
Insulin-Responsive Transcription Factors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Insulin and the Insulin Receptor
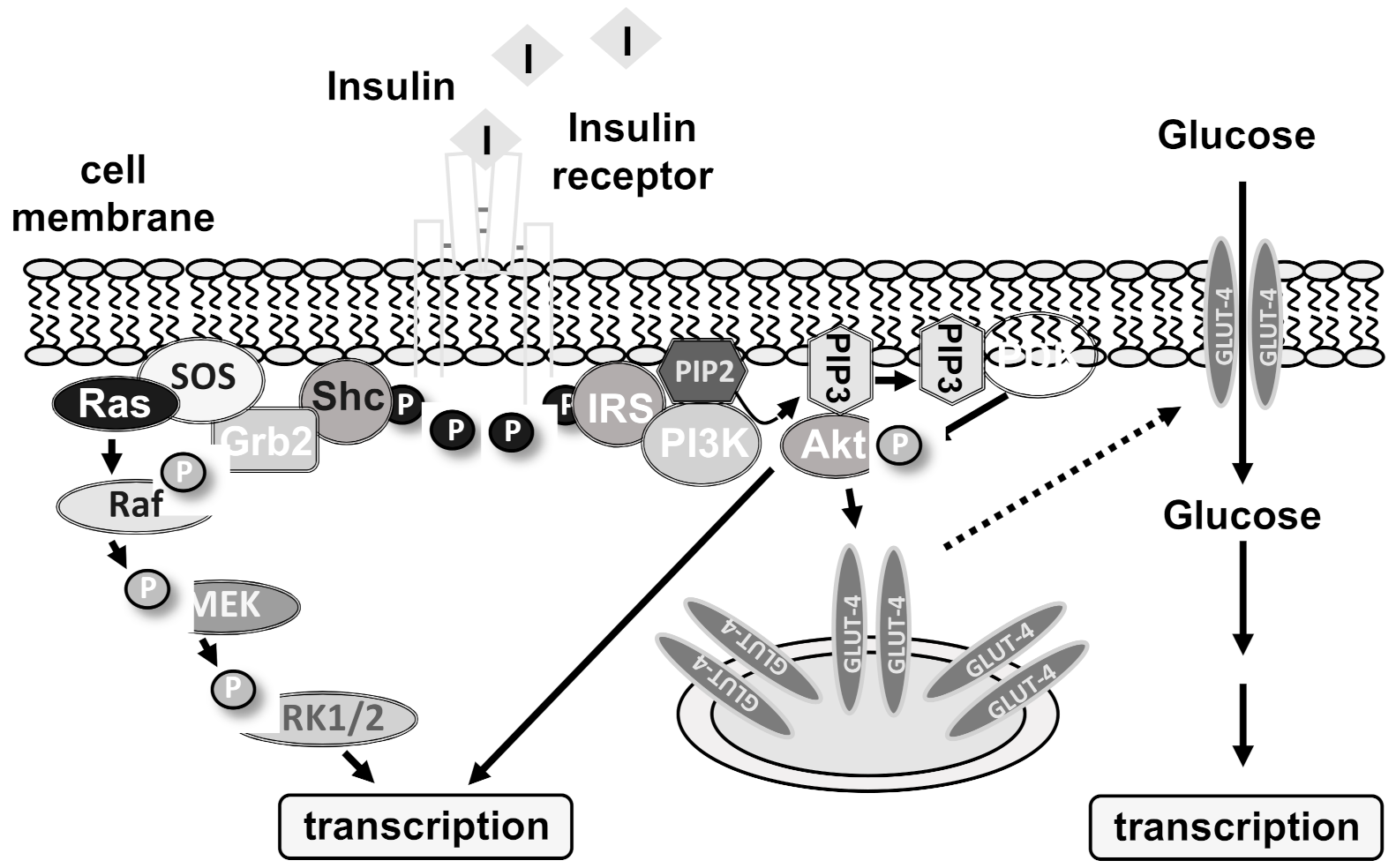
2. Insulin Receptor-Induced Intracellular Signaling Cascades
3. Transcription Factors under Control of Insulin Receptor Signaling
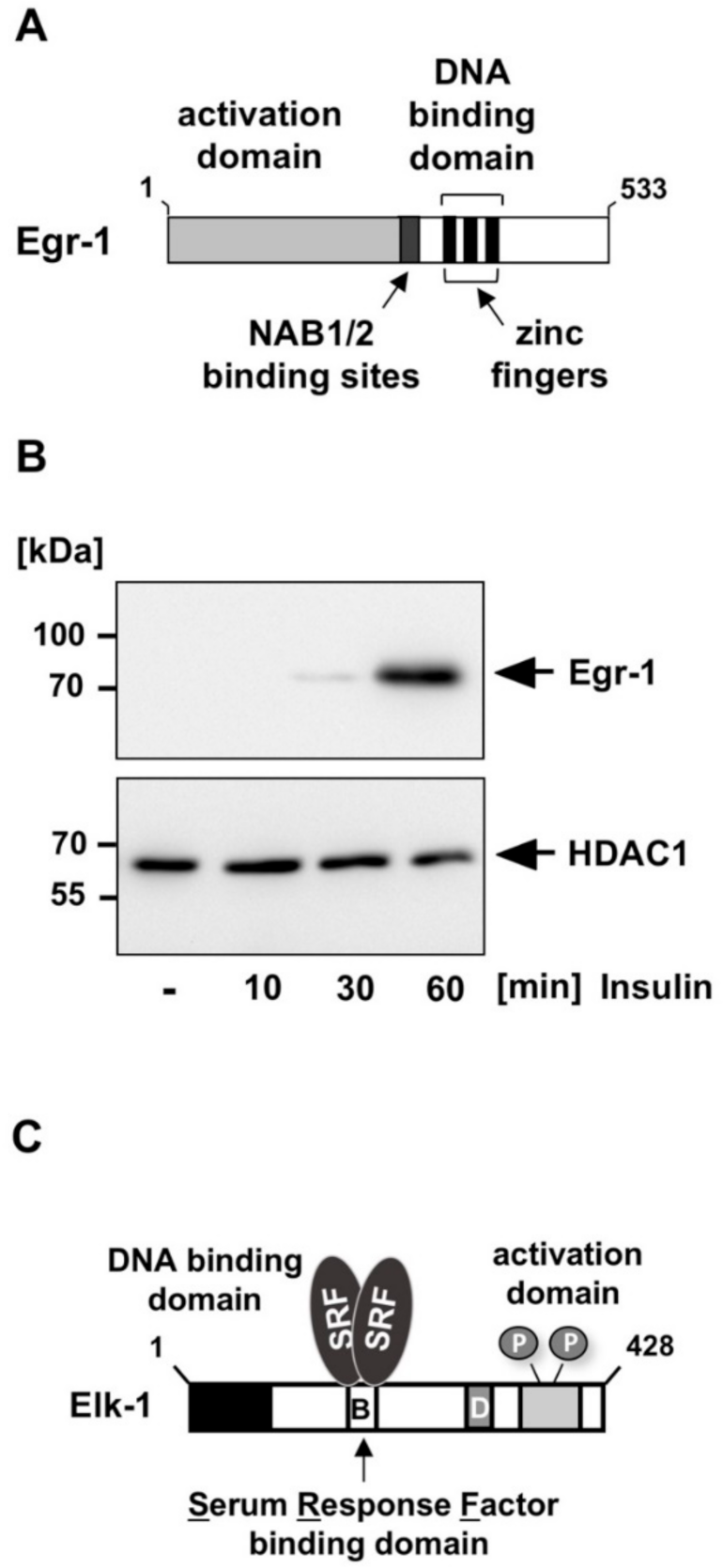
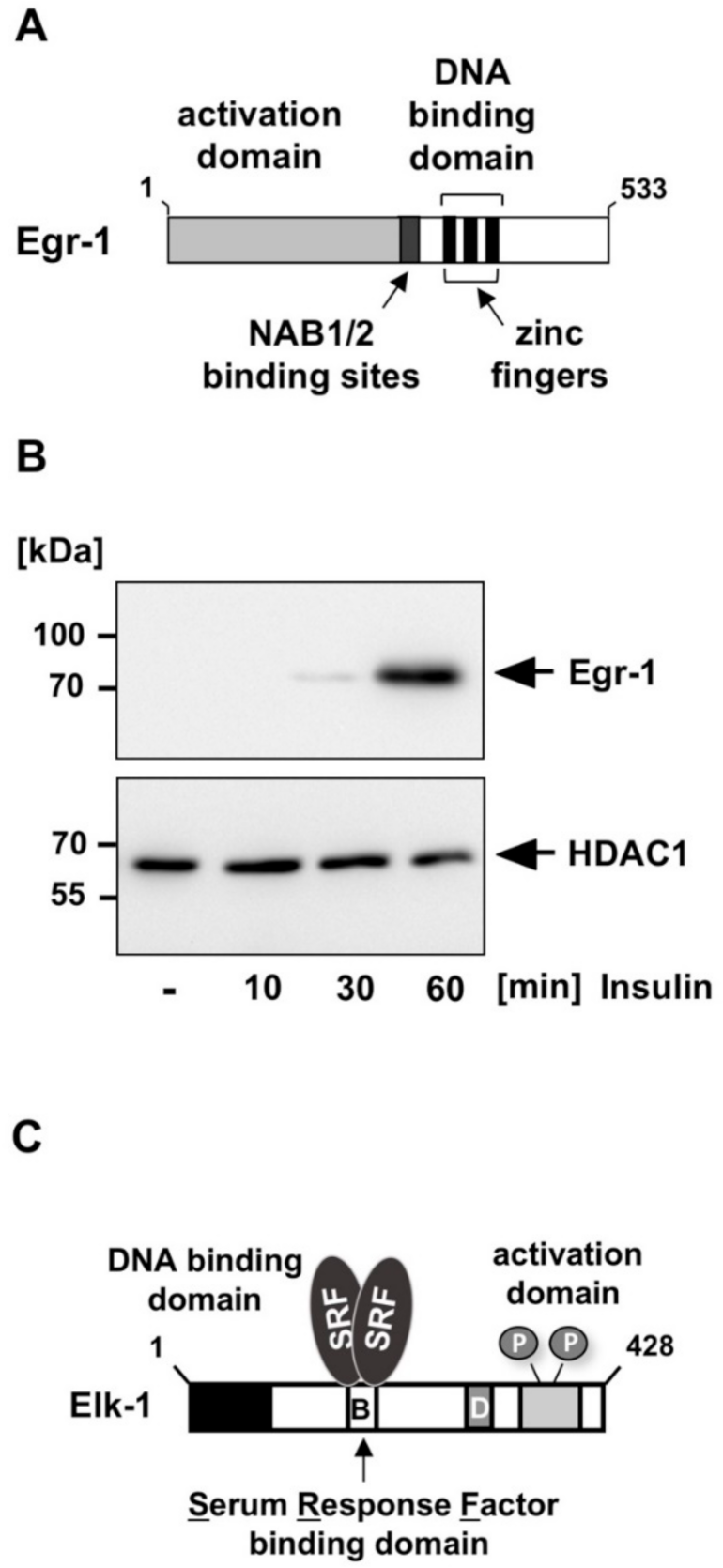
3.1. The Elk-1-Egr-1 Axis
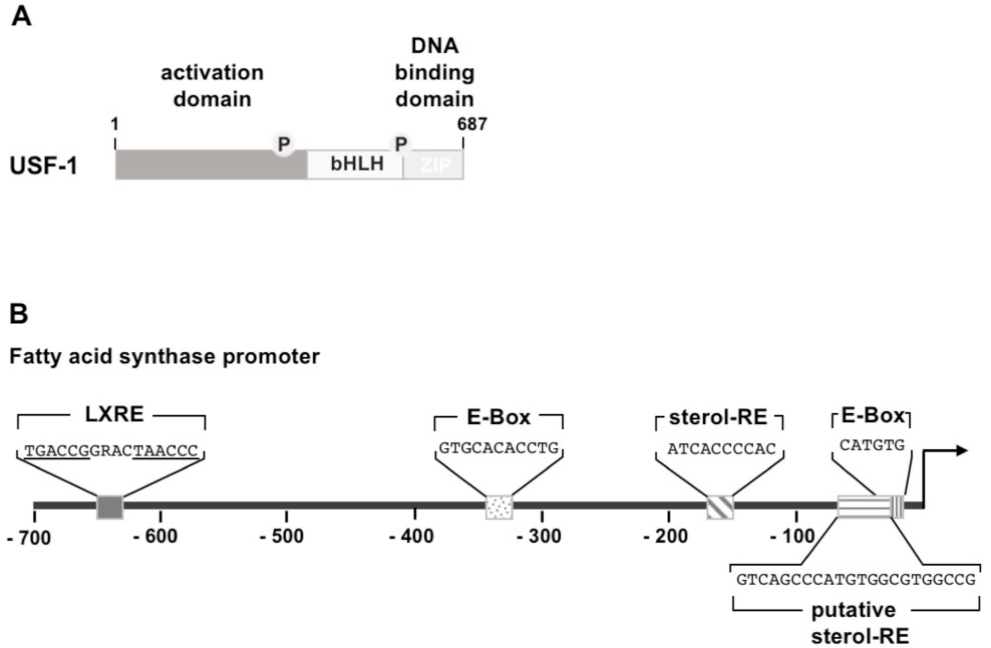
3.2. Upstream Stimulatory Factor (USF)
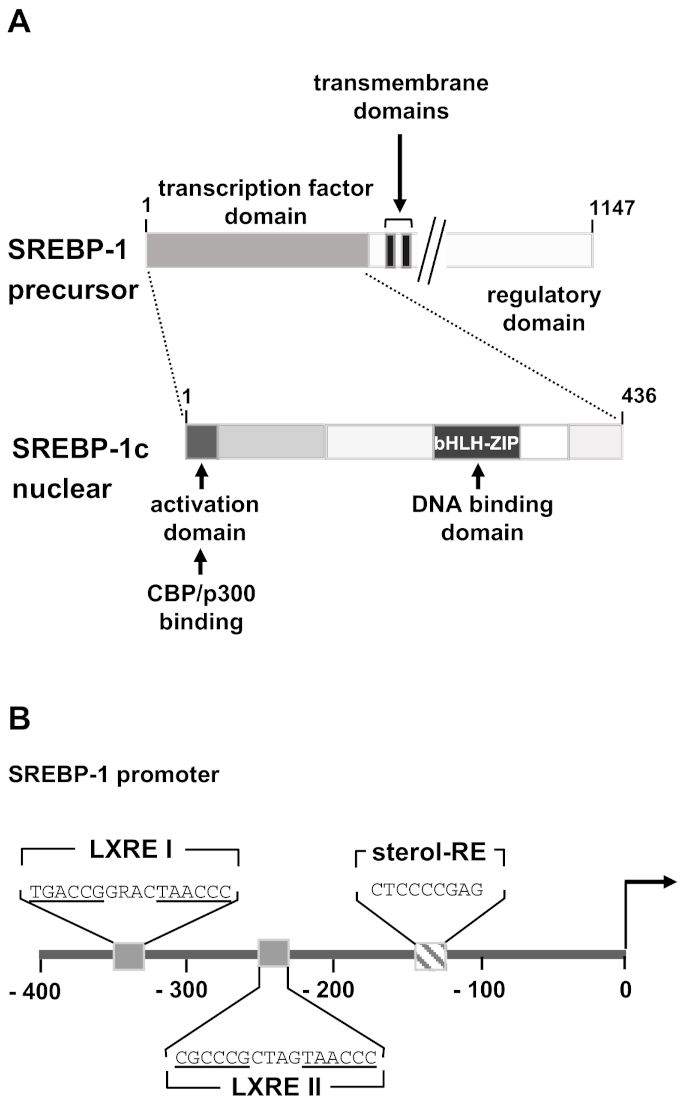
3.3. Sterol Regulatory Element-Binding Protein-1c (SREBP-1c)
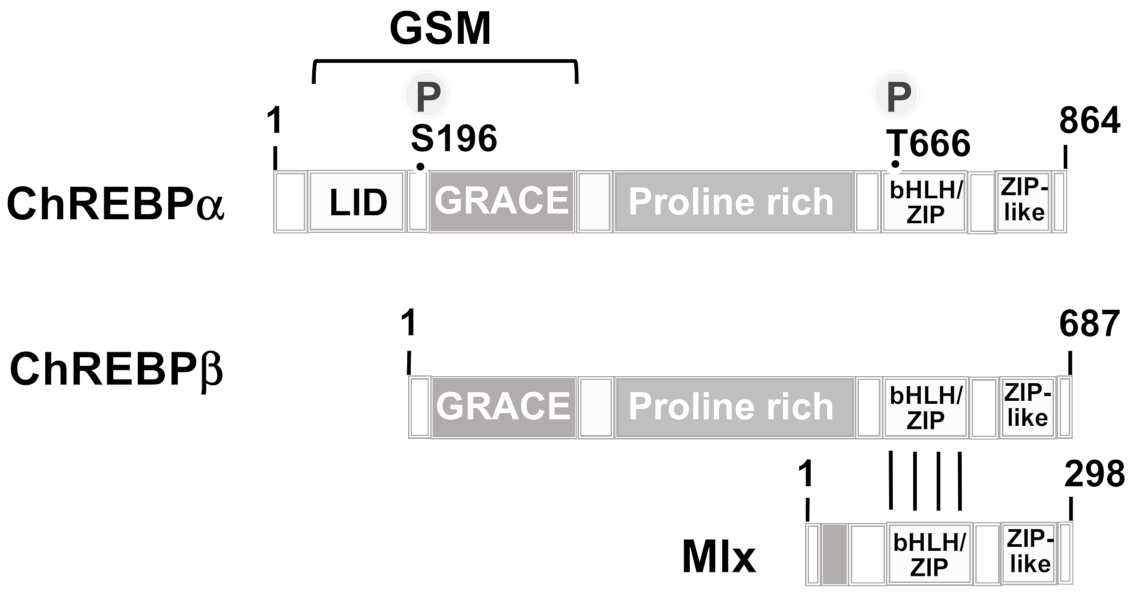
3.4. Carbohydrate Response Element-Binding Protein (ChREBP)
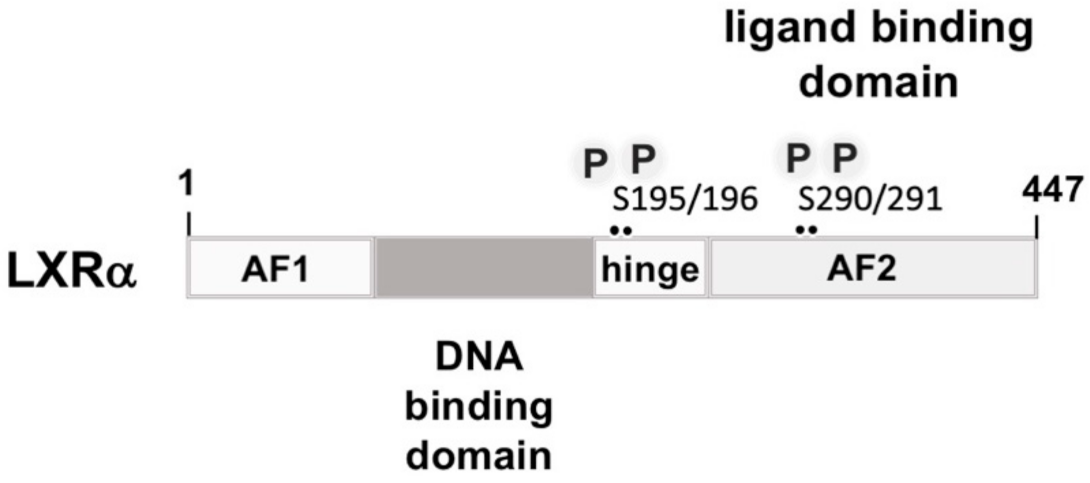
3.5. Liver X Receptor
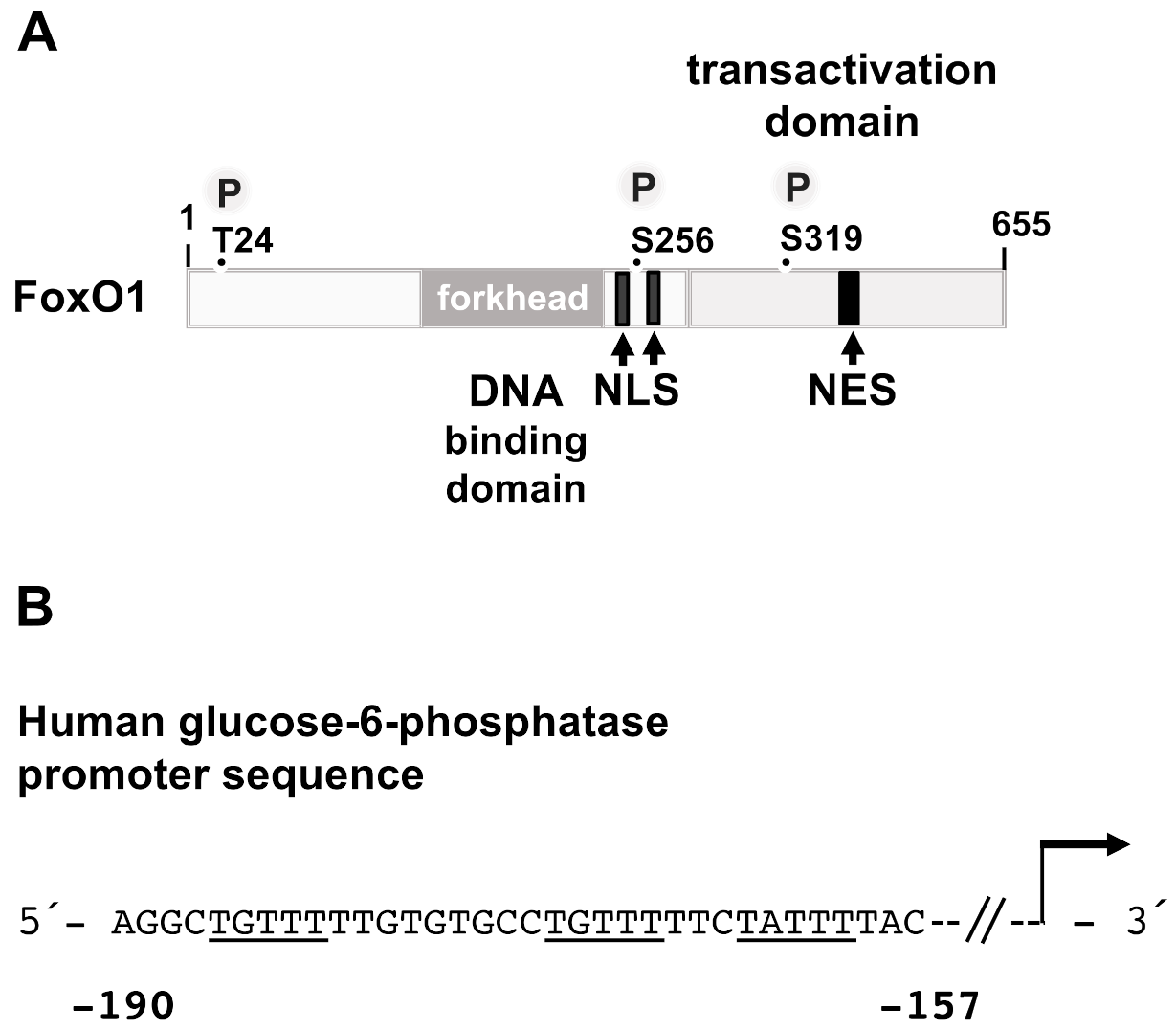
3.6. FoxO1
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell. Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Kullmann, S.; Kleinridders, A.; Small, D.M.; Fritsche, A.; Häring, H.-H.; Preissl, H.; Heni, M. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020, 8, 524–534. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Jhun, B.H.; Haruta, T.; Meinkoth, J.L.; Leitner, J.W.; Draznin, B.; Saltiel, A.R.; Pang, L.; Sasaoka, T.; Olefsky, J.M. Signal transduction pathways leading to insulin-induced early gene induction. Biochemistry 1995, 34, 7996–8004. [Google Scholar] [CrossRef]
- Harada, S.; Smith, R.M.; Smith, J.A.; White, M.F.; Jarett, L. Insulin-induced egr-1 and c-fos expression in 32D cells requires insulin receptor, Shc, and mitogen-activated protein kinase, but not insulin receptor substrate-1 and phosphatidylinositol 3-kinase activation. J. Biol. Chem. 1996, 271, 30222–30226. [Google Scholar] [CrossRef] [Green Version]
- Mohtar, O.; Ozdemir, C.; Roy, D.; Shantaram, D.; Emili, A.; Kandror, K.V. Egr1 mediates the effect of insulin on leptin transcription in adipocytes. J. Biol. Chem. 2019, 294, 5784–5789. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Wagner, L.; Ulrich, M.; Rössler, O.G. Immediate-early transcriptional response to insulin receptor stimulation. Biochem. Pharmacol. 2021, 192, 114696. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.; Thiel, G. Epidermal growth factor and thrombin induced proliferation of immortalized human keratinocytes is coupled to the synthesis of Egr-1, a zinc finger transcriptional regulator. J. Cell. Biochem. 2002, 85, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Rössler, O.G.; Thiel, G. Brain-derived neurotrophic factor, epidermal growth factor, or A-Raf induced growth of HaCaT keratinocytes requires extracellular signal-regulated kinase. Am. J. Physiol. Cell Physiol. 2004, 286, C1118–C1129. [Google Scholar] [CrossRef]
- Mayer, S.I.; Rössler, O.G.; Endo, T.; Charnay, P.; Thiel, G. Epidermal growth factor-induced proliferation of astrocytes requires Egr transcription factors. J. Cell Sci. 2009, 122, 3340–3350. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.; Bach, K.; Thiel, G. Extracellular signal-regulated protein kinases Erk1/Erk2 stimulate expression and biological activity of the transcriptional regulator Egr-1. Biol. Chem. 2001, 382, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Hohl, M.; Al-Sarraj, A.; Vinson, C.; Thiel, G. Transcriptional activation of the Egr-1 gene mediated by tetradecanoylphorbol acetate and extracellular signal-regulated protein kinase. Arch. Biochem. Biophys. 2005, 438, 36–52. [Google Scholar] [CrossRef]
- Liao, Y.; Shikapwashya, O.N.; Shteyer, E.; Dieckgraefe, B.K.; Hruz, P.W.; Rudnick, D.A. Delayed hepatocellular mitotic progression and impaired liver regeneration in early growth response-1-deficient mice. J. Biol. Chem. 2004, 279, 43107–43116. [Google Scholar] [CrossRef] [Green Version]
- Müller, I.; Rössler, O.G.; Wittig, C.; Menger, M.D.; Thiel, G. Critical role of Egr transcription factors in regulating insulin biosynthesis, blood glucose homeostasis and islet size. Endocrinology 2012, 153, 3040–3053. [Google Scholar] [CrossRef] [Green Version]
- Abdelaziz, R.B.; Chehida, A.B.; Azzouz, H.; Boudabbous, H.; Lascols, O.; Turkia, H.B.; Tebib, N. A novel homozygous missense mutation in the insulin receptor gene results in an atypical presentation of Rabson-Mendenhall syndrome. Eur. J. Med. Genet. 2016, 59, 16–19. [Google Scholar] [CrossRef]
- Mayer, S.I.; Thiel, G. Calcium influx into MIN6 insulinoma cells induces expression of Egr-1 involving extracellular signal-regulated protein kinase and the transcription factors Elk-1 and CREB. Eur. J. Cell Biol. 2009, 88, 19–33. [Google Scholar] [CrossRef]
- Mayer, S.I.; Müller, I.; Mannebach, S.; Endo, T.; Thiel, G. Signal transduction of pregnenolone sulfate in insulinoma cells. Activation of Egr-1 expression involving TRPM3, voltage-gated calcium channels, ERK, and ternary complex factors. J. Biol. Chem. 2011, 286, 10084–10096. [Google Scholar] [CrossRef] [Green Version]
- Thiel, G.; Backes, T.M.; Guethlein, L.A.; Rössler, O.G. Critical protein-protein interactions determine the biological activity of Elk-1, a master regulator of stimulus-induced gene transcription. Molecules 2021, 26, 6125. [Google Scholar] [CrossRef]
- Müller, I.; Endo, T.; Thiel, G. Regulation of AP-1 activity in glucose-stimulated insulinoma cells. J. Cell. Biochem. 2010, 110, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Keim, A.; Thiel, G. Regulation of immediate-early gene transcription following activation of Gaq-coupled designer receptors. J. Cell. Biochem. 2013, 114, 681–696. [Google Scholar] [CrossRef]
- Lesch, A.; Hui, X.; Lipp, P.; Thiel, G. Transient receptor potential melastatin-3 (TRPM3)-induced activation of AP-1 requires Ca2+ ions and the transcription factors c-Jun, ATF2, and ternary complex factor. Mol. Pharmacol. 2015, 87, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, G.; Lesch, A.; Keim, A. Transcriptional response to calcium-sensing receptor stimulation. Endocrinology 2012, 153, 4716–4728. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rössler, O.G. Resveratrol stimulates AP-1-regulated gene transcription. Mol. Nutr. Food Res. 2014, 58, 1402–1413. [Google Scholar] [CrossRef]
- Krook, A.; Whitehead, J.P.; Dobson, S.P.; Griffiths, M.R.; Ouwens, M.; Baker, C.; Hayward, A.C.; Sen, S.K.; Maassen, J.A.; Siddle, K.; et al. Two naturally occurring insulin receptor tyrosine kinase domain mutants provide evidence that phosphoinositide 3-kinase activation alone is not sufficient for the mediation of insulin´s metabolic and mitogenic effects. J. Biol. Chem. 1997, 272, 30208–30214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesch, A.; Backes, T.M.; Langfermann, D.S.; Rössler, O.G.; Laschke, M.W.; Thiel, G. Ternary complex factor regulates pancreatic islet size and blood glucose homeostasis in transgenic mice. Pharmacol. Res. 2020, 159, 104983. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Ricci, R.; Kenner, L.; Zenz, R.; David, J.P.; Rath, M.; Wagner, E.F. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell 2003, 112, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Backes, T.M.; Langfermann, D.S.; Lesch, A.; Rössler, O.G.; Laschke, M.W.; Vinson, C.; Thiel, G. Regulation and function of AP-1 in insulinoma cells and pancreatic b-cells. Biochem. Pharmacol. 2021, 193, 114748. [Google Scholar] [CrossRef]
- Wang, W.; Huang, L.; Huang, Y.; Yin, J.-W.; Berk, A.J.; Friedman, J.M.; Wang, G. Mediator MED23 links insulin signaling to the adipogenesis transcription cascade. Dev. Cell 2009, 16, 764–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré-D´Amaré, A.; Pognonec, P.; Roeder, R.G.; Burley, S.K. Structure and function of the b/HLH/Z domain of USF. EMBO J. 1994, 13, 180–189. [Google Scholar] [CrossRef]
- Griffin, M.J.; Wong, R.H.F.; Pandya, N.; Sul, H.S. Direct interaction between USF and SREBP-1c mediates synergistic activation of the fatty-acid synthase promoter. J. Biol. Chem. 2007, 282, 5453–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latasa, M.J.; Griffin, M.J.; Moon, Y.S.; Kang, C.; Sul, H.S. Occupancy and function of the -150 sterol regulatory element and -65 E-box in nutritional regulation of the fatty acid synthase gene in living animals. Mol. Cell. Biol. 2003, 23, 5896–5907. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.H.F.; Sul, H.S. Insulin signaling in fatty acid and fat synthesis: A transcriptional perspective. Curr. Opin. Pharmacol. 2010, 10, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Casado, M.; Vallet, V.S.; Kahn, A.; Vaulont, S. Essential role in vivo of upstream stimulatory factors for a normal dietary response of the fatty acid synthase gene in the liver. J. Biol. Chem. 1999, 274, 2009–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.H.F.; Chang, I.; Hudak, C.S.S.; Hyun, S.; Kwan, H.-K.; Sul, H.S. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 2009, 136, 1056–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Viscarra, J.; Kim, S.-J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, K.A.R.; Ulven, S.M.; Schuster, G.U.; Steineger, H.; Andresen, S.M.; Gustafsson, J.-A.; Nebb, H.I. Liver X receptors as insulin-mediating factors in fatty acid and cholesterol biosynthesis. J. Biol. Chem. 2002, 277, 10691–10697. [Google Scholar] [CrossRef] [Green Version]
- Dorotea, D.; Koya, D.; Ha, H. Recent insights into SREBP as a direct mediator of kidney fibrosis via lipid-independent pathways. Front. Pharmacol. 2020, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Pacot, C.; Dugail, I.; Lemarchand, P.; Guichard, C.; le Lièpvre, X.; Befrthelier-Lubrano, C.; Spiegelman, B.; Ferré, P.; Foufelle, F. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol. Cell. Biol. 1999, 19, 3760–3768. [Google Scholar] [CrossRef] [Green Version]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef]
- Liang, G.; Yang, J.; Horton, J.D.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 2002, 277, 9520–9528. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Guichard, C.; Ferré, P.; Foufelle, F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc. Natl. Acad. Sci. USA 1999, 96, 12737–12742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Shyy, J.Y.-J. Sterol regulatory element-binding protein 1 is negatively modulated by PKA phosphorylation. Am. J. Physiol Cell Physiol. 2006, 290, C1477–C1486. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Shimango, H.; Inoue, N.; Nakagawa, Y.; Matsuzaka, T.; Takahashi, A.; Yahagi, N.; Sone, H.; Suzuki, H.; Toyoshima, H.; et al. Protein kinase A suppresses sterol regulatory element-binding protein -1C expression via phosphorylation of liver X receptor in the liver. J. Biol. Chem. 2007, 282, 11687–11695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengoechea-Alonso, M.T.; Ericsson, J. A phosphorylation cascade controls the degradation of active SREBP1. J. Biol. Chem. 2009, 284, 5885–5895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponugoti, B.; Kim, D.-H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.-Y.; Chiang, C.-M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1a and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234. [Google Scholar] [CrossRef]
- Azzout-Marniche, D.; Bécard, D.; Guichard, C.; Foretz, M.; Ferré, P.; Foufelle, F. Insulin effects on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytes. Biochem. J. 2000, 350, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, B.D.; Bobard, A.; Hainault, I.; Bossard, P.; Foufelle, F. Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element-binding protein-1c. Proc. Natl. Acad. Sci. USA 2005, 102, 791–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, T.; Shimano, H.; Amemiya-Kuso, M.; Yahagi, N.; Hasty, A.H.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Ilzuka, Y.; Ohashi, K.; et al. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol. Cell Biol. 2001, 21, 2991–3000. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, K.; Wu, S.-Y.; Chiang, C.-M.; Samols, D.; Hanson, R.W. SREBP-1c and Sp1 interact to regulate transcription of the gene for phosphoenolpyruvate carboxykinase (GTP) in the liver. J. Biol. Chem. 2004, 279, 15385–15395. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1a interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Klip, A.; McGraw, T.E.; James, D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postic, C.; Dentin, R.; Denechaud, P.-D.; Girard, J. ChREBP, a transcriptional regulator of glucose and lipid mechanism. Annu. Rev. Nutr. 2007, 27, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel insights into ChREBP regulation and function. Trends Endocrinol. Metab. 2013, 24, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Baraille, F.; Planchais, J.; Dentin, R.; Guilmeau, S.; Postic, C. Integration of ChREBP-mediated glucose sensing into whole body metabolism. Physiology (Bethesda) 2015, 30, 428–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Wahed, A.; Guilmeau, S.; Postic, C. Sweet Sixteenth for ChREBP: Established roles and future goals. Cell Metab. 2017, 26, 324–341. [Google Scholar] [CrossRef]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP·Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Tsatsos, N.G.; Towle, H.C. Direct role of ChREBP·Mlx in regulating hepatic glucose-responsive genes. J. Biol. Chem. 2005, 280, 12019–12027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.-M.; Liu, Z.; Towle, H.C. Two CACGTG motifs with proper spacing dictate the carbohydrate regulation of hepatic gene transcription. J. Biol. Chem. 1995, 270, 21991–21997. [Google Scholar] [CrossRef] [Green Version]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sae-Lee, C.; Moolsuwan, K.; Chan, L.; Poungvarin, N. ChREBP regulates itself and metabolic genes implicated in lipid accumulation in b-cell line. PLoS ONE 2016, 11, e0147411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Ge, Q.; Pawlosky, R.; Wynn, R.M.; Veech, R.L.; Uyeda, K. Metabolite regulation of nucleo-cytosolic trafficking of carbohydrate response element-binding protein (ChREBP). Role of ketone bodies. J. Biol. Chem. 2013, 288, 28358–28367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.-R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.T.; et al. Metabolite regulation of nuclear localization of carbohydrate-response element-binding protein (ChREBP). Role of AMP as an allosteric inhibitor. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Lizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef] [Green Version]
- Rufo, C.; Teran-Garcia, M.; Nakamura, M.T.; Koo, S.-H.; Towle, H.C.; Clarke, S.D. Involvement of a unique carbohydrate-responsive factor in the glucose regulation of rat liver fatty-acid synthase gene. J. Biol. Chem. 2001, 276, 21969–21975. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response-element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.-S.; Kim, D.; Lee, Y.S.; Kim, H.-J.; Han, J.-Y.; Im, S.-S.; Chong, H.K.; Kwon, Y.-H.; Cho, Y.-H.; Kim, W.K.; et al. Integrated expression profiling and genome-wide analysis of ChREBP targets reveals the dual role of ChREBP in glucose-regulated gene expression. PLoS ONE 2011, 6, e22544. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Aryal, P.; Wen, J.; Syed, I.; Vazirani, R.P.; Moraes-Vieira, P.M.; Camporez, J.P.; Gallop, M.R.; Perry, R.J.; Peroni, O.D.; et al. Absence of carbohydrate response element binding protein in adipocytes causes systemic insulin resistance and impairs glucose transport. Cell Rep. 2017, 21, 1021–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poungvarin, N.; Lee, J.K.; Yechoor, V.K.; Li, M.V.; Assavapokee, T.; Suksaranjit, P.; Thepsongwajja, J.J.; Saha, P.K.; Oka, K.; Chan, L. Carbohydrate response element-binding protein (ChREBP) plays a pivotal role in beta cell glucotoxicity. Diabetologia 2012, 55, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tontonoz, P. Liver X receptor in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Peet, D.J.; Turlay, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.-M.A.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXR and LXR. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, J.-Y.; Repa, J.J. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Barthel, A.; Schmoll, D.; Unterman, T.G. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metabol. 2005, 16, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, D.; Walker, K.S.; Alessi, D.R.; Grempler, R.; Burchell, A.; Guo, S.; Walther, R.; Unterman, T.G. Regulation of glucose-6-phosphatase gene expression by protein kinase B and the forkhead transcription factor FKHR. J. Biol. Chem. 2000, 275, 36324–36333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, J.; Kitamura, T.; Silver, D.L.; Accili, D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Investig. 2001, 108, 1359–1367. [Google Scholar] [CrossRef]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; DePinho, R.A.; Accili, D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metabol. 2007, 6, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altomonte, J.; Richter, A.; Harbaran, S.; Suriawinata, J.; Nakae, J.; Thung, S.N.; Meseck, M.; Accili, D.; Dong, H. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E718–E728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bu, S.Y.; Mashek, M.T.; O-Sullivan, I.; Sibai, Z.; Khan, S.A.; Ilkayeva, O.; Newgard, C.B.; Mashek, D.G.; Unterman, T.G. Integrated regulation of hepatic lipid and glucose metabolism by adipose triacylglycerol lipase and FoxO proteins. Cell Rep. 2016, 15, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Zhang, W.; Patil, S.; Chauhan, B.; Guo, S.; Powell, D.R.; Le, J.; Klotsas, A.; Matika, R.; Xiao, X.; Franks, R.; et al. FoxO1 regulates multiple pathways in the liver. Effects on gluconeogenic, glycolytic, and lipogenic gene expression. J. Biol. Chem. 2006, 281, 10105–10117. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, S.J.; Azimifar, S.B.; Mann, M. High-thoughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotech. 2015, 33, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Daitoko, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of Foxo1 alters its DNA-binding activity and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar] [CrossRef] [Green Version]
- Banks, A.S.; Kim-Muller, J.Y.; Mastracci, T.L.; Kofler, N.M.; Qiang, L.; Haeusler, R.A.; Jurczak, M.J.; Laznik, D.; Heinrich, G.; Samuel, V.T.; et al. Dissociation of the glucose and lipid regulatory functions of FoxO1 by targeted knockin of acetylation-defective alleles in mice. Cell Metabol. 2011, 14, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Nakae, J.; Cao, Y.; Daitoku, H.; Fukamizu, A.; Ogawa, W.; Yano, Y.; Hayashi, Y. The LXXLL motif of murine forkhead transcription factor FoxO1 mediates SIRT1-dependent transcriptional activity. J. Clin. Investig. 2006, 116, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, P.; English, T.; Karki, S.; Qiang, L.; Tao, R.; Kim, J.; Luo, Z.; Farmer, S.R.; Kandror, K.V. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J. Lipid Res. 2011, 52, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Qiao, A.; Ke, Y.; Kong, X.; Liang, J.; Wang, R.; Ouyang, X.; Zuo, J.; Chang, Y.; Fang, F. FoxO1 represses LXRa-mediated transcriptional activity of SREBP-1c promoter in HepG2 cells. FEBS Lett. 2010, 584, 4330–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Zhang, W.; O-Sullivan, I.; Williams, J.B.; Dong, Q.; Park, E.A.; Raghow, R.; Unterman, T.G.; Elam, M.B. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factor Sp1 and SREBP-1c. J. Biol. Chem. 2012, 287, 20132–20143. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiel, G.; Guethlein, L.A.; Rössler, O.G. Insulin-Responsive Transcription Factors. Biomolecules 2021, 11, 1886. https://doi.org/10.3390/biom11121886
Thiel G, Guethlein LA, Rössler OG. Insulin-Responsive Transcription Factors. Biomolecules. 2021; 11(12):1886. https://doi.org/10.3390/biom11121886
Chicago/Turabian StyleThiel, Gerald, Lisbeth A. Guethlein, and Oliver G. Rössler. 2021. "Insulin-Responsive Transcription Factors" Biomolecules 11, no. 12: 1886. https://doi.org/10.3390/biom11121886
APA StyleThiel, G., Guethlein, L. A., & Rössler, O. G. (2021). Insulin-Responsive Transcription Factors. Biomolecules, 11(12), 1886. https://doi.org/10.3390/biom11121886