlas of 15 major organs. Genome Biol. 2020, 21, 294. [Google Scholar] [CrossRef]
- Fan, J.; Slowikowski, K.; Zhang, F. Single-cell transcriptomics in cancer: Computational challenges and opportunities. Exp. Mol. Med. 2020, 52, 1452–1465. [Google Scholar] [CrossRef]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, S.; Min, M.; Ni, Y.; Lu, Z.; Sun, X.; Wu, J.; Liu, B.; Ying, X.; Liu, Y. Dissecting transcriptional heterogeneity in primary gastric adenocarcinoma by single cell RNA sequencing. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tan, Y.; Fang, C.; Zhou, J.; Wang, Y.; Zhao, K.; Jin, W.; Wu, Y.; Liu, X.; Liu, X.; et al. Single-cell RNA-seq reveals RAD51AP1 as a potent mediator of EGFRvIII in human glioblastomas. Aging 2019, 11, 7707–7722. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, S.; Yu, J.; Li, Y.; Zhang, X.-Y.; Yang, L.; Zhang, H.; Hou, Q.; Jiang, M.; Brunicardi, F.C.; et al. Single-cell Transcriptome Analyses Reveal Molecular Signals to Intrinsic and Acquired Paclitaxel Resistance in Esophageal Squamous Cancer Cells. Cancer Lett. 2018, 420, 156–167. [Google Scholar] [CrossRef]
- Tanaka, N.; Katayama, S.; Reddy, A.; Nishimura, K.; Niwa, N.; Hongo, H.; Ogihara, K.; Kosaka, T.; Mizuno, R.; Kikuchi, E.; et al. Single-cell RNA-seq analysis reveals the platinum resistance gene COX7B and the surrogate marker CD63. Cancer Med. 2018, 7, 6193–6204. [Google Scholar] [CrossRef]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef]
- Andor, N.; Lau, B.T.; Catalanotti, C.; Sathe, A.; Kubit, M.; Chen, J.; Blaj, C.; Cherry, A.; Bangs, C.D.; Grimes, S.M.; et al. Joint single cell DNA-seq and RNA-seq of gastric cancer cell lines reveals rules of in vitro evolution. NAR Genom. Bioinform. 2020, 2, lqaa016. [Google Scholar] [CrossRef]
- Borriello, L.; Seeger, R.C.; Asgharzadeh, S.; DeClerck, Y.A. More than the genes, the tumor microenvironment in neuroblastoma. Cancer Lett. 2016, 380, 304–314. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O. Recent insights into the biology of neuroblastoma. Int. J. Cancer 2014, 135, 2249–2261. [Google Scholar] [CrossRef] [PubMed]
- Sokol, E.; Desai, A.V. The Evolution of Risk Classification for Neuroblastoma. Children 2019, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Rajbhandari, P.; Lopez, G.; Capdevila, C.; Salvatori, B.; Yu, J.; Rodriguez-Barrueco, R.; Martinez, D.; Yarmarkovich, M.; Weichert-Leahey, N.; Abraham, B.J.; et al. Cross-Cohort Analysis Identifies a TEAD4-MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma. Cancer Discov. 2018, 8, 582–599. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC-Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Harenza, J.L.; Diamond, M.A.; Adams, R.N.; Song, M.M.; Davidson, H.L.; Hart, L.S.; Dent, M.H.; Fortina, P.; Reynolds, C.P.; Maris, J.M. Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Sci. Data 2017, 4, 170033. [Google Scholar] [CrossRef]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef]
- Upton, K.; Modi, A.; Patel, K.; Kendsersky, N.M.; Conkrite, K.L.; Sussman, R.T.; Way, G.P.; Adams, R.N.; Sacks, G.I.; Fortina, P.; et al. Epigenomic profiling of neuroblastoma cell lines. Sci. Data 2020, 7, 116. [Google Scholar] [CrossRef]
- Paull, E.O.; Aytes, A.; Jones, S.J.; Subramaniam, P.S.; Giorgi, F.M.; Douglass, E.F.; Tagore, S.; Chu, B.; Vasciaveo, A.; Zheng, S.; et al. A modular master regulator landscape controls cancer transcriptional identity. Cell 2021, 184, 334–351.e20. [Google Scholar] [CrossRef]
- Cooper CO2 Concentration and pH Control in the Cell Culture Laboratory. Available online: https://www.phe-culturecollections.org.uk/news/ecacc-news/co2-concentration-and-ph-control-in-the-cell-culture-laboratory.aspx (accessed on 10 October 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Lopez-Garcia, G.; Giorgi, F.M. corto: A lightweight R package for gene network inference and master regulator analysis. Bioinformatics 2020, 36, 3916–3917. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene regulatory network inference resources: A practical overview. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194430. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Skalniak, A.; Boratyn, E.; Tyrkalska, S.D.; Horwacik, I.; Durbas, M.; Lastowska, M.; Jura, J.; Rokita, H. Expression of the monocyte chemotactic protein-1-induced protein 1 decreases human neuroblastoma cell survival. Oncol. Rep. 2014, 31, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.R.; Haug, B.H.; Buechner, J.; Tømte, E.; Løkke, C.; Flaegstad, T.; Einvik, C. Conditional expression of retrovirally delivered anti-MYCN shRNA as an in vitro model system to study neuronal differentiation in MYCN-amplified neuroblastoma. BMC Dev. Biol. 2011, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Lemma, S.; Avnet, S.; Meade, M.J.; Chano, T.; Baldini, N. Validation of Suitable Housekeeping Genes for the Normalization of mRNA Expression for Studying Tumor Acidosis. Int. J. Mol. Sci. 2018, 19, 2930. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-H.; Wu, H.-Y.; Gao, J.; Wang, X.-H.; Gao, T.-H.; Zhang, S.-F. FGF represses metastasis of neuroblastoma regulated by MYCN and TGF-β1 induced LMO1 via control of let-7 expression. Brain Res. 2019, 1704, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intra-tumoral copper modulates PD-L1 expression and influences tumor immune evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Islam, S.; Watanabe, H. Versican: A Dynamic Regulator of the Extracellular Matrix. J. Histochem. Cytochem. 2020, 68, 763–775. [Google Scholar] [CrossRef]
- Tsubouchi, T.; Fisher, A.G. Chapter Seven-Reprogramming and the Pluripotent Stem Cell Cycle. In Current Topics in Developmental Biology; Heard, E., Ed.; Epigenetics and Development; Academic Press: Cambridge, MA, USA, 2013; Volume 104, pp. 223–241. [Google Scholar]
- BE(2)-C ATCC ® CRL-2268TM. Available online: https://www.lgcstandards-atcc.org/Products/All/CRL-2268.aspx?geo_country=it (accessed on 10 October 2020).
- Cellosaurus Cell Line Kelly (CVCL_2092). Available online: https://web.expasy.org/cellosaurus/CVCL_2092 (accessed on 10 October 2020).
- Salt, M.B.; Bandyopadhyay, S.; McCormick, F. Epithelial-to-mesenchymal transition rewires the molecular path to PI3K-dependent proliferation. Cancer Discov. 2014, 4, 186–199. [Google Scholar] [CrossRef]
- Montano, N.; Cenci, T.; Martini, M.; D’Alessandris, Q.G.; Pelacchi, F.; Ricci-Vitiani, L.; Maira, G.; De Maria, R.; Larocca, L.M.; Pallini, R. Expression of EGFRvIII in Glioblastoma: Prognostic Significance Revisited. Neoplasia 2011, 13, 1113–1121. [Google Scholar] [CrossRef]
- Ma, Y.; Sun, S.; Shang, X.; Keller, E.T.; Chen, M.; Zhou, X. Integrative differential expression and gene set enrichment analysis using summary statistics for scRNA-seq studies. Nat. Commun. 2020, 11, 1585. [Google Scholar] [CrossRef]
- Behjati Ardakani, F.; Kattler, K.; Heinen, T.; Schmidt, F.; Feuerborn, D.; Gasparoni, G.; Lepikhov, K.; Nell, P.; Hengstler, J.; Walter, J.; et al. Prediction of single-cell gene expression for transcription factor analysis. GigaScience 2020, 9, giaa113. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.M. Gene network reverse engineering: The Next Generation. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194523. [Google Scholar] [CrossRef] [PubMed]
- Vasilevski, A.; Giorgi, F.M.; Bertinetti, L.; Usadel, B. LASSO modeling of the Arabidopsis thaliana seed/seedling transcriptome: A model case for detection of novel mucilage and pectin metabolism genes. Mol. BioSyst. 2012, 8, 2566–2574. [Google Scholar] [CrossRef]
- Kocak, H.; Ackermann, S.; Hero, B.; Kahlert, Y.; Oberthuer, A.; Juraeva, D.; Roels, F.; Theissen, J.; Westermann, F.; Deubzer, H.; et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 2013, 4, e586. [Google Scholar] [CrossRef]
- Ambrosio, S.; Saccà, C.D.; Amente, S.; Paladino, S.; Lania, L.; Majello, B. Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene 2017, 36, 6701–6711. [Google Scholar] [CrossRef]
- Kumar, A.; Shaha, C. RBX1-mediated ubiquitination of SESN2 promotes cell death upon prolonged mitochondrial damage in SH-SY5Y neuroblastoma cells. Mol. Cell. Biochem. 2018, 446, 1–9. [Google Scholar] [CrossRef]
- Selmi, A.; de Saint-Jean, M.; Jallas, A.-C.; Garin, E.; Hogarty, M.D.; Bénard, J.; Puisieux, A.; Marabelle, A.; Valsesia-Wittmann, S. TWIST1 is a direct transcriptional target of MYCN and MYC in neuroblastoma. Cancer Lett. 2015, 357, 412–418. [Google Scholar] [CrossRef]
- Susanti, S.; Iwasaki, H.; Inafuku, M.; Taira, N.; Oku, H. Mechanism of arctigenin-mediated specific cytotoxicity against human lung adenocarcinoma cell lines. Phytomedicine 2013, 21, 39–46. [Google Scholar] [CrossRef]
- Zhang, F.; Mai, S.-R.; Zhang, L. Circ-ZNF264 Promotes the Growth of Glioma Cells by Upregulating the Expression of miR-4493 Target Gene Apelin. J. Mol. Neurosci. 2019, 69, 75–82. [Google Scholar] [CrossRef]
- Weber, S.; Koschade, S.E.; Hoffmann, C.M.; Dubash, T.D.; Giessler, K.M.; Dieter, S.M.; Herbst, F.; Glimm, H.; Ball, C.R. The notch target gene HEYL modulates metastasis forming capacity of colorectal cancer patient-derived spheroid cells in vivo. BMC Cancer 2019, 19, 1181. [Google Scholar] [CrossRef] [PubMed]
- Cosi, I.; Pellecchia, A.; De Lorenzo, E.; Torre, E.; Sica, M.; Nesi, G.; Notaro, R.; De Angioletti, M. ETV4 promotes late development of prostatic intraepithelial neoplasia and cell proliferation through direct and p53-mediated downregulation of p21. J. Hematol. Oncol. 2020, 13, 112. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Lüscher, B.; Eisenman, R.N. Myc and Max associate in vivo. Genes Dev. 1992, 6, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Buettner, F.; Pratanwanich, N.; McCarthy, D.J.; Marioni, J.C.; Stegle, O. f-scLVM: Scalable and versatile factor analysis for single-cell RNA-seq. Genome Biol. 2017, 18, 212. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, X.; Weichert-Leahey, N.; Dong, Z.; Zhang, C.; Lopez, G.; Tao, T.; He, S.; Wood, A.C.; Oldridge, D.; et al. LMO1 Synergizes with MYCN to Promote Neuroblastoma Initiation and Metastasis. Cancer Cell 2017, 32, 310–323.e5. [Google Scholar] [CrossRef]
- Le Grand, M.; Mukha, A.; Püschel, J.; Valli, E.; Kamili, A.; Vittorio, O.; Dubrovska, A.; Kavallaris, M. Interplay between MycN and c-Myc regulates radioresistance and cancer stem cell phenotype in neuroblastoma upon glutamine deprivation. Theranostics 2020, 10, 6411–6429. [Google Scholar] [CrossRef]
- Thiele, C. Neuroblastoma Cell Lines. J. Hum. Cell Cult. 1998, 1, 21–53. [Google Scholar]
- Mercatelli, D.; Ray, F.; Giorgi, F.M. Pan-Cancer and Single-Cell Modeling of Genomic Alterations Through Gene Expression. Front. Genet. 2019, 10, 671. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | p-Value | Average Log Fold Change | Fraction of Expressing Cells in Cluster 1 | Fraction of Expressing Cells in Cluster 2 | Adjusted p-Value |
|---|---|---|---|---|---|
| RPSA | 1.30 × 10−149 | −0.92656 | 0.998 | 1 | 2.05 × 10−145 |
| RPL35A | 4.43 × 10−123 | 0.482077 | 1 | 1 | 7.00 × 10−119 |
| VCAN | 7.56 × 10−123 | −0.74714 | 0.268 | 0.962 | 1.19 × 10−118 |
| RPL15 | 3.18 × 10−116 | −0.59664 | 0.998 | 1 | 5.01 × 10−112 |
| RPL29 | 3.00 × 10−115 | −0.41375 | 1 | 1 | 4.73 × 10−111 |
| TMA7 | 5.28 × 10−111 | −0.53624 | 0.995 | 1 | 8.33 × 10−107 |
| SAMD11 | 2.40 × 10−108 | −0.72644 | 0.805 | 0.99 | 3.79 × 10−104 |
| RPL11 | 1.11 × 10−106 | −0.53941 | 1 | 1 | 1.75 × 10−102 |
| PPP1R14A | 8.54 × 10−105 | 0.899921 | 0.945 | 0.428 | 1.35 × 10−100 |
| MAGEA4 | 2.41 × 10−104 | 0.562087 | 0.899 | 0.333 | 3.80 × 10−100 |
| RPL32 | 4.36 × 10−102 | −0.43999 | 1 | 1 | 6.88 × 10−98 |
| SRM | 2.21 × 10−101 | −0.58422 | 0.986 | 1 | 3.49 × 10−97 |
| RPL22 | 1.89 × 10−97 | −0.4962 | 1 | 1 | 2.98 × 10−93 |
| CDKAL1 | 2.70 × 10−96 | −0.64301 | 0.412 | 0.933 | 4.27 × 10−92 |
| RPL14 | 2.26 × 10−95 | −0.52412 | 1 | 1 | 3.57 × 10−91 |
| RPL38 | 2.68 × 10−94 | 0.437493 | 1 | 1 | 4.23 × 10−90 |
| ENO1 | 6.25 × 10−90 | −0.53807 | 0.998 | 1 | 9.86 × 10−86 |
| RPLP0 | 1.50 × 10−89 | −0.30715 | 1 | 1 | 2.37 × 10−85 |
| TMEM98 | 1.62 × 10−85 | 0.543221 | 0.892 | 0.474 | 2.56 × 10−81 |
| RPL26L1 | 2.01 × 10−84 | 0.503347 | 0.984 | 0.95 | 3.17 × 10−80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercatelli, D.; Balboni, N.; Palma, A.; Aleo, E.; Sanna, P.P.; Perini, G.; Giorgi, F.M. Single-Cell Gene Network Analysis and Transcriptional Landscape of MYCN-Amplified Neuroblastoma Cell Lines. Biomolecules 2021, 11, 177. https://doi.org/10.3390/biom11020177
Mercatelli D, Balboni N, Palma A, Aleo E, Sanna PP, Perini G, Giorgi FM. Single-Cell Gene Network Analysis and Transcriptional Landscape of MYCN-Amplified Neuroblastoma Cell Lines. Biomolecules. 2021; 11(2):177. https://doi.org/10.3390/biom11020177
Chicago/Turabian StyleMercatelli, Daniele, Nicola Balboni, Alessandro Palma, Emanuela Aleo, Pietro Paolo Sanna, Giovanni Perini, and Federico Manuel Giorgi. 2021. "Single-Cell Gene Network Analysis and Transcriptional Landscape of MYCN-Amplified Neuroblastoma Cell Lines" Biomolecules 11, no. 2: 177. https://doi.org/10.3390/biom11020177
APA StyleMercatelli, D., Balboni, N., Palma, A., Aleo, E., Sanna, P. P., Perini, G., & Giorgi, F. M. (2021). Single-Cell Gene Network Analysis and Transcriptional Landscape of MYCN-Amplified Neuroblastoma Cell Lines. Biomolecules, 11(2), 177. https://doi.org/10.3390/biom11020177