
The Pathophysiology and Treatment of Essential Tremor: The Role of Adenosine and Dopamine Receptors in Animal Models


Abstract
1. Introduction
2. Essential Tremor (ET)
2.1. Epidemiology
2.2. ET Etiology
2.2.1. Genetic Background
2.2.2. Environmental Factors
2.3. The Pathophysiology of ET
2.3.1. The Neurodegenerative Hypothesis
2.3.2. The Central Oscillatory Network Hypothesis
2.3.3. The GABAergic Hypothesis
2.4. The Current Treatment of ET
2.4.1. Pharmacological Therapy
2.4.2. Neurosurgical Methods
2.5. Animal Models of ET
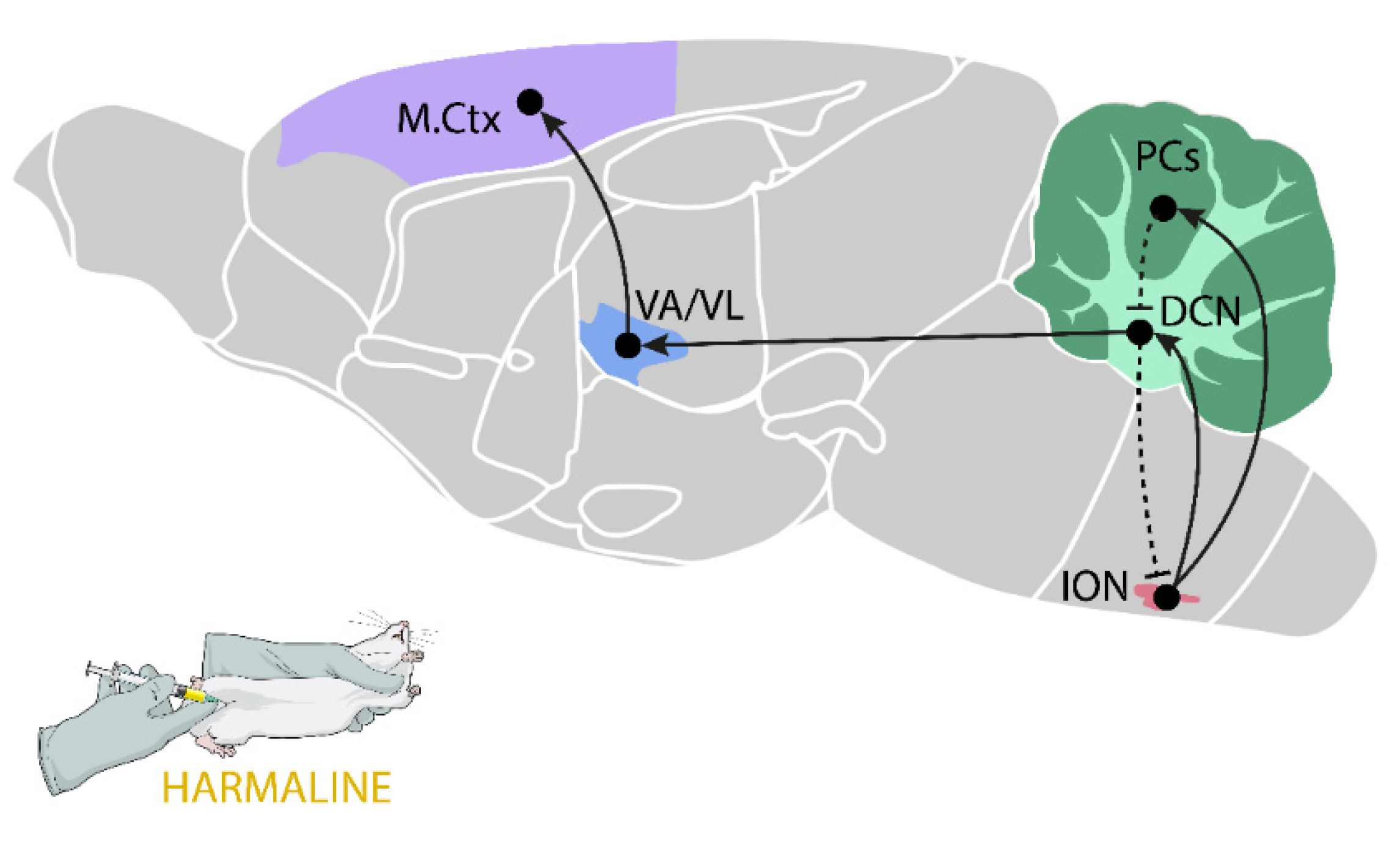
2.5.1. Harmaline-Induced Tremor
2.5.2. Genetic Models
3. The Dopaminergic System and Dopamine Receptors—The Role in ET
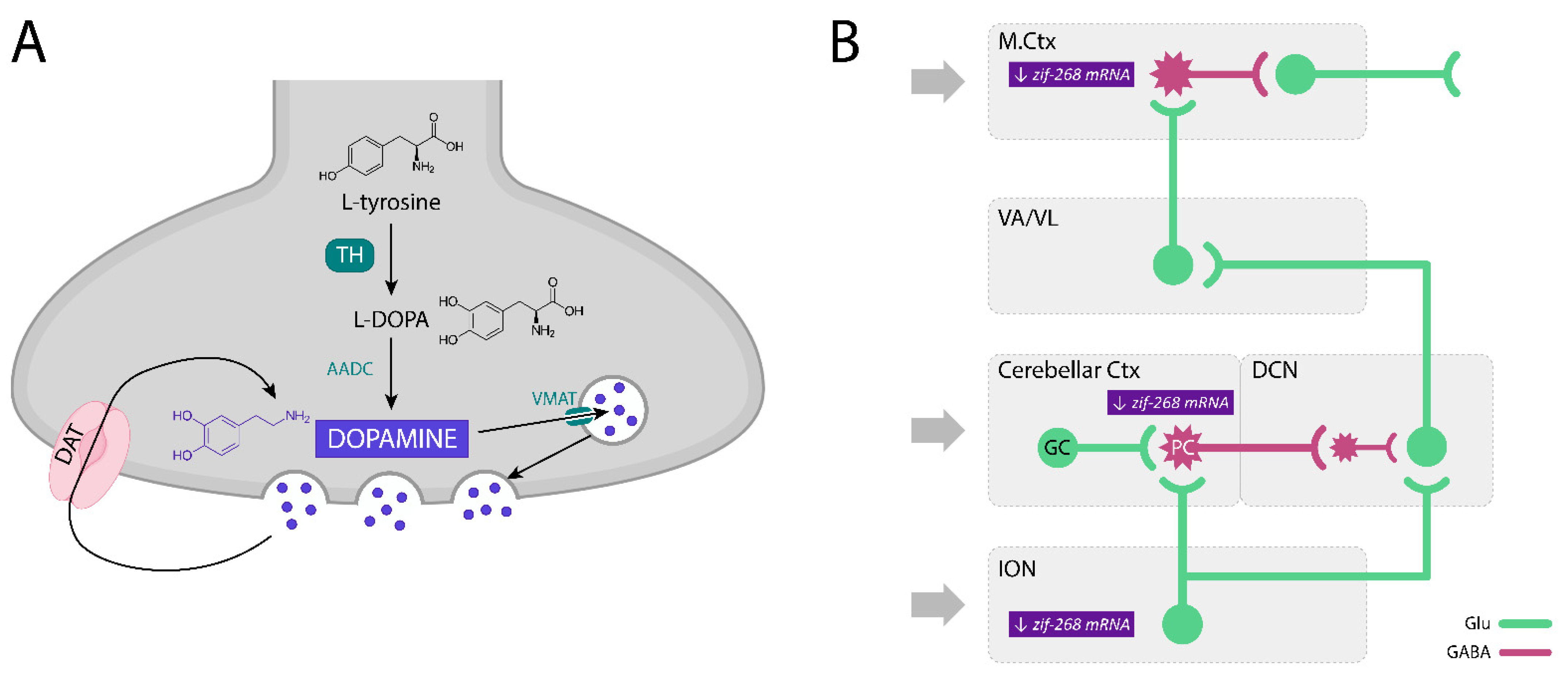
3.1. The Participation of Dopamine and Its Receptors in the Modulation of ET and Harmaline-Induced Tremor
3.2. Involvement of Dopamine D3 Receptors in the Harmaline-Induced Tremor
4. Adenosine and Its Receptors—The Role in ET
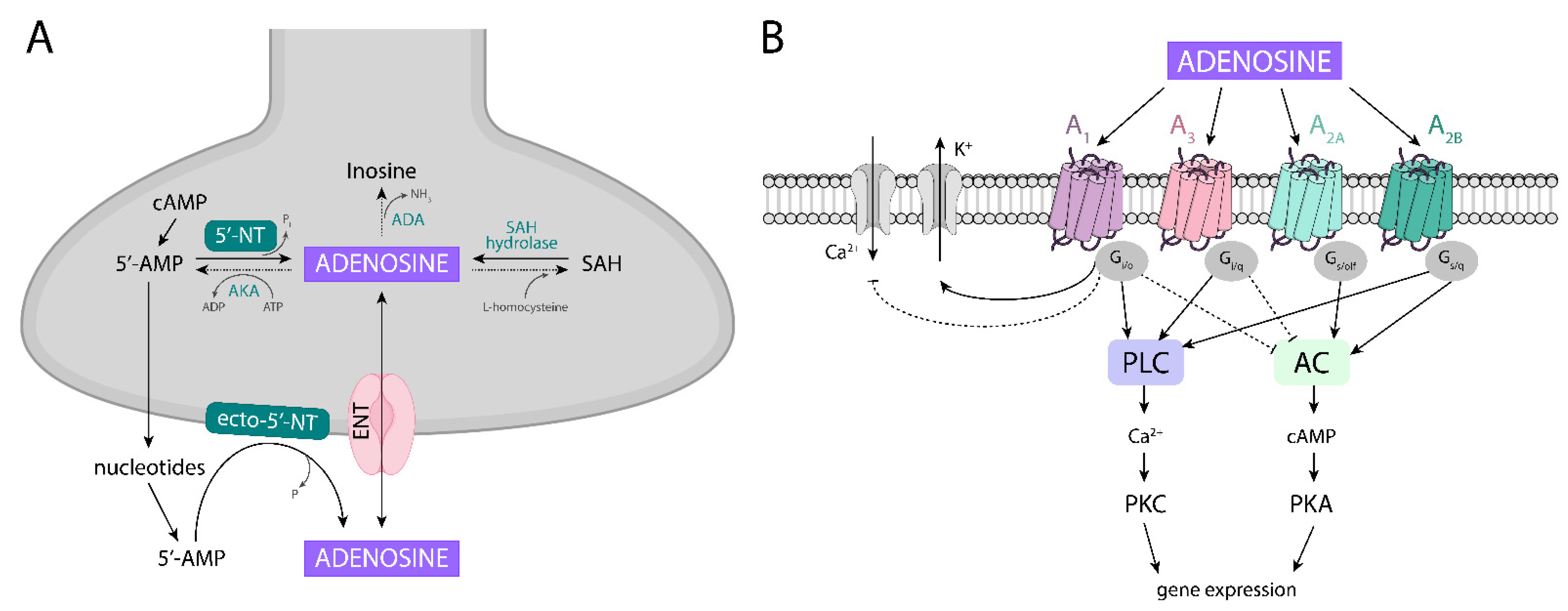
4.1. Adenosine Metabolism
4.2. Adenosine Receptors, Their Localization, and Homomeric and Heteromeric Complexes
4.3. The Involvement of Adenosine and Its Receptors in Tremor Modulation
4.4. Adenosine A1 Receptors and Their Role in Harmaline-Induced Tremor
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatia, K.P.; Bain, P.; Bajaj, N.; Elble, R.J.; Hallett, M.; Louis, E.D.; Raethjen, J.; Stamelou, M.; Testa, C.M.; Deuschl, G.; et al. Consensus Statement on the classification of tremors, from the task force on tremor of the International Parkinson and Movement Disorder Society. Mov. Disord. 2017, 33, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Jankovic, J.; Hallett, M. Tremors. In Principles and Practice of Movement Disorders; Elsevier Inc.: Amsterdam, The Netherlands, 2011; pp. 389–414. ISBN 9781437723694. [Google Scholar]
- Hass, P.L. Differentiation and Diagnosis of Jaundice. AACN Adv. Crit. Care 1999, 10, 433–441. [Google Scholar] [CrossRef]
- Hallett, M. Classification and treatment of tremor. JAMA-J. Am. Med. Assoc. 1991, 266, 1115–1117. [Google Scholar] [CrossRef]
- Deuschl, G.; Krack, P.; Lauk, M.; Timmer, J. Clinical Neurophysiology of Tremor. J. Clin. Neurophysiol. 1996, 13, 110–121. [Google Scholar] [CrossRef]
- Louis, E.D.; Broussolle, E.; Goetz, C.G.; Krack, P.; Kaufmann, P.; Mazzoni, P. Historical underpinnings of the term essential tremor in the late 19th century. Neurology 2008, 71, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Elble, R.J. What is Essential Tremor? Curr. Neurol. Neurosci. Rep. 2013, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Laviță, S.I.; Aro, R.; Kiss, B.; Manto, M.; Duez, P. The Role of β-Carboline Alkaloids in the Pathogenesis of Essential Tremor. Cerebellum 2015, 15, 276–284. [Google Scholar] [CrossRef]
- Louis, E.D. Non-motor symptoms in essential tremor: A review of the current data and state of the field. Park. Relat. Disord. 2015, 22, S115–S118. [Google Scholar] [CrossRef]
- Sinoff, G.; Badarny, S. Mild Cognitive Impairment, Dementia, and Affective Disorders in Essential Tremor: A Prospective Study. Tremor Other Hyperkinet. Mov. 2014, 4. [Google Scholar] [CrossRef]
- Chandran, V.; Pal, P.K.; Reddy, J.Y.C.; Thennarasu, K.; Yadav, R.; Shivashankar, N. Non-motor features in essential tremor. Acta Neurol. Scand. 2011, 125, 332–337. [Google Scholar] [CrossRef]
- Louis, E.D.; Benito-León, J.; Bermejo-Pareja, F.; On behalf of the Neurological Disorders in Central Spain (NEDICES) Study Group. Self-reported depression and anti-depressant medication use in essential tremor: Cross-sectional and prospective analyses in a population-based study. Eur. J. Neurol. 2007, 14, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.D.; McCreary, M. How Common is Essential Tremor? Update on the Worldwide Prevalence of Essential Tremor. Tremor Other Hyperkinet. Mov. 2021, 11, 28. [Google Scholar] [CrossRef]
- Bellows, S.T.; Jankovic, J. Phenotypic Features of Isolated Essential Tremor, Essential Tremor Plus, and Essential Tremor-Parkinson’s Disease in a Movement Disorders Clinic. Tremor Other Hyperkinet. Mov. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.D.; Ottman, R. Study of possible factors associated with age of onset in essential tremor. Mov. Disord. 2006, 21, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, F.; Haubenberger, D.; Galpern, W.R.; Gwinn, K.; Veer, A.V.; White, S.; Bhatia, K.; Adler, C.H.; Eidelberg, D.; Ondo, W.; et al. Knowledge gaps and research recommendations for essential tremor. Park. Relat. Disord. 2016, 33, 27–35. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.; Alonso-Navarro, H.; García-Martín, E.; Álvarez, I.; Pastor, P.; Agúndez, J. Genomic Markers for Essential Tremor. Pharmaceuticals 2021, 14, 516. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, D.; Frederiksen, H.; Moises, H.; Kopper, F.; Deuschl, G.; Christensen, K. High concordance for essential tremor in monozygotic twins of old age. Neurology 2004, 62, 208–211. [Google Scholar] [CrossRef]
- Tanner, C.M.; Goldman, S.M.; Lyons, K.E.; Aston, D.A.; Tetrud, J.W.; Welsh, M.D.; Langston, J.W.; Koller, W.C. Essential tremor in twins: An assessment of genetic vs. environmental determinants of etiology. Neurology 2001, 57, 1389–1391. [Google Scholar] [CrossRef]
- Critchley, M. OBSERVATIONS ON ESSENTIAL (HEREDOFAMILIAL) TREMOR. Brain 1949, 72, 113–139. [Google Scholar] [CrossRef]
- Gulcher, J.R.; Jónsson, Þ.; Kong, A.; Kristjánsson, K.; Frigge, M.L.; Kárason, A.; Einarsdóttir, I.E.; Stefansson, H.; Einarsdóttir, A.S.; Sigurdardðttir, S.; et al. Mapping of a familial essential tremor gene, FET1, to chromosome 3q13. Nat. Genet. 1997, 17, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.J.; Pho, L.T.; Nee, L.E. A gene (ETM) for essential tremor maps to chromosome2p22-p25. Mov. Disord. 1997, 12, 859–864. [Google Scholar] [CrossRef]
- Shatunov, A.; Sambuughin, N.; Jankovic, J.; Elble, R.; Lee, H.S.; Singleton, A.B.; Dagvadorj, A.; Ji, J.; Zhang, Y.; Kimonis, V.E.; et al. Genomewide scans in North American families reveal genetic linkage of essential tremor to a region on chromosome 6p23. Brain 2006, 129, 2318–2331. [Google Scholar] [CrossRef]
- Hicks, J.E.; Konidari, I.; Scott, B.L.; Stajich, J.M.; Ashley-Koch, A.E.; Gilbert, J.R.; Scott, W.K. Linkage of familial essential tremor to chromosome 5q35. Mov. Disord. 2016, 31, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Jeanneteau, F.; Funalot, B.; Jankovic, J.; Deng, H.; Lagarde, J.-P.; Lucotte, G.; Sokoloff, P. A functional variant of the dopamine D3 receptor is associated with risk and age-at-onset of essential tremor. Proc. Natl. Acad. Sci. USA 2006, 103, 10753–10758. [Google Scholar] [CrossRef] [PubMed]
- Kuhlenbäumer, G.; Hopfner, F.; Deuschl, G. Genetics of essential tremor: Meta-analysis and review. Neurology 2014, 82, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.D. Etiology of essential tremor: Should we be searching for environmental causes? Mov. Disord. 2001, 16, 822–829. [Google Scholar] [CrossRef]
- Louis, E.D.; Benito-León, J.; Moreno-García, S.; Vega, S.; Romero, J.P.; Bermejo-Pareja, F.; Gerbin, M.; Viner, A.S.; Factor-Litvak, P.; Jiang, W.; et al. Blood harmane (1-methyl-9H-pyrido[3,4-b]indole) concentration in essential tremor cases in Spain. NeuroToxicology 2012, 34, 264–268. [Google Scholar] [CrossRef][Green Version]
- Louis, E.D.; Zheng, W.; Mao, X.; Shungu, D.C. Blood harmane is correlated with cerebellar metabolism in essential tremor: A pilot study. Neurology 2007, 69, 515–520. [Google Scholar] [CrossRef]
- Louis, E.D.; Factor-Litvak, P.; Liu, X.; Vonsattel, J.-P.G.; Galecki, M.; Jiang, W.; Zheng, W. Elevated brain harmane (1-methyl-9H-pyrido[3,4-b]indole) in essential tremor cases vs. controls. NeuroToxicology 2013, 38, 131–135. [Google Scholar] [CrossRef]
- Helmich, R.C.; Toni, I.; Deuschl, G.; Bloem, B.R. The Pathophysiology of Essential Tremor and Parkinson’s Tremor. Curr. Neurol. Neurosci. Rep. 2013, 13, 1–10. [Google Scholar] [CrossRef]
- Uusisaari, M.Y.; Knopfel, T. Diversity of Neuronal Elements and Circuitry in the Cerebellar Nuclei. Cerebellum 2012, 11, 420–421. [Google Scholar] [CrossRef]
- Uusisaari, M.Y.; Knopfel, T. GABAergic synaptic communication in the GABAergic and non-GABAergic cells in the deep cerebellar nuclei. Neuroscience 2008, 156, 537–549. [Google Scholar] [CrossRef]
- Sharifi, S.; Nederveen, A.J.; Booij, J.; van Rootselaar, A.-F. Neuroimaging essentials in essential tremor: A systematic review. NeuroImage: Clin. 2014, 5, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.D. Essential tremor: Evolving clinicopathological concepts in an era of intensive post-mortem enquiry. Lancet Neurol. 2010, 9, 613–622. [Google Scholar] [CrossRef]
- Jimenez-Shahed, J.; Jankovic, J. Exploring the relationship between essential tremor and Parkinson’s disease. Park. Relat. Disord. 2007, 13, 67–76. [Google Scholar] [CrossRef]
- Louis, E.D.; Lee, M.; Babij, R.; Ma, K.; Cortés, E.; Vonsattel, J.-P.G.; Faust, P.L. Reduced Purkinje cell dendritic arborization and loss of dendritic spines in essential tremor. Brain 2014, 137, 3142–3148. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.D.; Faust, P.L.; Ma, K.J.; Yu, M.; Cortes, E.; Vonsattel, J.-P.G. Torpedoes in the Cerebellar Vermis in Essential Tremor Cases vs. Controls. Cerebellum 2011, 10, 812–819. [Google Scholar] [CrossRef]
- Louis, E.D.; Faust, P.L.; Vonsattel, J.-P.G.; Honig, L.S.; Rajput, A.; Rajput, A.; Pahwa, R.; Lyons, K.E.; Ross, W.G.; Elble, R.J.; et al. Torpedoes in Parkinson’s disease, Alzheimer’s disease, essential tremor, and control brains. Mov. Disord. 2009, 24, 1600–1605. [Google Scholar] [CrossRef]
- Louis, E.D.; Faust, P.L.; Vonsattel, J.-P.G.; Honig, L.S.; Rajput, A.; Robinson, C.A.; Pahwa, R.; Lyons, K.E.; Ross, G.; Borden, S.; et al. Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain 2007, 130, 3297–3307. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.-H.; Erickson-Davis, C.; Gillman, A.; Faust, P.L.; Vonsattel, J.-P.G.; Louis, E.D. Increased number of heterotopic Purkinje cells in essential tremor. J. Neurol. Neurosurg. Psychiatry 2010, 82, 1038–1040. [Google Scholar] [CrossRef]
- Louis, E.D.; Kuo, S.-H.; Tate, W.; Kelly, G.C.; Gutierrez, J.; Cortes, E.P.; Vonsattel, J.-P.G.; Faust, P.L. Heterotopic Purkinje Cells: A Comparative Postmortem Study of Essential Tremor and Spinocerebellar Ataxias 1, 2, 3, and 6. Cerebellum 2017, 17, 104–110. [Google Scholar] [CrossRef]
- Axelrad, J.; Louis, E.D.; Honig, L.S.; Flores, I.; Ross, G.W.; Pahwa, R.; Lyons, K.E.; Faust, P.L.; Vonsattel, J.P.G. Reduced Purkinje Cell Number in Essential Tremor. Arch. Neurol. 2008, 65, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Choe, M.; Cortés, E.; Vonsattel, J.-P.G.; Kuo, S.-H.; Faust, P.L.; Louis, E.D. Purkinje cell loss in essential tremor: Random sampling quantification and nearest neighbor analysis. Mov. Disord. 2016, 31, 393–401. [Google Scholar] [CrossRef]
- Rajput, A.H.; Adler, C.H.; Shill, A.H.; Rajput, A. Essential tremor is not a neurodegenerative disease. Neurodegener. Dis. Manag. 2012, 2, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Bs, C.S.; Shill, H.A.; Dugger, B.; Hentz, J.G.; Adler, C.H.; Jacobson, S.A.; Driver-Dunckley, E.; Beach, T.G. Essential tremor is not associated with cerebellar Purkinje cell loss. Mov. Disord. 2014, 29, 496–500. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Louis, E.D.; Faust, P.L.; Koeppen, A.H.; Vonsattel, J.-P.G.; Kuo, S.-H. Abnormal climbing fibre-Purkinje cell synaptic connections in the essential tremor cerebellum. Brain 2014, 137, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.-H.; Lin, C.-Y.; Wang, J.; Sims, P.; Pan, M.-K.; Liou, J.-Y.; Lee, D.; Tate, W.; Kelly, G.; Louis, E.D.; et al. Climbing fiber-Purkinje cell synaptic pathology in tremor and cerebellar degenerative diseases. Acta Neuropathol. 2016, 133, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.-H.; Tang, G.; Louis, E.D.; Ma, K.; Babji, R.; Balatbat, M.; Cortes, E.; Vonsattel, J.-P.G.; Yamamoto, A.; Sulzer, D.; et al. Lingo-1 expression is increased in essential tremor cerebellum and is present in the basket cell pinceau. Acta Neuropathol. 2013, 125, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Erickson-Davis, C.R.; Faust, P.L.; Vonsattel, J.-P.G.; Gupta, S.; Honig, L.S.; Louis, E.D. “Hairy Baskets” Associated With Degenerative Purkinje Cell Changes in Essential Tremor. J. Neuropathol. Exp. Neurol. 2010, 69, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.D.; Vonsattel, J.; Honig, L.S.; Ross, G.W.; Lyons, K.E.; Pahwa, R. Neuropathologic findings in essential tremor. Neurology 2006, 66, 1756–1759. [Google Scholar] [CrossRef] [PubMed]
- Shill, H.A.; Adler, C.H.; Sabbagh, M.N.; Connor, D.J.; Caviness, J.N.; Hentz, J.G.; Beach, T.G. Pathologic findings in prospectively ascertained essential tremor subjects. Neurology 2008, 70, 1452–1455. [Google Scholar] [CrossRef]
- Louis, E.D.; Faust, P.L. Essential tremor pathology: Neurodegeneration and reorganization of neuronal connections. Nat. Rev. Neurol. 2020, 16, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Holtbernd, F.; Shah, N.J. Imaging the Pathophysiology of Essential Tremor—A Systematic Review. Front. Neurol. 2021, 12. [Google Scholar] [CrossRef]
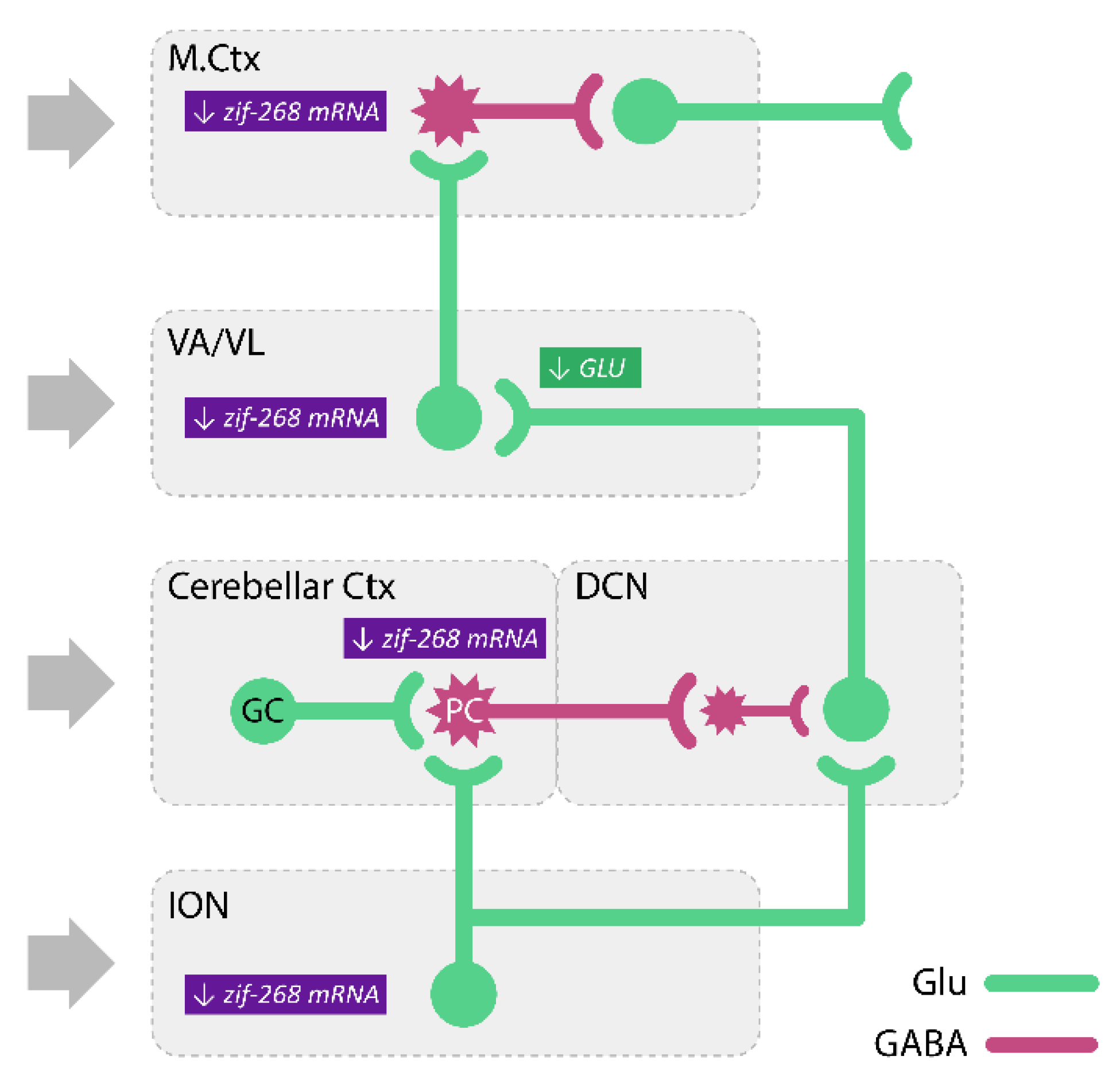
- Baranyi, M.; Vizi, E.S. Change in the concentrations of amino acids in CSF and serum of patients with essential tremor. J. Neural Transm. 1996, 103, 555–560. [Google Scholar] [CrossRef]
- Barbagallo, G.; Arabia, G.; Novellino, F.; Nisticò, R.; Salsone, M.; Morelli, M.; Rocca, F.; Quattrone, A.; Caracciolo, M.; Sabatini, U.; et al. Increased glutamate + glutamine levels in the thalamus of patients with essential tremor: A preliminary proton MR spectroscopic study. Park. Relat. Disord. 2018, 47, 57–63. [Google Scholar] [CrossRef]
- Nagy, J.I.; Pereda, A.E.; Rash, J.E. Electrical synapses in mammalian CNS: Past eras, present focus and future directions. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1860, 102–123. [Google Scholar] [CrossRef]
- Beitz, A.J.; Saxon, D. Harmaline-induced climbing fiber activation causes amino acid and peptide release in the rodent cerebellar cortex and a unique temporal pattern of Fos expression in the olivo-cerebellar pathway. J. Neurocytol. 2004, 33, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Hallett, M.; Dubinsky, R.M. Glucose metabolism in the brain of patients with essential tremor. J. Neurol. Sci. 1993, 114, 45–48. [Google Scholar] [CrossRef]
- Boecker, H.; Wills, A.J.; Ceballos-Baumann, A.; Samuel, M.; Thompson, P.D.; Findley, L.J.; Brooks, D. The effect of ethanol on alcohol-responsive essential tremor: A positron emission tomography study. Ann. Neurol. 1996, 39, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Lv, F.; Luo, T.; Cheng, O.; Liao, W.; Sheng, K.; Wang, X.; Wu, F.; Hu, Y.; Luo, J.; et al. Abnormal Regional Homogeneity in Patients with Essential Tremor Revealed by Resting-State Functional MRI. PLoS ONE 2013, 8, e69199. [Google Scholar] [CrossRef]
- Schnitzler, A.; Münks, C.; Butz, M.; Timmermann, L.; Gross, J. Synchronized brain network associated with essential tremor as revealed by magnetoencephalography. Mov. Disord. 2009, 24, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Bucher, S.F.; Seelos, K.C.; Dodel, R.C.; Reiser, M.; Oertel, W.H. Activation mapping in essential tremor with functional magnetic resonance imaging. Ann. Neurol. 1997, 41, 32–40. [Google Scholar] [CrossRef]
- Raethjen, J.; Lindemann, M.; Schmajohann, H.; Wenzelburger, R.; Pfister, G.; Deuschl, G. Multiple oscillators are causing Parkinson and essential tremor. Mov. Disord. 2000, 15, 84–94. [Google Scholar] [CrossRef]
- Raethjen, J.; Govindan, R.B.; Kopper, F.; Muthuraman, M.; Deuschl, G. Cortical Involvement in the Generation of Essential Tremor. J. Neurophysiol. 2007, 97, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Buijink, A.W.G.; Van Der Stouwe, A.M.M.; Broersma, M.; Sharifi, S.; Groot, P.F.C.; Speelman, J.D.; Maurits, N.M.; Van Rootselaar, A.-F. Motor network disruption in essential tremor: A functional and effective connectivity study. Brain 2015, 138, 2934–2947. [Google Scholar] [CrossRef]
- Dupuis, M.J.-M.; Evrard, F.L.; Jacquerye, P.G.; Picard, G.R.; Lermen, O.G. Disappearance of essential tremor after stroke. Mov. Disord. 2010, 25, 2884–2887. [Google Scholar] [CrossRef] [PubMed]
- Chahine, L.M.; Ghosh, D. Essential Tremor after Ipsilateral Cerebellar Hemispherectomy: Support for the Thalamus as the Central Oscillator. J. Child Neurol. 2009, 24, 861–864. [Google Scholar] [CrossRef]
- Gironell, A. The GABA Hypothesis in Essential Tremor: Lights and Shadows. Tremor Other Hyperkinet. Mov. 2014, 4. [Google Scholar] [CrossRef]
- Pahapill, P.A.; Levy, R.; Dostrovsky, J.O.; Davis, K.D.; Rezai, A.R.; Tasker, R.R.; Lozano, A. Tremor arrest with thalamic microinjections of muscimol in patients with essential tremor. Ann. Neurol. 1999, 46, 249–252. [Google Scholar] [CrossRef]
- Paris-Robidas, S.; Brochu, E.; Sintes, M.; Emond, V.; Bousquet, M.; Vandal, M.; Pilote, M.; Tremblay, C.; Di Paolo, T.; Rajput, A.H.; et al. Defective dentate nucleus GABA receptors in essential tremor. Brain 2011, 135, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Shill, H.A.; Adler, C.H.; Beach, T.G.; Lue, L.-F.; Caviness, J.N.; Sabbagh, M.N.; Sue, L.I.; Walker, D.G. Brain biochemistry in autopsied patients with essential tremor. Mov. Disord. 2011, 27, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Boecker, H.; Weindl, A.; Brooks, D.J.; Ceballos-Baumann, A.O.; Liedtke, C.; Miederer, M.; Sprenger, T.; Wagner, K.J.; Miederer, I. GABAergic Dysfunction in Essential Tremor: An 11C-Flumazenil PET Study. J. Nucl. Med. 2010, 51, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Gironell, A.; Figueiras, F.P.; Pagonabarraga, J.; Herance, J.R.; Sedano, B.M.P.; Trampal, C.; Gispert, J.D. Gaba and serotonin molecular neuroimaging in essential tremor: A clinical correlation study. Park. Relat. Disord. 2012, 18, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Buijink, A.W.G.; Prent, N.; Puts, N.A.; Schrantee, A.; Potters, W.V.; van Rootselaar, A.-F. GABA, Glutamate, and NAA Levels in the Deep Cerebellar Nuclei of Essential Tremor Patients. Front. Neurol. 2021, 12, 75. [Google Scholar] [CrossRef]
- Louis, E.D.; Hernandez, N.; Dyke, J.P.; Ma, R.E.; Dydak, U. In Vivo Dentate Nucleus Gamma-aminobutyric Acid Concentration in Essential Tremor vs. Controls. Cerebellum 2017, 17, 165–172. [Google Scholar] [CrossRef]
- Tapper, S.; Göransson, N.; Lundberg, P.; Tisell, A.; Zsigmond, P. A pilot study of essential tremor: Cerebellar GABA+/Glx ratio is correlated with tremor severity. Cerebellum Ataxias 2020, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gövert, F.; Becktepe, J.; Deuschl, G. Current concepts of essential tremor. Rev. Neurol. 2016, 172, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Hedera, P.; Cibulčík, F.; Davis, T.L. Pharmacotherapy of Essential Tremor. J. Cent. Nerv. Syst. Dis. 2013, 5, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Abila, B.; Wilson, J.; Marshall, R.; Richens, A. The tremorolytic action of beta-adrenoceptor blockers in essential, physiological and isoprenaline-induced tremor is mediated by beta- adrenoceptors located in a deep peripheral compartment. Br. J. Clin. Pharmacol. 1985, 20, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Song, I.-U.; Ha, S.-W.; Yang, Y.-S.; Chung, Y.-A. Differences in Regional Glucose Metabolism of the Brain Measured with F-18-FDG-PET in Patients with Essential Tremor According to Their Response to Beta-Blockers. Korean J. Radiol. 2015, 16, 967–972. [Google Scholar] [CrossRef]
- Thanvi, B.; Lo, N.; Robinson, T. Essential tremor—the most common movement disorder in older people. Age Ageing 2006, 35, 344–349. [Google Scholar] [CrossRef]
- Ondo, W.G. Current and Emerging Treatments of Essential Tremor. Neurol. Clin. 2020, 38, 309–323. [Google Scholar] [CrossRef]
- Alonso-Navarro, H.; García-Martín, E.; Agundez, J.; Jiménez-Jiménez, F.J. Current and Future Neuropharmacological Options for the Treatment of Essential Tremor. Curr. Neuropharmacol. 2020, 18, 518–537. [Google Scholar] [CrossRef]
- Ondo, W.G.; Jankovic, J.; Connor, G.S.; Pahwa, R.; Elble, R.; Stacy, M.A.; Koller, W.C.; Schwarzman, L.; Wu, S.-C.; Hulihan, J.F. Topiramate in essential tremor: A double-blind, placebo-controlled trial. Neurology 2006, 66, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.M.; Shim, H.; Christophersen, P.; Wulff, H. Pharmacology of Small- and Intermediate-Conductance Calcium-Activated Potassium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 219–240. [Google Scholar] [CrossRef]
- Mittal, S.O.; Jog, M.; Lee, J.; Jabbari, B. Novel Botulinum Toxin Injection Protocols for Parkinson Tremor and Essential Tremor—the Yale Technique and Sensor-Based Kinematics Procedure for Safe and Effective Treatment. Tremor Other Hyperkinet. Mov. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Elble, R.J.; Shih, L.; Cozzens, J.W. Surgical treatments for essential tremor. Expert Rev. Neurother. 2018, 18, 303–321. [Google Scholar] [CrossRef]
- Vaillancourt, D.E.; Sturman, M.M.; Metman, L.V.; Bakay, R.A.; Corcos, D.M. Deep brain stimulation of the VIM thalamic nucleus modifies several features of essential tremor. Neurology 2003, 61, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Baizabal-Carvallo, J.F.; Kagnoff, M.N.; Jimenez-Shahed, J.; Fekete, R.; Jankovic, J. The safety and efficacy of thalamic deep brain stimulation in essential tremor: 10 years and beyond. J. Neurol. Neurosurg. Psychiatry 2013, 85, 567–572. [Google Scholar] [CrossRef]
- Eobeso, I.; Cerasa, A.; Quattrone, A. The Effectiveness of Transcranial Brain Stimulation in Improving Clinical Signs of Hyperkinetic Movement Disorders. Front. Neurosci. 2016, 9, 486. [Google Scholar] [CrossRef] [PubMed]
- Popa, T.; Russo, M.; Vidailhet, M.; Roze, E.; Lehéricy, S.; Bonnet, C.; Apartis, E.; Legrand, A.; Marais, L.; Meunier, S.; et al. Cerebellar rTMS stimulation may induce prolonged clinical benefits in essential tremor, and subjacent changes in functional connectivity: An open label trial. Brain Stimul. 2013, 6, 175–179. [Google Scholar] [CrossRef]
- Witjas, T.; Carron, R.; Boutin, E.; Eusebio, A.; Azulay, J.P.; Régis, J. Essential tremor: Update of therapeutic strategies (medical treatment and gamma knife thalamotomy). Rev. Neurol. 2016, 172, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Shaw, J.D.; Allison, K.G.; Staffetti, J.S.; Okun, M.S.; Sullivan, K.L. Update on Treatment of Essential Tremor. Curr. Treat. Options Neurol. 2013, 15, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Jung, N.Y.; Na, Y.C.; Chang, J.W. Four-year follow-up results of magnetic resonance-guided focused ultrasound thalamotomy for essential tremor. Mov. Disord. 2018, 34, 727–734. [Google Scholar] [CrossRef]
- Kim, M.; Jung, N.Y.; Park, C.K.; Chang, W.S.; Jung, H.H.; Chang, J.W. Comparative Evaluation of Magnetic Resonance-Guided Focused Ultrasound Surgery for Essential Tremor. Ster. Funct. Neurosurg. 2017, 95, 279–286. [Google Scholar] [CrossRef]
- Martin, F.C.; Le, A.T.; Handforth, A. Harmaline-induced tremor as a potential preclinical screening method for essential tremor medications. Mov. Disord. 2005, 20, 298–305. [Google Scholar] [CrossRef]
- Handforth, A. Harmaline Tremor: Underlying Mechanisms in a Potential Animal Model of Essential Tremor. Tremor Other Hyperkinet. Mov. 2012, 2. [Google Scholar] [CrossRef]
- Lee, J.; Kim, I.; Lee, J.; Knight, E.; Cheng, L.; Kang, S.I.; Jang, N.P.; Chang, S.-Y. Development of Harmaline-induced Tremor in a Swine Model. Tremor Other Hyperkinet. Mov. 2018, 8. [Google Scholar] [CrossRef]
- Wilms, H.; Sievers, J.; Deuschl, G. Animal models of tremor. Mov. Disord. 1999, 14, 557–571. [Google Scholar] [CrossRef]
- Miwa, H. Rodent models of tremor. Cerebellum 2007, 6, 66–72. [Google Scholar] [CrossRef]
- Grella, B.; Dukat, M.; Young, R.; Teitler, M.; Herrick-Davis, K.; Gauthier, C.B.; Glennon, A.R. Investigation of hallucinogenic and related β-carbolines. Drug Alcohol Depend. 1998, 50, 99–107. [Google Scholar] [CrossRef]
- Sugihara, I.; Lang, E.J.; Llinás, R. Serotonin Modulation of Inferior Olivary Oscillations and Synchronicity: A Multiple-electrode Study in the Rat Cerebellum. Eur. J. Neurosci. 1995, 7, 521–534. [Google Scholar] [CrossRef]
- Robertson, H.A. Harmaline-induced tremor: The benzodiazepine receptor as a site of action. Eur. J. Pharmacol. 1980, 67, 129–132. [Google Scholar] [CrossRef]
- Schweri, M.; Cain, M.; Cook, J.; Paul, S.; Skolnick, P. Blockade of 3-carbomethoxy-β-carboline induced seizures by diazepam and the benzodiazepine antagonists, Ro 15-1788 and CGS 8216. Pharmacol. Biochem. Behav. 1982, 17, 457–460. [Google Scholar] [CrossRef]
- Du, W.; Aloyo, V.J.; Harvey, A.J. Harmaline competitively inhibits [3H]MK-801 binding to the NMDA receptor in rabbit brain. Brain Res. 1997, 770, 26–29. [Google Scholar] [CrossRef]
- Park, Y.-G.; Park, H.-Y.; Lee, C.J.; Choi, S.; Jo, S.; Choi, H.; Kim, Y.-H.; Shin, H.-S.; Llinas, R.R.; Kim, D. CaV3.1 is a tremor rhythm pacemaker in the inferior olive. Proc. Natl. Acad. Sci. USA 2010, 107, 10731–10736. [Google Scholar] [CrossRef]
- Wang, G.; Fowler, S.C. Concurrent quantification of tremor and depression of locomotor activity induced in rats by harmaline and physostigmine. Psychopharmacology 2001, 158, 273–280. [Google Scholar] [CrossRef]
- Miwa, H.; Kubo, T.; Suzuki, A.; Kihira, T.; Kondo, T. A species-specific difference in the effects of harmaline on the rodent olivocerebellar system. Brain Res. 2006, 1068, 94–101. [Google Scholar] [CrossRef]
- De Montigny, C.; Lamarre, Y. Effects Produced by Local Applications of Harmaline in the Inferior Olive. Can. J. Physiol. Pharmacol. 1975, 53, 845–849. [Google Scholar] [CrossRef]
- Elble, R.J. Animal models of action tremor. Mov. Disord. 2008, 13, 35–39. [Google Scholar] [CrossRef]
- Lamarre, Y.; de Montigny, C.; Dumont, M.; Weiss, M. Harmaline-induced rhythmic activity of cerebellar and lower brain stem neurons. Brain Res. 1971, 32, 246–250. [Google Scholar] [CrossRef]
- Gołembiowska, K.; Berghauzen-Maciejewska, K.; Górska, A.; Kamińska, K.; Ossowska, K. A partial lesion of the substantia nigra pars compacta and retrorubral field decreases the harmaline-induced glutamate release in the rat cerebellum. Brain Res. 2013, 1537, 303–311. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, C. Microcircuitry and function of the inferior olive. Trends Neurosci. 1998, 21, 391–400. [Google Scholar] [CrossRef]
- Haroian, A.J.; Massopust, L.C.; Young, P.A. Cerebellothalamic projections in the rat: An autoradiographic and degeneration study. J. Comp. Neurol. 1981, 197, 217–236. [Google Scholar] [CrossRef]
- Nieoullon, A.; Kerkerian, L.; Dusticier, N. High affinity glutamate uptake in the red nucleus and ventrolateral thalamus after lesion of the cerebellum in the adult cat: Biochemical evidence for functional changes in the deafferented structures. Exp. Brain Res. 1984, 55, 409–419. [Google Scholar] [CrossRef]
- Manto, M.; Laute, M.-A. A possible mechanism for the beneficial effect of ethanol in essential tremor. Eur. J. Neurol. 2008, 15, 697–705. [Google Scholar] [CrossRef]
- Kosmowska, B.; Ossowska, K.; Konieczny, J.; Lenda, T.; Berghauzen-Maciejewska, K.; Wardas, J. Inhibition of Excessive Glutamatergic Transmission in the Ventral Thalamic Nuclei by a Selective Adenosine A1 Receptor Agonist, 5′-Chloro-5′-Deoxy-(±)-ENBA Underlies its Tremorolytic Effect in the Harmaline-Induced Model of Essential Tremor. Neuroscience 2020, 429, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-P.; Wang, Y.-W.; Qi, S.-L.; Zhang, Y.-P.; Deng, G.; Ding, W.-Z.; Ma, C.; Lin, Q.-Y.; Guan, H.-D.; Liu, W.; et al. Analogous β-Carboline Alkaloids Harmaline and Harmine Ameliorate Scopolamine-Induced Cognition Dysfunction by Attenuating Acetylcholinesterase Activity, Oxidative Stress, and Inflammation in Mice. Front. Pharmacol. 2018, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.; Richard, J.; Pilapil, C.; Nair, N. Antagonists of excitatory amino acids and cyclic guanosine monophosphate in cerebellum. Neuropharmacology 1982, 21, 1235–1238. [Google Scholar] [CrossRef]
- Eblen, F.; Löschmann, P.-A.; Wüllner, U.; Turski, L.; Klockgether, T. Effects of 7-nitroindazole, NG-nitro-l-arginine, and D-CPPene on harmaline-induced postural tremor, N-methyl-d-aspartate-induced seizures, and lisuride-induced rotations in rats with nigral 6-hydroxydopamine lesions. Eur. J. Pharmacol. 1996, 299, 9–16. [Google Scholar] [CrossRef]
- Du, W.; Harvey, A.J. Harmaline-induced tremor and impairment of learning are both blocked by dizocilpine in the rabbit. Brain Res. 1997, 745, 183–188. [Google Scholar] [CrossRef]
- Paterson, N.E.; Malekiani, S.A.; Foreman, M.M.; Olivier, B.; Hanania, T. Pharmacological characterization of harmaline-induced tremor activity in mice. Eur. J. Pharmacol. 2009, 616, 73–80. [Google Scholar] [CrossRef]
- Iseri, P.K.; Karson, A.; Gullu, K.M.; Akman, O.; Kokturk, S.; Yardýmoglu, M.; Erturk, S.; Ates, N. The effect of memantine in harmaline-induced tremor and neurodegeneration. Neuropharmacology 2011, 61, 715–723. [Google Scholar] [CrossRef]
- Mignani, S.; Bohme, G.A.; Birraux, G.; Boireau, A.; Jimonet, P.; Damour, D.; Genevois-Borella, A.; Debono, M.-W.; Pratt, J.; Vuilhorgne, M.; et al. 9-Carboxymethyl-5 H,10H-imidazo[1,2-a]indeno[1,2-e]pyrazin-4-one-2-carbocylic Acid (RPR117824): Selective Anticonvulsive and Neuroprotective AMPA Antagonist. Bioorg. Med. Chem. 2002, 10, 1627–1637. [Google Scholar] [CrossRef]
- Simantov, R.; Snyder, S.H.; Oster-Granite, M.-L. Harmaline-induced tremor in the rat: Abolition by 3-acetylpyridine destruction of cerebellar climbing fibers. Brain Res. 1976, 114, 144–151. [Google Scholar] [CrossRef]
- Bin Tian, J.; Bishop, A.G. Stimulus-dependent activation of c-Fos in neurons and glia in the rat cerebellum. J. Chem. Neuroanat. 2001, 23, 157–170. [Google Scholar] [CrossRef]
- Miwa, H.; Nishi, K.; Fuwa, T.; Mizuno, Y. Differential expression of c-fos following administration of two tremorgenic agents: Harmaline and oxotremorine. Neuroreport 2000, 11, 2385–2390. [Google Scholar] [CrossRef] [PubMed]
- Kosmowska, B.; Ossowska, K.; Głowacka, U.; Wardas, J. Tremorolytic effect of 5′-chloro-5′-deoxy-(±)-ENBA, a potent and selective adenosine A1 receptor agonist, evaluated in the harmaline-induced model in rats. CNS Neurosci. Ther. 2017, 23, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Llinás, R.; Mühlethaler, M. Electrophysiology of guinea-pig cerebellar nuclear cells in the in vitro brain stem-cerebellar preparation. J. Physiol. 1988, 404, 241–258. [Google Scholar] [CrossRef]
- Battista, A.F.; Nakatani, S.; Goldstein, M.; Anagnoste, B. Effect of harmaline in monkeys with central nervous system lesions. Exp. Neurol. 1970, 28, 513–524. [Google Scholar] [CrossRef]
- Bekar, L.; Libionka, W.; Tian, G.-F.; Xu, Q.; Torres, A.; Wang, X.; Lovatt, D.; Williams, E.; Takano, T.; Schnermann, J.; et al. Adenosine is crucial for deep brain stimulation–mediated attenuation of tremor. Nat. Med. 2007, 14, 75–80. [Google Scholar] [CrossRef]
- Lee, J.; Chang, S.-Y. Altered Primary Motor Cortex Neuronal Activity in a Rat Model of Harmaline-Induced Tremor During Thalamic Deep Brain Stimulation. Front. Cell. Neurosci. 2019, 13, 448. [Google Scholar] [CrossRef]
- Lamarre, Y.; Weiss, M. Harmaline-induced rhythmic activity of alpha and gamma motoneurons in the cat. Brain Res. 1973, 63, 430–434. [Google Scholar] [CrossRef]
- Kralic, J.E.; Criswell, H.E.; Osterman, J.L.; O’Buckley, T.K.; Wilkie, M.E.; Matthews, D.B.; Hamre, K.; Breese, G.R.; Homanics, G.E.; Morrow, A.L. Genetic essential tremor in gamma-aminobutyric acidA receptor alpha1 subunit knockout mice. J. Clin. Investig. 2005, 115, 774–779. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kralic, J.E.; Korpi, E.R.; O’Buckley, T.K.; Homanics, G.E.; Morrow, A.L. Molecular and Pharmacological Characterization of GABAAReceptor α1 Subunit Knockout Mice. J. Pharmacol. Exp. Ther. 2002, 302, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-K.; Li, Y.-S.; Wong, S.-B.; Ni, C.-L.; Wang, Y.-M.; Liu, W.-C.; Lu, L.-Y.; Lee, J.-C.; Cortes, E.P.; Vonsattel, J.-P.G.; et al. Cerebellar oscillations driven by synaptic pruning deficits of cerebellar climbing fibers contribute to tremor pathophysiology. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Miyazaki, T.; Yamasaki, M.; Takeuchi, T.; Sakimura, K.; Mishina, M.; Watanabe, M. Ablation of Glutamate Receptor GluR 2 in Adult Purkinje Cells Causes Multiple Innervation of Climbing Fibers by Inducing Aberrant Invasion to Parallel Fiber Innervation Territory. J. Neurosci. 2010, 30, 15196–15209. [Google Scholar] [CrossRef]
- Handforth, A.; Lang, E.J. Increased Purkinje Cell Complex Spike and Deep Cerebellar Nucleus Synchrony as a Potential Basis for Syndromic Essential Tremor. A Review and Synthesis of the Literature. Cerebellum 2020, 20, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Kosmowska, B.; Ossowska, K.; Wardas, J. Pramipexole Reduces zif-268 mRNA Expression in Brain Structures involved in the Generation of Harmaline-Induced Tremor. Neurochem. Res. 2020, 45, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.-M.; Espinoza, S.; Gainetdinov, R.R. Dopamine receptors-IUPHAR Review 13. Br. J. Pharmacol. 2014, 172, 1–23. [Google Scholar] [CrossRef]
- Foil, B. Le Dopamine Receptors. BIOTREND Rev. 2010, 6, 1–11. [Google Scholar] [CrossRef]
- Panagopoulos, N.T.; Papadopoulos, G.C.; Matsokis, N.A. Dopaminergic innervation and binding in the rat cerebellum. Neurosci. Lett. 1991, 130, 208–212. [Google Scholar] [CrossRef]
- Kizer, J.; Palkovits, M.; Brownstein, M. The projections of the A8, A9 and A10 dopaminergic cell bodies: Evidence for a nigral-hypothalamic-median eminence dopaminergic pathway. Brain Res. 1976, 108, 363–370. [Google Scholar] [CrossRef]
- Ikai, Y.; Takada, M.; Shinonaga, Y.; Mizuno, N. Dopaminergic and non-dopaminergic neurons in the ventral tegmental area of the rat project, respectively, to the cerebellar cortex and deep cerebellar nuclei. Neuroscience 1992, 51, 719–728. [Google Scholar] [CrossRef]
- Barili, P.; Bronzetti, E.; Ricci, A.; Zaccheo, D.; Amenta, F. Microanatomical localization of dopamine receptor protein immunoreactivity in the rat cerebellar cortex. Brain Res. 2000, 854, 130–138. [Google Scholar] [CrossRef]
- Díaz, J.; Pilon, C.; Le Foll, B.; Gros, C.; Triller, A.; Schwartz, J.-C.; Sokoloff, P. Dopamine D3 Receptors Expressed by All Mesencephalic Dopamine Neurons. J. Neurosci. 2000, 20, 8677–8684. [Google Scholar] [CrossRef]
- Barik, S.; de Beaurepaire, R. Evidence for a functional role of the dopamine D3 receptors in the cerebellum. Brain Res. 1996, 737, 347–350. [Google Scholar] [CrossRef]
- Barik, S.; de Beaurepaire, R. Dopamine D3 modulation of locomotor activity and sleep in the nucleus accumbens and in lobules 9 and 10 of the cerebellum in the rat. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 718–726. [Google Scholar] [CrossRef]
- Kolasiewicz, W.; Maj, J.; Ossowska, K. The involvement of cerebellar dopamine D3 receptors in locomotor activity of rats. J. Neural Transm. 2008, 115, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Takada, M.; Sugimoto, T.; Hattori, T. Tyrosine hydroxylase immunoreactivity in cerebellar Purkinje cells of the rat. Neurosci. Lett. 1993, 150, 61–64. [Google Scholar] [CrossRef]
- Voogd, J. The anatomy of the cerebellum. Trends Neurosci. 1998, 21, 370–375. [Google Scholar] [CrossRef]
- Berthie, B.; Axelrad, H.; Verney, C.; Marc, M.E.; Axelrad, H.; Verney, C.; Marc, M.E. A small population of Purkinje cells in the posterior vermis is specifically labeled by a tyrosine hydroxylase antibody. In Serotonin, the Cerebellum and Ataxia; Trovillas, P., Fuxe, K., Eds.; Raven Press: New York, NY, USA, 1993; pp. 121–127. [Google Scholar]
- Kim, Y.S.; Shin, J.H.; Hall, F.; Linden, D.J. Dopamine Signaling Is Required for Depolarization-Induced Slow Current in Cerebellar Purkinje Cells. J. Neurosci. 2009, 29, 8530–8538. [Google Scholar] [CrossRef]
- Ishibashi, T.; Wakabayashi, J.; Ohno, Y. 7-Hydroxy-N,N’-di-n-propyl-2-aminotetraline, a Preferential Dopamine D3 Agonist, Induces c-fos mRNA Expression in the Rat Cerebellum. Jpn. J. Pharmacol. 2002, 89, 309–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heman, P.; Barcia, C.; Gómez, A.; Ros Gómez, C.M.; Ros Bernal, F.; Yuste Jiménez, J.E.; de Pablos, V.; Fernandez-Villalba, E.; Cárdenas, R.T.; Ezquerro, M.H. Nigral degeneration correlates with persistent activation of cerebellar Purkinje cells in MPTP-treated monkeys. Histol. Histopathol. 2012, 89–94. [Google Scholar] [CrossRef]
- Kolasiewicz, W.; Kuter, K.; Berghauzen, K.; Nowak, P.; Schulze, G.; Ossowska, K. 6-OHDA injections into A8–A9 dopaminergic neurons modelling early stages of Parkinson’s disease increase the harmaline-induced tremor in rats. Brain Res. 2012, 1477, 59–73. [Google Scholar] [CrossRef]
- Bostan, A.C.; Dum, R.P.; Strick, P.L. The basal ganglia communicate with the cerebellum. Proc. Natl. Acad. Sci. USA 2010, 107, 8452–8456. [Google Scholar] [CrossRef] [PubMed]
- Bouthenet, M.-L.; Souil, E.; Martres, M.-P.; Sokoloff, P.; Giros, B.; Schwartz, J.-C. Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: Comparison with dopamine D2 receptor mRNA. Brain Res. 1991, 564, 203–219. [Google Scholar] [CrossRef]
- Rajput, A.H.; Rozdilsky, B.; Kish, S.; Hornykiewicz, O. Parkinson’s disease association with essential tremor. Mov. Disord. 1990, 5 (Suppl. 1), 1414. [Google Scholar]
- Asenbaum, S.; Pirker, W.; Angelberger, P.; Bencsits, G.; Pruckmayer, M.; Brücke, T. [123 I]β-CIT and SPECT in essential tremor and Parkinson’s disease. J. Neural Transm. 1998, 105, 1213–1228. [Google Scholar] [CrossRef]
- Benamer, T.S.; Patterson, J.; Grosset, D.G.; Booij, J.; De Bruin, K.; Van Royen, E.; Speelman, J.D.; Horstink, M.H.; Sips, H.J.; Dierckx, A.R.; et al. Accurate differentiation of parkinsonism and essential tremor using visual assessment of [123I]-FP-CIT SPECT imaging: The [123I]-FP-CIT study group. Mov. Disord. 2000, 15, 503–510. [Google Scholar] [CrossRef]
- Isaias, I.U.; Marotta, G.; Hirano, S.; Canesi, M.; Benti, R.; Righini, A.; Tang, C.; Cilia, R.; Pezzoli, G.; Eidelberg, D.; et al. Imaging essential tremor. Mov. Disord. 2010, 25, 679–686. [Google Scholar] [CrossRef]
- Antonini, A.; Moresco, R.M.; Gobbo, C.; De Notaris, R.; Panzacchi, A.; Barone, P.; Calzetti, S.; Negrotti, A.; Pezzoli, G.; Fazio, F. The status of dopamine nerve terminals in Parkinson’s disease and essential tremor: A PET study with the tracer [11-C]FE-CIT. Neurol. Sci. 2001, 22, 47–48. [Google Scholar] [CrossRef]
- Di Giuda, D.; Camardese, G.; Bentivoglio, A.R.; Cocciolillo, F.; Guidubaldi, A.; Pucci, L.; Bruno, I.; Janiri, L.; Giordano, A.; Fasano, A. Dopaminergic dysfunction and psychiatric symptoms in movement disorders: A 123I-FP-CIT SPECT study. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1937–1948. [Google Scholar] [CrossRef]
- Waln, O.; Wu, Y.; Perlman, R.; Wendt, J.; Van, A.K.; Jankovic, J. Dopamine transporter imaging in essential tremor with and without parkinsonian features. J. Neural Transm. 2015, 122, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, G.; Arabia, G.; Morelli, M.; Nisticò, R.; Novellino, F.; Salsone, M.; Rocca, F.; Quattrone, A.; Caracciolo, M.; Sabatini, U.; et al. Thalamic neurometabolic alterations in tremulous Parkinson’s disease: A preliminary proton MR spectroscopy study. Park. Relat. Disord. 2017, 43, 78–84. [Google Scholar] [CrossRef]
- Gerasimou, G.; Costa, D.C.; Papanastasiou, E.; Bostanjiopoulou, S.; Arnaoutoglou, M.; Moralidis, E.; Aggelopoulou, T.; Gotzamani-Psarrakou, A. SPECT study with I-123-Ioflupane (DaTSCAN) in patients with essential tremor. Is there any correlation with Parkinson’s disease? Ann. Nucl. Med. 2012, 26, 337–344. [Google Scholar] [CrossRef]
- Isaias, I.U.; Canesi, M.; Benti, R.; Gerundini, P.; Cilia, R.; Pezzoli, G.; Antonini, A. Striatal dopamine transporter abnormalities in patients with essential tremor. Nucl. Med. Commun. 2008, 29, 349–353. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, Y.-P.; Liu, X.-D.; Chen, Z.-P.; Yang, L.-Q.; Liu, C.-J.; Xiang, J.-D.; Su, H.-L. 99mTc-TRODAT-1 SPECT study in early Parkinson’s disease and essential tremor. Acta Neurol. Scand. 2005, 112, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Breit, S.; Reimold, M.; Reischl, G.; Klockgether, T.; Wullner, U. [11C]d-threo-methylphenidate PET in patients with Parkinson’s disease and essential tremor. J. Neural Transm. 2005, 113, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J.; Playford, E.D.; Ibanez, V.; Sawle, G.V.; Thompson, P.D.; Findley, L.J.; Marsden, C.D. Isolated tremor and disruption of the nigrostriatal dopaminergic system: An 18F-dopa PET study. Neurology 1992, 42, 1554. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, M.; Amthauer, H.; Klaffke, S.; Arnold, G.; Wernecke, K.-D.; Kupsch, A.; Felix, R.; Venz, S. Combined 123I-FP-CIT and 123I-IBZM SPECT for the diagnosis of parkinsonian syndromes: Study on 72 patients. J. Neural Transm. 2004, 112, 677–692. [Google Scholar] [CrossRef]
- Herceg, M.; Nagy, F.; Pal, E.; Janszky, J.; Késmárky, I.; Komoly, S.; Kovacs, N. Pramipexole May Be an Effective Treatment Option in Essential Tremor. Clin. Neuropharmacol. 2012, 35, 73–76. [Google Scholar] [CrossRef]
- Kim, H.; Sablin, S.O.; Ramsay, R. Inhibition of Monoamine Oxidase A by β-Carboline Derivatives. Arch. Biochem. Biophys. 1997, 337, 137–142. [Google Scholar] [CrossRef]
- Ossowska, K.; Głowacka, U.; Kosmowska, B.; Wardas, J. Apomorphine enhances harmaline-induced tremor in rats. Pharmacol. Rep. 2015, 67, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Kosmowska, B.; Wardas, J.; Głowacka, U.; Ananthan, S.; Ossowska, K. Pramipexole at a Low Dose Induces Beneficial Effect in the Harmaline-induced Model of Essential Tremor in Rats. CNS Neurosci. Ther. 2015, 22, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Pogarell, O.; Gasser, T.; Van Hilten, J.J.; Spieker, S.; Pollentier, S.; Meier, D.; Oertel, W.H. Pramipexole in patients with Parkinson’s disease and marked drug resistant tremor: A randomised, double blind, placebo controlled multicentre study. J. Neurol. Neurosurg. Psychiatry 2002, 72, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Chen, J.-F.; Fredholm, B.B. Adenosine signaling and function in glial cells. Cell Death Differ. 2009, 17, 1071–1082. [Google Scholar] [CrossRef]
- Daré, E.; Schulte, G.; Karovic, O.; Hammarberg, C.; Fredholm, B.B. Modulation of glial cell functions by adenosine receptors. Physiol. Behav. 2007, 92, 15–20. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Chen, J.-F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.-M. Adenosine and Brain Function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Mittal, A.; Kumar, P.; Alhayani, D.M.; Al-Aboudi, A. Therapeutic Potential of Agonists and Antagonists of A1, A2a, A2b and A3 Adenosine Receptors. Curr. Pharm. Des. 2019, 25, 2892–2905. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Simola, N.; Wardas, J. Adenosine A2A receptors: Localization and function. In The Adenosinergic System: A Non-Dopaminergic Target in Parkinson’s Disease; Morelli, M., Simola, N., Wardas, J., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2015; pp. 1–25. [Google Scholar]
- Latini, S.; Pedata, F. Adenosine in the central nervous system: Release mechanisms and extracellular concentrations. J. Neurochem. 2008, 79, 463–484. [Google Scholar] [CrossRef]
- Bonan, C.D. Ectonucleotidases and nucleotide/nucleoside transporters as pharmacological targets for neurological disorders. CNS Neurol Disord. Drug Targets 2012, 11, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, F.E.; Damaraju, V.L.; Graham, K.; Yao, S.Y.; Baldwin, S.A.; Cass, C.E.; Young, J.D. Molecular Biology of Nucleoside Transporters and their Distributions and Functions in the Brain. Curr. Top. Med. Chem. 2011, 11, 948–972. [Google Scholar] [CrossRef] [PubMed]
- Dunwiddie, T.V.; Diao, L.; Kim, H.O.; Jiang, J.-L.; Jacobson, K. Activation of Hippocampal Adenosine A3Receptors Produces a Desensitization of A1Receptor-Mediated Responses in Rat Hippocampus. J. Neurosci. 1997, 17, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Latini, S.; Bordoni, F.; Pedata, F.; Corradetti, R. Extracellular adenosine concentrations duringin vitroischaemia in rat hippocampal slices. Br. J. Pharmacol. 1999, 127, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Sperlagh, B. The Role of Extracellular Adenosine in Chemical Neurotransmission in the Hippocampus and Basal Ganglia: Pharmacological and Clinical Aspects. Curr. Top. Med. Chem. 2011, 11, 1034–1046. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Schindler, M.; Harris, C.A.; Hayes, B.; Papotti, M.; Humphrey, P.P. Immunohistochemical localization of adenosine A1 receptors in human brain regions. Neurosci. Lett. 2001, 297, 211–215. [Google Scholar] [CrossRef]
- Ochiishi, T.; Chen, L.; Yukawa, A.; Saitoh, Y.; Sekino, Y.; Arai, T.; Nakata, H.; Miyamoto, H. Cellular localization of adenosine A1 receptors in rat forebrain: Immunohistochemical analysis using adenosine A1 receptor-specific monoclonal antibody. J. Comp. Neurol. 1999, 411, 301–316. [Google Scholar] [CrossRef]
- Sebastião, A.M.; Ribeiro, J.A. Adenosine Receptors and the Central Nervous System. Handb. Exp. Pharmacol. 2009, 193, 471–534. [Google Scholar] [CrossRef]
- Fastbom, J.; Pazos, A.; Palacios, J. The distribution of adenosine a1 receptors and 5’-nucleotidase in the brain of some commonly used experimental animals. Neuroscience 1987, 22, 813–826. [Google Scholar] [CrossRef]
- Dixon, A.K.; Gubitz, A.K.; Sirinathsinghji, D.J.; Richardson, P.J.; Freeman, T. Tissue distribution of adenosine receptor mRNAs in the rat. Br. J. Pharmacol. 1996, 118, 1461–1468. [Google Scholar] [CrossRef]
- Biber, K.; Klotz, K.-N.; Berger, M.; Gebicke-Härter, P.J.; Van Calker, D. Adenosine A1Receptor-Mediated Activation of Phospholipase C in Cultured Astrocytes Depends on the Level of Receptor Expression. J. Neurosci. 1997, 17, 4956–4964. [Google Scholar] [CrossRef] [PubMed]
- Gebicke-Haerter, P.J.; Christoffel, F.; Timmer, J.; Northoff, H.; Berger, M.; VAN Calker, D. Both Adenosine a1- and a2-Receptors are Required to Stimulate Microglial Proliferation. Neurochem. Int. 1996, 29, 37–42. [Google Scholar] [CrossRef]
- Othman, T.; Yan, H.; Rivkees, S.A. Oligodendrocytes express functional A1 adenosine receptors that stimulate cellular migration. Glia 2003, 44, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Rivkees, S.A.; Price, S.L.; Zhou, F.C. Immunohistochemical detection of A1 adenosine receptors in rat brain with emphasis on localization in the hippocampal formation, cerebral cortex, cerebellum, and basal ganglia. Brain Res. 1995, 677, 193–203. [Google Scholar] [CrossRef]
- Alexander, S.; Reddington, M. The cellular localization of adenosine receptors in rat neostriatum. Neuroscience 1989, 28, 645–651. [Google Scholar] [CrossRef]
- Ferré, S.; O’Connor, W.T.; Svenningsson, P.; Björklund, L.; Lindberg, J.; Tinner, B.; Strömberg, I.; Goldstein, M.; Ögren, S.O.; Ungerstedt, U.; et al. Dopamine D1Receptor-mediated Facilitation of GABAergic Neurotransmission in the Rat Strioentopeduncular Pathway and its Modulation by Adenosine A1Receptor-mediated Mechanisms. Eur. J. Neurosci. 1996, 8, 1545–1553. [Google Scholar] [CrossRef]
- Cunha, R. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: Different roles, different sources and different receptors. Neurochem. Int. 2001, 38, 107–125. [Google Scholar] [CrossRef]
- Rebola, N.; Pinheiro, P.; Oliveira, C.; Malva, J.; Cunha, R.A. Subcellular localization of adenosine A1 receptors in nerve terminals and synapses of the rat hippocampus. Brain Res. 2003, 987, 49–58. [Google Scholar] [CrossRef]
- Deb, P.K.; Deka, S.; Borah, P.; Abed, S.; Klotz, K.-N. Medicinal Chemistry and Therapeutic Potential of Agonists, Antagonists and Allosteric Modulators of A1 Adenosine Receptor: Current Status and Perspectives. Curr. Pharm. Des. 2019, 25, 2697–2715. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A.; Milusheva, E.; Vizi, E.S.; Ribeiro, J.A.; Sebastião, A.M. Excitatory and inhibitory effects of A1 and A2A adenosine receptor activation on the electrically evoked [3H]acetylcholine release from different areas of the rat hippocampus. J. Neurochem. 1994, 63, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Rebola, N.; Canas, P.; Oliveira, C.; Cunha, R. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 2005, 132, 893–903. [Google Scholar] [CrossRef]
- Rosin, D.L.; Robeva, A.; Woodard, R.L.; Guyenet, P.G.; Linden, J. Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J. Comp. Neurol. 1998, 401, 163–186. [Google Scholar] [CrossRef]
- Svenningsson, P.; Le Moine, C.; Fisone, G.; Fredholm, B.B. Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog. Neurobiol. 1999, 59, 355–396. [Google Scholar] [CrossRef]
- Svenningsson, P.; Le Moine, C.; Aubert, I.; Burbaud, P.; Fredholm, B.; Bloch, B. Cellular distribution of adenosine A2A receptor mrna in the primate striatum. J. Comp. Neurol. 1998, 399, 229–240. [Google Scholar] [CrossRef]
- Rosin, D.L.; Hettinger, B.D.; Lee, A.; Linden, J. Anatomy of adenosine A2A receptors in brain: Morphological substrates for integration of striatal function. Neurology 2003, 61, S12–S18. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Williams, M. Direct autoradiographic localization of adenosine A2 receptors in the rat brain using the A2-selective agonist, [3H]CGS 21680. Eur. J. Pharmacol. 1989, 168, 243–246. [Google Scholar] [CrossRef]
- Hettinger, B.D.; Lee, A.; Linden, J.; Rosin, D.L. Ultrastructural localization of adenosine A2A receptors suggests multiple cellular sites for modulation of GABAergic neurons in rat striatum. J. Comp. Neurol. 2001, 431, 331–346. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Dos Santos-Rodrigues, A.; Schwarzschild, M.A.; Chen, J.-F.; Cunha, R.A.; Agostinho, P. Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia 2012, 60, 702–716. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Agostinho, P.; Cunha, R.A.; Chen, J.-F. Antagonistic Interaction between Adenosine A2A Receptors and Na+/K+-ATPase- 2 Controlling Glutamate Uptake in Astrocytes. J. Neurosci. 2013, 33, 18492–18502. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, S.; Fisone, G.; Moresco, R.; Cunha, R.; Ferre, S. Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 2007, 83, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Augood, S.J.; Emson, P.C. Adenosine A2a receptor mRNA is expressed by enkephalin cells but not by somatostatin cells in rat striatum: A co-expression study. Mol. Brain Res. 1994, 22, 204–210. [Google Scholar] [CrossRef]
- Fink, J.; Weaver, D.; Rivkees, S.A.; Peterfreund, R.A.; Pollack, A.E.; Adler, E.M.; Reppert, S.M. Molecular cloning of the rat A2 adenosine receptor: Selective co-expression with D2 dopamine receptors in rat striatum. Mol. Brain Res. 1992, 14, 186–195. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Jacobs, O.; Vanderhaeghen, J.-J. Striatal Restricted Adenosine A2Receptor (RDC8) Is Expressed by Enkephalin but Not by Substance P Neurons: An In Situ Hybridization Histochemistry Study. J. Neurochem. 1991, 57, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, C.; Luján, R.; Uchigashima, M.; Simoes, A.P.; Lerner, T.N.; Borycz, J.; Kachroo, A.; Canas, P.M.; Orru, M.; Schwarzschild, M.A.; et al. Key Modulatory Role of Presynaptic Adenosine A2AReceptors in Cortical Neurotransmission to the Striatal Direct Pathway. Sci. World J. 2009, 9, 1321–1344. [Google Scholar] [CrossRef]
- Rodrigues, R.J.; Alfaro, T.M.; Rebola, N.; Oliveira, C.R.; Cunha, R.A. Co-localization and functional interaction between adenosine A2A and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J. Neurochem. 2004, 92, 433–441. [Google Scholar] [CrossRef]
- Ciruela, F.; Casadó, V.; Rodrigues, R.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic Control of Striatal Glutamatergic Neurotransmission by Adenosine A1-A2A Receptor Heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef]
- Kurokawa, M.; Kirk, I.P.; Kirkpatrick, K.A.; Kase, H.; Richardson, P.J. Inhibition by KF17837 of adenosine A2A receptor-mediated modulation of striatal GABA and ACh release. Br. J. Pharmacol. 1994, 113, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; James, S.; Reddington, M.; Richardson, P.J. Both A1and A2aPurine Receptors Regulate Striatal Acetylcholine Release. J. Neurochem. 1990, 55, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Koga, K.; Kase, H.; Nakamura, J.; Kuwana, Y. Adenosine A2a Receptor-Mediated Modulation of Striatal Acetylcholine Release In Vivo. J. Neurochem. 2002, 66, 1882–1888. [Google Scholar] [CrossRef]
- Feoktistov, I.; Biaggioni, I. Adenosine A2B receptors. Pharmacol. Rev. 1997, 49, 381–402. [Google Scholar] [PubMed]
- Burnstock, G.; Verkhratsky, A. Receptors for Purines and Pyrimidines. Pharmacol. Rev. 2012, 119–244. [Google Scholar] [CrossRef]
- Pierce, K.; Furlong, T.J.; Selbie, L.A.; Shine, J. Molecular cloning and expression of an adenosine A2b receptor from human brain. Biochem. Biophys. Res. Commun. 1992, 187, 86–93. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Brand, A.; Vissiennon, Z.; Eschke, D.; Nieber, K. Adenosine A1 and A3 receptors mediate inhibition of synaptic transmission in rat cortical neurons. Neuropharmacology 2000, 40, 85–95. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Ijzerman, A.; Jacobson, K.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Hammarberg, C.; Schulte, G.; Fredholm, B.B. Evidence for functional adenosine A3 receptors in microglia cells. J. Neurochem. 2003, 86, 1051–1054. [Google Scholar] [CrossRef]
- Wittendorp, M.C.; Boddeke, H.W.; Biber, K. Adenosine A3 receptor-induced CCL2 synthesis in cultured mouse astrocytes. Glia 2004, 46, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Shearman, L.P.; Weaver, D.R. [125I]4-Aminobenzyl-5′-N-methylcarboxamidoadenosine ([125I]AB-MECA) labels multiple adenosine receptor subtypes in rat brain. Brain Res. 1997, 745, 10–20. [Google Scholar] [CrossRef]
- Rivkees, S.A.; Thevananther, S.; Hao, H. Are A3 adenosine receptors expressed in the brain? NeuroReport 2000, 11, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Ciruela, F. Functional and Neuroprotective Role of Striatal Adenosine A2AReceptor Heterotetramers. J. Caffeine Adenosine Res. 2019, 9, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cordomí, A.; del Torrent, C.L.; Lillo, A.; Serrano-Marín, J.; Navarro, G.; Pardo, L. Structure and function of adenosine receptor heteromers. Cell. Mol. Life Sci. 2021, 78, 3957–3968. [Google Scholar] [CrossRef]
- Fuxe, K.; Marcellino, D.; Borroto-Escuela, D.O.; Guescini, M.; Fernández-Dueñas, V.; Tanganelli, S.; Rivera, A.; Ciruela, F.; Agnati, L.F. Adenosine–Dopamine Interactions in the Pathophysiology and Treatment of CNS Disorders. CNS Neurosci. Ther. 2010, 16, e18–e42. [Google Scholar] [CrossRef]
- Ciruela, F.; Ferre, S.; Casadó, V.; Cortes, A.; Cunha, R.A.; Lluis, C.; Franco, R. Heterodimeric adenosine receptors: A device to regulate neurotransmitter release. Cell. Mol. Life Sci. 2006, 63, 2427–2431. [Google Scholar] [CrossRef] [PubMed]
- Ginés, S.; Hillion, J.; Torvinen, M.; Le Crom, S.; Casadó, V.; Canela, E.I.; Rondin, S.; Lew, J.Y.; Watson, S.; Zoli, M.; et al. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc. Natl. Acad. Sci. USA 2000, 97, 8606–8611. [Google Scholar] [CrossRef]
- Kamikubo, Y.; Tabata, T.; Sakairi, H.; Hashimoto, Y.; Sakurai, T. Complex formation and functional interaction between adenosine A1 receptor and type-1 metabotropic glutamate receptor. J. Pharmacol. Sci. 2015, 128, 125–130. [Google Scholar] [CrossRef]
- Yoshioka, K.; Saitoh, O.; Nakata, H. Heteromeric association creates a P2Y-like adenosine receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7617–7622. [Google Scholar] [CrossRef]
- Hillion, J.; Canals, M.; Torvinen, M.; Casadó, V.; Scott, R.; Terasmaa, A.; Hansson, A.; Watson, S.; Olah, M.E.; Mallol, J.; et al. Coaggregation, Cointernalization, and Codesensitization of Adenosine A2A Receptors and Dopamine D2Receptors. J. Biol. Chem. 2002, 277, 18091–18097. [Google Scholar] [CrossRef]
- Torvinen, M.; Marcellino, D.; Canals, M.; Agnati, L.F.; Lluis, C.; Franco, R.; Fuxe, K. Adenosine A2A Receptor and Dopamine D3 Receptor Interactions: Evidence of Functional A2A/D3 Heteromeric Complexes. Mol. Pharmacol. 2004, 67, 400–407. [Google Scholar] [CrossRef]
- Díaz-Cabiale, Z.; Vivó, M.; Del Arco, A.; O’Connor, W.T.; Harte, M.K.; Müller, C.E.; Martínez, E.; Popoli, P.; Fuxe, K.; Ferré, S. Metabotropic glutamate mGlu5 receptor-mediated modulation of the ventral striopallidal GABA pathway in rats. Interactions with adenosine A2A and dopamine D2 receptors. Neurosci. Lett. 2002, 324, 154–158. [Google Scholar] [CrossRef]
- Carriba, P.; Ortiz, O.; Patkar, K.; Justinova, Z.; Stroik, J.; Themann, A.; Müller, C.; Woods, A.S.; Hope, B.; Ciruela, F.; et al. Striatal Adenosine A2A and Cannabinoid CB1 Receptors Form Functional Heteromeric Complexes that Mediate the Motor Effects of Cannabinoids. Neuropsychopharmacology 2007, 32, 2249–2259. [Google Scholar] [CrossRef]
- Cunha, R.A. Neuroprotection by adenosine in the brain: From A1 receptor activation to A2A receptor blockade. Purinergic Signal. 2005, 1, 111–134. [Google Scholar] [CrossRef] [PubMed]
- Manev, H.; Favaron, M.; Guidotti, A.; Costa, E. Delayed increase of Ca2+ influx elicited by glutamate: Role in neuronal death. Mol. Pharmacol. 1989, 36, 106–112. [Google Scholar] [PubMed]
- Wardas, J. Neuroprotective role of adenosine in the CNS. Pol. J. Pharmacol. 2003, 54, 313–326. [Google Scholar]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A Receptors in the Brain: Current Research and Their Role in Neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef] [PubMed]
- Simola, N.; Fenu, S.; Baraldi, P.G.; Tabrizi, M.A.; Morelli, M. Blockade of adenosine A2A receptors antagonizes parkinsonian tremor in the rat tacrine model by an action on specific striatal regions. Exp. Neurol. 2004, 189, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Collins-Praino, L.E.; Paul, N.E.; Rychalsky, K.L.; Hinman, J.R.; Chrobak, J.J.; Senatus, P.B.; Salamone, J.D. Pharmacological and Physiological Characterization of the Tremulous Jaw Movement Model of Parkinsonian Tremor: Potential Insights into the Pathophysiology of Tremor. Front. Syst. Neurosci. 2011, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Sandyk, R. Essential tremor-successful treatment with aminophylline. A case report. S. Afr. Med. J. 1983, 64, 639. [Google Scholar] [PubMed]
- Buss, D.C.; Marshall, R.W.; Milligan, N.; McQueen, I.; Compston, D.A.S.; Routledge, P.A. The effect of intravenous aminophylline on essential tremor. Br. J. Clin. Pharmacol. 1997, 43, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Mally, J. Aminophylline and Essential Tremor. Lancet 1989, 334, 278–279. [Google Scholar] [CrossRef]
- Mally, J.; Stone, T. Efficacy of an adenosine antagonist, theophylline, in essential tremor: Comparison with placebo and propranolol. J. Neurol. Sci. 1995, 132, 129–132. [Google Scholar] [CrossRef]
- Mally, J.; Stone, T.W. Potential role of adenosine antagonist therapy in pathological tremor disorders. Pharmacol. Ther. 1996, 72, 243–250. [Google Scholar] [CrossRef]
- Mally, J.; Stone, T.W. The effect of theophylline on essential tremor: The possible role of GABA. Pharmacol. Biochem. Behav. 1991, 39, 345–349. [Google Scholar] [CrossRef]
- Louis, E.D.; Ba, E.C.J.; Ba, L.A.; Luchsinger, J.A.; Factor-Litvak, P.; Parides, M. Semiquantitative study of current coffee, caffeine, and ethanol intake in essential tremor cases and controls. Mov. Disord. 2004, 19, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.-H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of Coffee and Caffeine Intake With the Risk of Parkinson Disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Prakash, K.; Fook-Choong, S.; Yuen, Y.; Tan, E. Exploring the relationship between caffeine intake and essential tremor. J. Neurol. Sci. 2006, 251, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Al-Deeb, S.; Al-Moutaery, K.; Arshaduddin, M.; Biary, N.; Tariq, M. Effect of acute caffeine on severity of harmaline induced tremor in rats. Neurosci. Lett. 2002, 325, 216–218. [Google Scholar] [CrossRef]
- Trotta, E.E.; Freire, G.L. Inhibition by caffeine of calcium uptake by brain microsomal vesicles. J. Pharm. Pharmacol. 1980, 32, 791–793. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Serrano, A.; Satrústegui, J. Caffeine-sensitive calcium stores in presynaptic nerve endings: A physiological role? Biochem. Biophys. Res. Commun. 1989, 161, 965–971. [Google Scholar] [CrossRef]
- Ferreira, J.J.; Mestre, T.A.; Lyons, K.E.; Benito-León, J.; Tan, E.; Abbruzzese, G.; Hallett, M.; Haubenberger, D.; Elble, R.; Deuschl, G.; et al. MDS evidence-based review of treatments for essential tremor. Mov. Disord. 2019, 34, 950–958. [Google Scholar] [CrossRef]
- Tasker, R.R. Deep brain stimulation is preferable to thalamotomy for tremor suppression. Surg. Neurol. 1998, 49, 145–153. [Google Scholar] [CrossRef]
- Chang, S.-Y.; Kim, I.; Marsh, M.P.; Jang, D.P.; Hwang, S.-C.; Van Gompel, J.J.; Goerss, S.J.; Kimble, C.J.; Bennet, K.E.; Garris, P.A.; et al. Wireless Fast-Scan Cyclic Voltammetry to Monitor Adenosine in Patients With Essential Tremor During Deep Brain Stimulation. Mayo Clin. Proc. 2012, 87, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Kiliman, B.; Humes, C.; Arai, A.C. Spontaneous activity in Purkinje cells: Multi-electrode recording from organotypic cerebellar slice cultures. Brain Res. 2008, 1218, 54–69. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Main Mechanism of Action | Therapeutic Effect | Main Side-Effects |
|---|---|---|---|
| First-line therapies Approved by the FDA, or supported by double-blinded, placebo controlled studies that meet the criteria for class I evidence (defined by USPSTF) | |||
| Propranolol | Non-selective β-adrenergic receptor antagonist Anti-tremor mechanism: probably acting on peripheral non-cardiac β2 receptors (for more information see Section 2.4.1). | Tremor reduction by 50–70% (>50% ET patients respond), mainly for tremor affecting upper extremities, and head tremor response islimited | Frequent, rather mild, occurs in >60% ET patients: hypotension, bradycardia, slow heartbeat, fatigue, dizziness, exertional dyspnea, erectile dysfunction, headaches, somnolence |
| Primidone | Derivative of barbituric acid, antiepileptic drug Anti-tremor mechanism: still unknown; interacts with voltage-gated sodium channels or opening/potentiating GABA receptors | Tremor reduction by 50–70% (30–50% ET patients respond) | In 22–72% ET patients: sedation, fatigue, drowsiness, dizziness, ataxia, confusion, nausea, loss of coordination, anorexia, nausea, vomiting, flu-like symptoms |
| Second-line therapies Supported by double-blinded, placebo-controlled trials that do not meet the requirements for class I evidence studies | |||
| Topiramate Most effective second-line treatment | Blockade of voltage-gated sodium channels, inhibition of high voltage-activated calcium channels Anti-tremor mechanism: not clear | Tremor improvement by 20–37% (30–40% response rate) | Paresthesias, difficulties with concentration, nausea, somnolence, fatigue, malaise, dyspepsia, weight loss, confusion, psychomotor slowing, abnormal taste perception, visual disturbances, nephrolithiasis Discontinuation of therapy because of adverse side-effects in approximately 30% of ET patients |
| Gabapentin Pregabalin | Structural analogs of GABA Inhibit α2δ subunits of voltage-gated calcium N-type channels; do not bind directly to GABA-A, GABA-B receptors Anti-tremor mechanism: not clear | Tremor improvement by 30–40% (approximately 30–50% response rate) | Sleepiness, dizziness, ataxia, nausea, weight gain in 30–40% of patients (mild) Discontinuation in cases of increased anxiety, insomnia, nausea, increased risk of depression and suicidal behavior |
| Alprazolam Clonazepam | Benzodiazepines Bind to GABA-A receptor complex, Cl- influx | Tremor improvement by 30–50% (>50% response rate) | Sedation, cognitive impairment, tolerance, dependency, abuse, withdrawal symptoms, side-effects in approximately 50% of ET patients |
| Atenolol | Competitive β1-adenergic antagonist | Only in patients responding to propranolol (37% tremor reduction); response rate similar to other β-blockers | Similar to propranolol (see above), without possible bronchospasm |
| Metoprolol | Competitive β1-adenergic antagonist | Equal efficacy to propranolol (single dose), but no effect on tremor after chronic use | Similar to propranolol (see above) |
| Nadolol Sotalol Indenolol Arotinolol Timolol | Non-selective β-blockers | Effective, but only in ET patients who responded previously to propranolol | Most common side-effect isreduced alertness, otherwise similar to propranolol (see above) Adverse effects in approximately 25% patients |
| Third-line therapies Based on open-label studies or case series | |||
| Nimodipine Flunarizine Nicardipine | Calcium channel blockers Binds to L-type voltage-gated Ca2+ channel | Tremor improvement by 50% (>50% response rate), based on studies on small numbers of ET patients | Most common: hypotension, oedema, headaches in 10–20% patients, dizziness, nausea, constipation, fatigue, palpitations and others (rather well-tolerated) |
| Clozapine | Atypical antipsychotic High affinity to D1, D4, 5HT2A, 5HT2C, 5HT6, 5HT7, α1- and α 2-adrenergic receptors, H1 and M1-M5 muscarinic receptors, weak affinity for D2 receptors | Tremor improvement by 50% (75% response rate), based on small clinical trials | Main: sedation, orthostatic hypotension, tachycardia, syncope, weight gain, metabolic syndrome (adverse effects in approximately 50% of ET patients) Possible: agranulocytosis, cardiomyopathy, tachycardia, risk of seizure, risk of neutropenia (not observed) |
| Olanzapine Quetiapine | Atypical antipsychotics High/moderate affinity to 5HT2A/C, 5HT6, 5HT7, D1-D4, H1, α1-adrenergic receptors, and M1-M5 muscarinic receptors | Tremor improvement by <50% | Insomnia, anxiety, headache, sedation, somnolence, dizziness, weight gain, orthostatic hypotension In long-term treatment there is an increased risk of parkinsonism |
| Imaging Study | Clinical Diagnosis, Subjects | Key Findings | References |
|---|---|---|---|
| Markers of DAT | |||
| [123I]β-CIT SPECT | ET (n = 32), controls (n = 30) | No difference in the striatum between ET and controls | Asenbaum et al. (1998) [161] |
| [123I]FP-CIT SPECT | ET (n = 27), controls (n = 35) | No alterations in ET patients vs. controls | Benamer et al. (2000) [162] |
| ET (n = 20), controls (n = 23) | No alterations in ET patients vs. controls | Isaias et al. (2010) [163] | |
| [11C]FE-CIT PET | ET (n = 5), controls (n = 8) | No difference between ET patients and controls | Antonini et al. (2001) [164] |
| [123I]ioflupane SPECT | ET (n = 15), controls (n = 17) | No alterations in ET patients vs. controls | Di Giuda et al. (2012) [165] |
| ET (n = 22), controls (n = 13) | No alterations in ET patients vs. controls | Waln et al. (2015) [166] | |
| ET (n = 12), controls (n = 10) | No alterations in ET with rest tremor vs. controls | Barbagallo et al. (2017) [167] | |
| ET (n = 28), controls (n = 28) | Mild striatal deficit in ET patients vs. control (less marked than in PD) | Gerasimou et al. (2012) [168] | |
| ET (n = 32, including 16 familial), controls (n = 31) | Mild striatal deficit in ET patients vs. control (less marked than in PD) | Isaias et al. (2008) [169] | |
| [99mTc]TRODAT-1 SPECT | ET (n = 12), control (n = 10) | No alterations in ET patients vs. controls | Wang et al. (2005) [170] |
| [11C]dMP PET | ET (n = 6), controls (n = 10) | No alterations in ET patients vs. controls | Breit et al. (2006) [171] |
| Presynaptic radioligand (measures DA synthesis) | |||
| [18F]DOPA PET | ET (n = 20, including 8 familial), controls (n = 30) | ↓ 13% uptake in putamen (familial ET) and ↓ 10% (sporadic ET) vs. control | Brooks et al. (1992) [172] |
| Postsynaptic (D2 R) | |||
| [123I]IBZM SPECT | ET (n = 11), no controls | No alterations in ET patients | Plotkin et al. (2005) [173] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosmowska, B.; Wardas, J. The Pathophysiology and Treatment of Essential Tremor: The Role of Adenosine and Dopamine Receptors in Animal Models. Biomolecules 2021, 11, 1813. https://doi.org/10.3390/biom11121813
Kosmowska B, Wardas J. The Pathophysiology and Treatment of Essential Tremor: The Role of Adenosine and Dopamine Receptors in Animal Models. Biomolecules. 2021; 11(12):1813. https://doi.org/10.3390/biom11121813
Chicago/Turabian StyleKosmowska, Barbara, and Jadwiga Wardas. 2021. "The Pathophysiology and Treatment of Essential Tremor: The Role of Adenosine and Dopamine Receptors in Animal Models" Biomolecules 11, no. 12: 1813. https://doi.org/10.3390/biom11121813
APA StyleKosmowska, B., & Wardas, J. (2021). The Pathophysiology and Treatment of Essential Tremor: The Role of Adenosine and Dopamine Receptors in Animal Models. Biomolecules, 11(12), 1813. https://doi.org/10.3390/biom11121813