
The Effects of Angiotensin II or Angiotensin 1-7 on Rat Pial Microcirculation during Hypoperfusion and Reperfusion Injury: Role of Redox Stress

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Treatments
2.3. Surgical Animal Preparation
2.4. Pial Microvascular Observation
2.5. ROS Production Detection
2.6. Statistical Analysis
3. Results
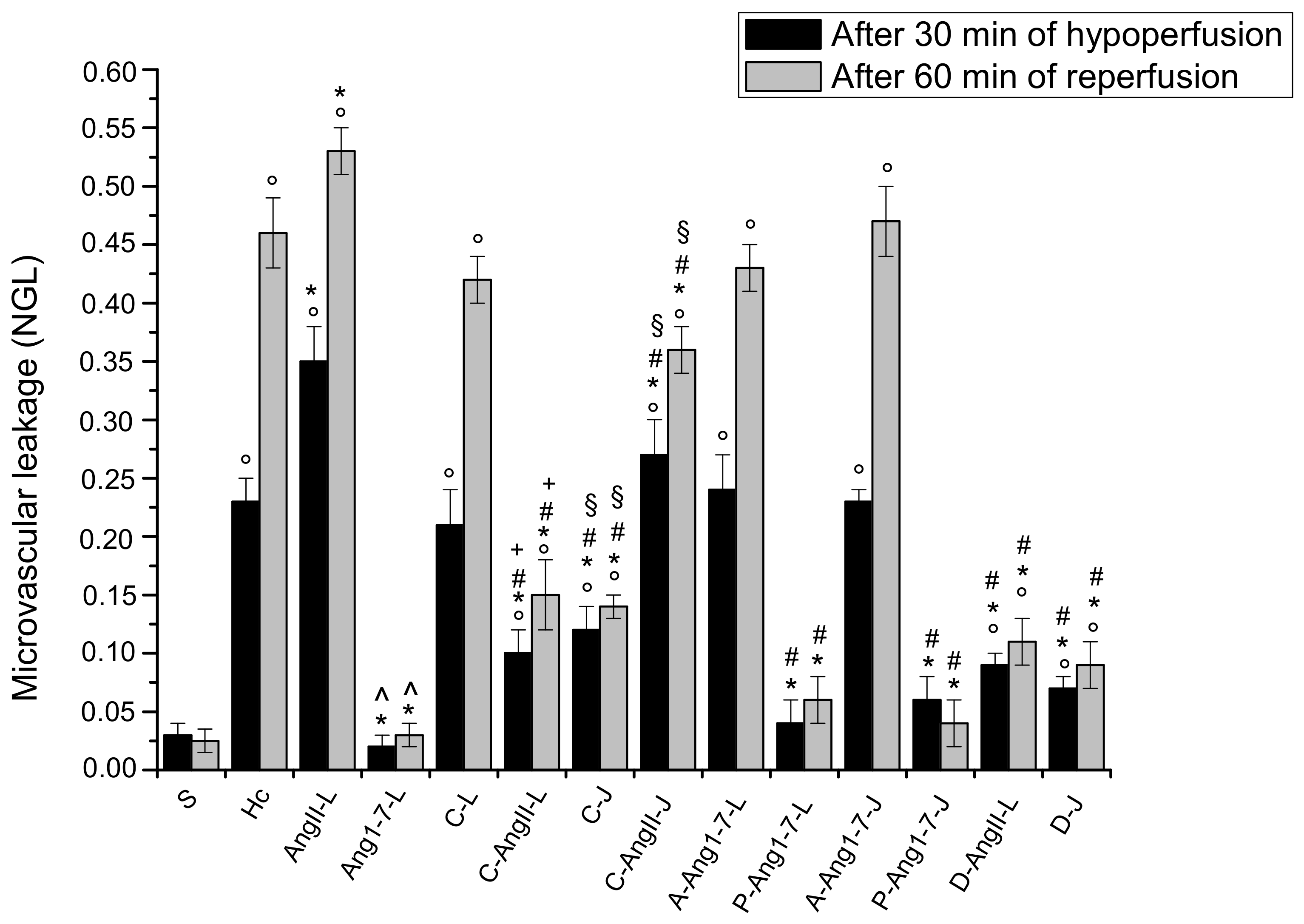
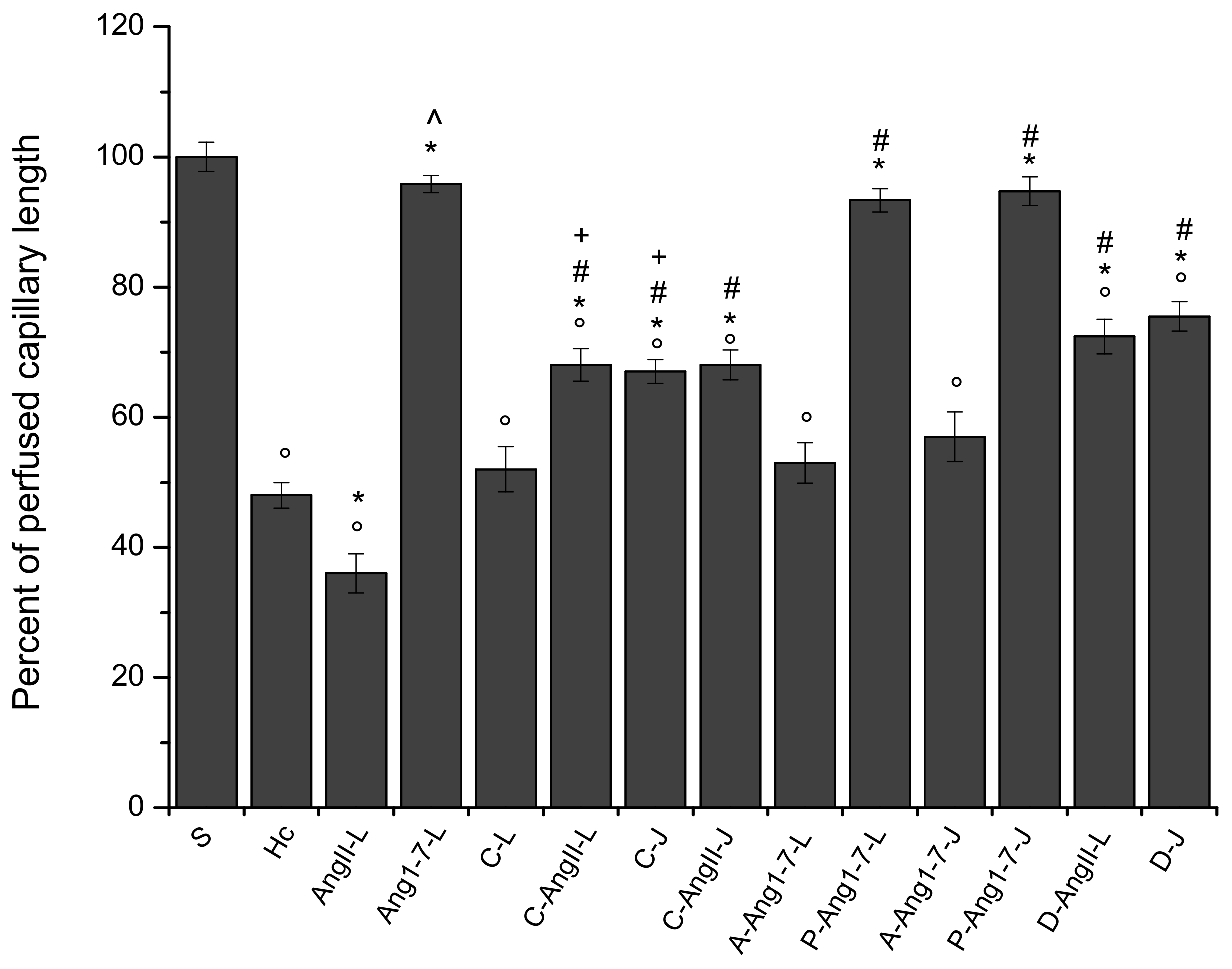
3.1. The Hypoperfusion and the Subsequent Reperfusion Caused Significant Microvascular Alterations
3.2. Local Application of Ang II Exacerbated Microvascular Damage Due to Hypoperfusion and Reperfusion
3.3. Local Application of Ang 1-7 Prevented Microvascular Damage Due to Hypoperfusion and Reperfusion
3.4. Jugular Vein Infusion of Ang II or Ang 1-7 Achieved Similar Results to Those Triggered by Local Application
3.5. Local Application of Candesartan Cilexetil Alone Did Not Significantly Reduce the Microvascular Damage Due to Hypoperfusion and Reperfusion
3.6. Local Application of Candesartan Cilexetil Prior to Local Angiotensin II Prevented Ang II Microvascular Damage Exacerbation
3.7. Jugular Infusion of Candesartan Cilexetil Protected the Pial Microcirculation from Damage Due to Hypoperfusion and Reperfusion
3.8. Jugular Infusion of Candesartan Cilexetil, Prior to Intravenous Angiotensin II, Partially Protected the Pial Microvasculature from the Damage Induced by Hypoperfusion and Reperfusion
3.9. Local Application of Mas Receptor Antagonist (A779) Prior to Local Ang 1-7 Abolished the Effects Induced by Ang 1-7
3.10. Local Application of AT2R Antagonist (PD123319) Prior to Local Ang 1-7 Did Not Change the Effects Induced by Ang 1-7
3.11. Jugular Infusion of A779 or PD123319 Prior to Intravenous Ang 1-7 Caused the Same Effects Observed with the Local Applications
3.12. Local Application of Diminazene Aceturate Prior to Ang II Prevented Ang II-Induced Microvascular Damage Exacerbation
3.13. Jugular Administration of Diminazene Aceturate Prevented the Microvascular Damage Due to Hypoperfusion and Reperfusion
3.14. No differences between Sexes Were Observed in Terms of Microvascular Damage Induced by Hypoperfusion–Reperfusion
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, M.; Mao, Y.; Ramirez, S.H.; Tuma, R.F.; Chabrashvili, T. Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience 2010, 171, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Panahpour, H.; Nekooeian, A.A.; Dehghani, G.A. Candesartan attenuates ischemic brain edema and protects the blood-brain barrier integrity from ischemia/reperfusion injury in rats. Iran Biomed. J. 2014, 18, 232–2238. [Google Scholar] [CrossRef]
- Unger, T. Inhibiting renin-angiotensin in the brain: The possible therapeutic implications. Blood Press 2001, 10, 12–16. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin converting enzyme2 (ACE2), SARSCoV2 and pathophysiology of coronavirus disease 2019 (COVID 19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256–294. [Google Scholar] [CrossRef] [PubMed]
- South, A.M.; Brady, T.M.; Flynn, J.T. ACE2, COVID-19, and ACE1 Inhibitor and ARB Use during the Pandemic: The Pediatric Perspective. Hypertension 2020, 76, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Gerbino, A.; Lionetti, V.; Miragoli, M.; Munaron, L.M.; Pagliaro, P.; Pasqua, T.; Penna, C.; Rocca, C.; Samaja, M.; et al. COVID-19-associated cardiovascular morbidity in older adults: A position paper from the Italian Society of Cardiovascular Research. GeroScience 2020, 42, 1021–1049. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Engelhorn, T.; Goerike, S.; Doerfler, A.; Okorn, C.; Forsting, M.; Heusch, G.; Schulz, R. The Angiotensin II Type 1–Receptor Blocker Candesartan Increases Cerebral Blood Flow, Reduces Infarct Size, and Improves Neurologic Outcome After Transient Cerebral Ischemia in Rats. J. Cerebral Blood Flow Metab. 2004, 24, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.H.; Dhaunsi, G.S.; Makki, B.M.; Qabazard, B.A.; Akhtar, S.; Benter, I.F. Characterization of Angiotensin-(1-7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacol. Res. 2012, 66, 269–275. [Google Scholar] [CrossRef]
- Benter, I.F.; Yousif, M.H.; Cojocel, C.; Al-Maghrebi, M.; Diz, D.I. Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H666–H672. [Google Scholar] [CrossRef]
- Gradman, A.H. AT (1)-receptor blockers: Differences that matter. J. Hum. Hypertens. 2000, 16, S9–S16. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Premilovac, D.; Attrill, E.; Rattigan, S.; Richards, S.M.; Kim, J.; Keske, M.A. Acute, local infusion of angiotensin II impairs microvascular and metabolic insulin sensitivity in skeletal muscle. Cardiovasc. Res. 2019, 115, 590–601. [Google Scholar] [CrossRef]
- Rukavina Mikusic, N.L.; Pineda, A.M.; Gironacci, M.M. Angiotensin-(1-7) and Mas receptor in the brain. Explor. Med. 2021, 2, 268–293. [Google Scholar] [CrossRef]
- Gomes, G.N.; Aires, M.M. Interaction of atrial natriuretic factor and angiotensin II in proximal HCO3-reabsorption. Am. J. Physiol. 1992, 262, F303–F308. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.J.; Shih, C.C.; Chang, K.Y.; Liao, M.H.; Liaw, W.J.; Wu, C.C.; Tsao, C.M. Angiotensin-(1-7) treatment blocks lipopolysaccharide-induced organ damage, platelet dysfunction, and IL-6 and nitric oxide production in rats. Sci. Rep. 2021, 11, 610. [Google Scholar] [CrossRef]
- Kohara, K.; Mikami, H.; Okuda, N.; Higaki, J.; Ogihara, T. Angiotensin blockade and the progression of renal damage in the spontaneously hypertensive rat. Hypertension 1993, 21, 975–979. [Google Scholar] [CrossRef]
- Sartório, C.L.; Pimentel, E.B.; Dos Santos, R.L.; Rouver, W.N.; Mill, J.G. Acute hypotensive effect of diminazene aceturate in spontaneously hypertensive rats: Role of NO and Mas receptor. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1723–1730. [Google Scholar] [CrossRef]
- Inada, Y.; Naka, T. Candesartan cilexetil: Pharmacological properties and protective effects against organ damage of a novel nonpeptide angiotensin II-receptor antagonist. Nihon Yakurigaku Zasshi 2002, 115, 151–160. [Google Scholar] [CrossRef][Green Version]
- Fabiani, M.E.; Dinh, D.T.; Nassis, L.; Casley, D.J.; Johnston, C.I. In vivo inhibition of angiotensin receptors in the rat kidney by candesartan cilexetil: A comparison with losartan. J. Hum. Hypertens. 1999, 13, S75–S80. [Google Scholar] [CrossRef]
- Watanabe, S. In vivo fluorometric measurement of cerebral oxidative stress using 2′-7′-dichlorofluorescein (DCF). Keio. J. Med. 1998, 47, 92–98. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gonçalves Maia, L.; Caldeira Ramos, M.; Fernandes, L.; Catelli de Carvalho, M.H.; Campagnole-Santos, M.J.; Souza dos Santos, R.A. Angiotensin-(1-7) antagonist A-779 attenuates the potentiation of bradykinin by captopril in rats. J. Cardiovasc. Pharmacol. 2004, 43, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Pezeshki, Z.; Nematbakhsh, M. Sex Differences in the Renal Vascular Responses of AT1 and Mas Receptors in Two-Kidney-One-Clip Hypertension. Int. J. Hypertens. 2021, 2021, 8820646. [Google Scholar] [CrossRef] [PubMed]
- Lyer, S.N.; Chappel, M.C.; Brosnihan, K.B.; Ferrario, C.M. Role of AT1 and AT2 receptors in the plasma clearance of angiotensin II. J. Cardiovasc. Pharmacol. 1998, 31, 464–469. [Google Scholar] [CrossRef]
- Reinecke, K.; Lucius, R.; Reinecke, A.; Rickert, U.; Herdegen, T.; Unger, T. Angiotensin II accelerates functional recovery in the rat sciatic nerve in vivo: Role of the AT2 receptor and the transcription factor NF-kappaB. FASEB J. 2003, 17, 2094–2096. [Google Scholar] [CrossRef]
- Lapi, D.; Vagnani, S.; Pignataro, G.; Esposito, E.; Paterni, M.; Colantuoni, A. Rat Pial Microvascular Responses to Transient Bilateral Common Carotid Artery Occlusion and Reperfusion: Quercetin’s Mechanism of Action. Front. Physiol. 2012, 27, 99. [Google Scholar] [CrossRef]
- Lapi, D.; Mastantuono, T.; Sapio, D.; Paterni, M.; Colantuoni, A. Pial microvascular responses induced by transient bilateral common carotid artery occlusion in Zucker rats. Clin. Hemorheol. Microcirc. 2013, 54, 415–429. [Google Scholar] [CrossRef]
- Mastantuono, T.; Starita, N.; Sapio, D.; D’Avanzo, S.A.; Di Maro, M.; Muscariello, E.; Paterni, M.; Colantuoni, A.; Lapi, D. The Effects of Vaccinium myrtillus Extract on Hamster Pial Microcirculation during Hypoperfusion-Reperfusion Injury. PLoS ONE 2016, 11, e0150659. [Google Scholar] [CrossRef]
- Lapi, D.; Marchiafava, P.L.; Colantuoni, A. Geometric characteristics of arterial network of rat pial microcirculation. J. Vasc. Res. 2008, 45, 69–77. [Google Scholar] [CrossRef]
- Lapi, D.; Sabatino, L.; Altobelli, G.G.; Mondola, P.; Cimini, V.; Colantuoni, A. Effects of propionyl-L-carnitine on ischemia-reperfusion injury in hamster cheek pouch microcirculation. Front. Physiol. 2010, 19, 132. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Nishimura, Y.; Ito, T.; Saavedra, J.M. Angiotensin II AT1 Blockade Normalizes Cerebrovascular Autoregulation and Reduces Cerebral Ischemia in Spontaneously Hypertensive Rats. Stroke 2000, 31, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- St. Paul, A.; Corbett, C.B.; Okune, R.; Autieri, M.V. Angiotensin II, Hypercholesterolemia, and Vascular Smooth Muscle Cells: A Perfect Trio for Vascular Pathology. Int. J. Mol. Sci. 2020, 21, 4525. [Google Scholar] [CrossRef]
- Jie, L.J.; Jiang, T.; Wu, L.; Gao, L.; Wang, Y.; Zhou, F.; Zhang, S.; Zhang, Y. The expression of angiotensin-converting enzyme 2-angiotensin-(1-7)-Mas receptor axis are upregulated after acute cerebral ischemic stroke in rats. Neuropeptides 2013, 47, 289–295. [Google Scholar] [CrossRef]
- Arroja, M.M.C.; Reid, E.; Roy, L.A.; Vallatos, A.V.; Holmes, W.M.; Nicklin, S.A.; Work, L.M.; McCabe, C. Assessing the effects of Ang-(1-7) therapy following transient middle cerebral artery occlusion. Sci. Rep. 2019, 9, 3154. [Google Scholar] [CrossRef]
- Arroja, M.M.C.; Emma Reid, M.; McCabe, C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp. Trans. Stroke Med. 2016, 8, 8. [Google Scholar] [CrossRef]
- Murphy, S.J.; McCullough, L.D.; Smith, J.M. Stroke in the Female: Role of Biological Sex and Estrogen. ILAR J. 2004, 45, 147–159. [Google Scholar] [CrossRef]
- Pagliaro, P.; Penna, C. ACE/ACE2 Ratio: A Key Also in 2019 Coronavirus Disease (Covid-19)? Front. Med. 2020, 7, 335. [Google Scholar] [CrossRef]
- Uhlig, S.; Yang, Y.; Waade, J.; Wittenberg, C.; Babendreyer, A.; Kuebler, W.M. Differential regulation of lung endothelial permeability in vitro and in situ. Cell Physiol. Biochem. 2014, 34, 1–19. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, G.Y. LPS induces permeability injury in lung microvascular endothelium via AT (1) receptor. Arch. Biochem. Biophys. 2005, 441, 75–83. [Google Scholar] [CrossRef]
- Sasaki, K.; Taniguchi, M.; Miyoshi, M.; Goto, O.; Sato, K.; Watanabe, T. Are transcription factors NF-B and AP-1 involved in the ANG II-stimulated production of proinflammatory cytokines induced by LPS in dehydrated rats? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1599–R1608. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sharma, D.; Li, G.; Zhao, Y. Effect of Angiotensin II Type 1 Receptor Antagonist, Losartan on Inflammatory Factor in Atherosclerotic Rabbits. Res. Cardiovasc. Med. 2013, 2, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Gao, L.; Lu, J.; Zhang, Y.D. ACE2-Ang-(1-7)-Mas Axis in Brain: A Potential Target for Prevention and Treatment of Ischemic Stroke. Curr. Neuropharmacol. 2013, 11, 209–217. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Zhang, Q.Q.; Tan, M.S.; Cao, L.; Wang, H.F.; Lu, J.; Gao, Q.; Zhang, Y.; et al. Angiotensin-(1-7) induces cerebral ischaemic tolerance by promoting brain angiogenesis in a Mas/eNOS-dependent pathway. Br. J. Pharmacol. 2014, 171, 4222–4232. [Google Scholar] [CrossRef]
- Mecca, A.P.; Regenhardt, R.W.; O’Connor, T.E.; Joseph, J.P.; Raizada, M.; Katovich, M.J.; Sumners, C. Cerebroprotection by angiotensin-(1-7) in endothelin-1-induced ischaemic stroke. Exp. Physiol. 2011, 96, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Leisman, D.E.; Deutschman, C.; Legrand, M. Facing COVID-19 in the ICU; vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med. 2020, 28, 1105–1108. [Google Scholar] [CrossRef]
- Maryam Malek, M.; Nematbakhsh, M. The Preventive Effects of Diminazene Aceturate in Renal Ischemia/Reperfusion Injury in Male and Female Rats. Adv. Prev. Med. 2014, 2014, 740647. [Google Scholar] [CrossRef]
- Boegehold, M.A.; Drenjancevic, I.; Lombard, J.H. Salt, Angiotensin II, Superoxide, and Endothelial Function. Compr. Physiol. 2015, 15, 215–254. [Google Scholar] [CrossRef]
- Waldner, M.J.; Baethmann, A.; Uhl, E.; Lehmberg, J. Bradykinin-induced leukocyte-and platelet-endothelium interactions in the cerebral microcirculation. Brain Res. 2012, 1448, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Goru, K.S.; Kadakol, A.; Malek, V.; Pandey, A.; Sharma, N.; Gaikwad, A.B. Diminazene aceturate prevents nephropathy by increasing glomerular ACE2 and AT2 receptor expression in a rat model of type 1 diabetes. Br. J. Pharmacol. 2017, 174, 3118–3130. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, J.; Cole-jeffrey, C.T.; Shenoy, V.; Espejo, A.; Hanna, M.; Song, C.; Pepine, C.J.; Katovich, M.J.; Raizada, M.K. Diminazene aceturate enhances angiotensin-converting enzyme 2 activity and attenuates ischemia-induced cardiac pathophysiology. Hypertension 2013, 62, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Oudit, Z.; Kassiri, C.; Jiang, P.P.; Liu, S.M.; Poutanen, J.M.; Penninger, J.; Butany, G.Y. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Lanza, K.; Perez, L.G.; Costa, L.B.; Cordeiro, T.M.; Palmeira, V.A.; Ribeiro, V.T.; Silva, A.C.S. Covid-19: The renin-angiotensin system imbalance hypothesis. Clin. Sci. 2020, 134, 1259–1264. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GROUPS | n | Sex | HYPOPERFUSION and REPERFUSION | TREATMENTS | |
|---|---|---|---|---|---|
| Local Application | Intravenous Administration | ||||
| S | 5 | M | no | no | no |
| Hc | 11 | M | yes | no | no |
| AngII-L | 11 | M | yes | Angiotensin II | no |
| Ang 1-7-L | 11 | M | yes | Angiotensin 1-7 | no |
| C-L | 11 | M | yes | Candersartan cilexetil | no |
| C-AngII-L | 11 | M | yes | Candersartan cilexetil plus Angiotensin II | no |
| A-Ang 1-7-L | 11 | M | yes | A779 plus Angiotensin 1-7 | no |
| P-Ang 1-7-L | 11 | M | yes | P123319 plus Angiotensin 1-7 | no |
| D-L | 11 | M | yes | Diminazene aceturate | no |
| D-AngII-L | 11 | M | yes | Diminazene aceturate plus Angiotensin II | no |
| AngII-J | 11 | M | yes | no | Angiotensin II |
| Ang 1-7-J | 11 | M | yes | no | Angiotensin 1-7 |
| C-J | 11 | M | yes | no | Candersartan cilexetil |
| C-AngII-J | 11 | M | yes | no | Candersartan cilexetil plus Angiotensin II |
| A-Ang 1-7-J | 11 | M | yes | no | A779 plus Angiotensin 1-7 |
| P-Ang 1-7 | 11 | M | yes | no | P123319 plus Angiotensin 1-7 |
| D-J | 11 | M | yes | no | Diminazene aceturate |
| D-AngII-J | 11 | M | yes | no | Diminazene aceturate plus Angiotensin II |
| AngII-LF | 11 | F | yes | Angiotensin II | no |
| Ang 1-7-LF | 11 | F | yes | Angiotensin 1-7 | no |
| AngII-JF | 11 | F | yes | no | Angiotensin II |
| Ang 1-7-JF | 11 | F | yes | no | Angiotensin 1-7 |
| ORDER | ARTERIOLES (n) | DIAMETER (μm) | LENGTH (μm) | RAT (n) |
|---|---|---|---|---|
| 5 | 35 | 72.4 ± 4.5 * | 987 ± 22 | 11 |
| 4 | 40 | 42.5 ± 3.3 * | 763 ± 31 | 11 |
| 3 | 105 | 31.8 ± 2.0 * | 1034 ±27 | 11 |
| 2 | 94 | 24.3 ± 1.2 * | 880 ± 35 | 11 |
| 1 | 58 | 15.7 ± 0.7 * | 310 ± 23 | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lapi, D.; Cammalleri, M.; Dal Monte, M.; Di Maro, M.; Santillo, M.; Belfiore, A.; Nasti, G.; Damiano, S.; Trio, R.; Chiurazzi, M.; et al. The Effects of Angiotensin II or Angiotensin 1-7 on Rat Pial Microcirculation during Hypoperfusion and Reperfusion Injury: Role of Redox Stress. Biomolecules 2021, 11, 1861. https://doi.org/10.3390/biom11121861
Lapi D, Cammalleri M, Dal Monte M, Di Maro M, Santillo M, Belfiore A, Nasti G, Damiano S, Trio R, Chiurazzi M, et al. The Effects of Angiotensin II or Angiotensin 1-7 on Rat Pial Microcirculation during Hypoperfusion and Reperfusion Injury: Role of Redox Stress. Biomolecules. 2021; 11(12):1861. https://doi.org/10.3390/biom11121861
Chicago/Turabian StyleLapi, Dominga, Maurizio Cammalleri, Massimo Dal Monte, Martina Di Maro, Mariarosaria Santillo, Anna Belfiore, Gilda Nasti, Simona Damiano, Rossella Trio, Martina Chiurazzi, and et al. 2021. "The Effects of Angiotensin II or Angiotensin 1-7 on Rat Pial Microcirculation during Hypoperfusion and Reperfusion Injury: Role of Redox Stress" Biomolecules 11, no. 12: 1861. https://doi.org/10.3390/biom11121861
APA StyleLapi, D., Cammalleri, M., Dal Monte, M., Di Maro, M., Santillo, M., Belfiore, A., Nasti, G., Damiano, S., Trio, R., Chiurazzi, M., De Conno, B., Serao, N., Mondola, P., Colantuoni, A., & Guida, B. (2021). The Effects of Angiotensin II or Angiotensin 1-7 on Rat Pial Microcirculation during Hypoperfusion and Reperfusion Injury: Role of Redox Stress. Biomolecules, 11(12), 1861. https://doi.org/10.3390/biom11121861