
Engineering a Pseudo-26-kDa Schistosoma Glutathione Transferase from bovis/haematobium for Structure, Kinetics, and Ligandin Studies

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Vector Construction
2.3. Protein Over-Expression
2.4. Affinity Chromatography Purification
2.5. GSH–CDNB Conjugation Assay
2.6. Far-UV Circular Dichroism
2.7. Fluorescence Spectroscopy
2.8. Isothermal Titration Calorimetry
2.9. Thermal Shift Assay
2.10. Homology Modelling and Induced Fit Ligand Docking
3. Results
3.1. Expression and Purification of Sbh26GST
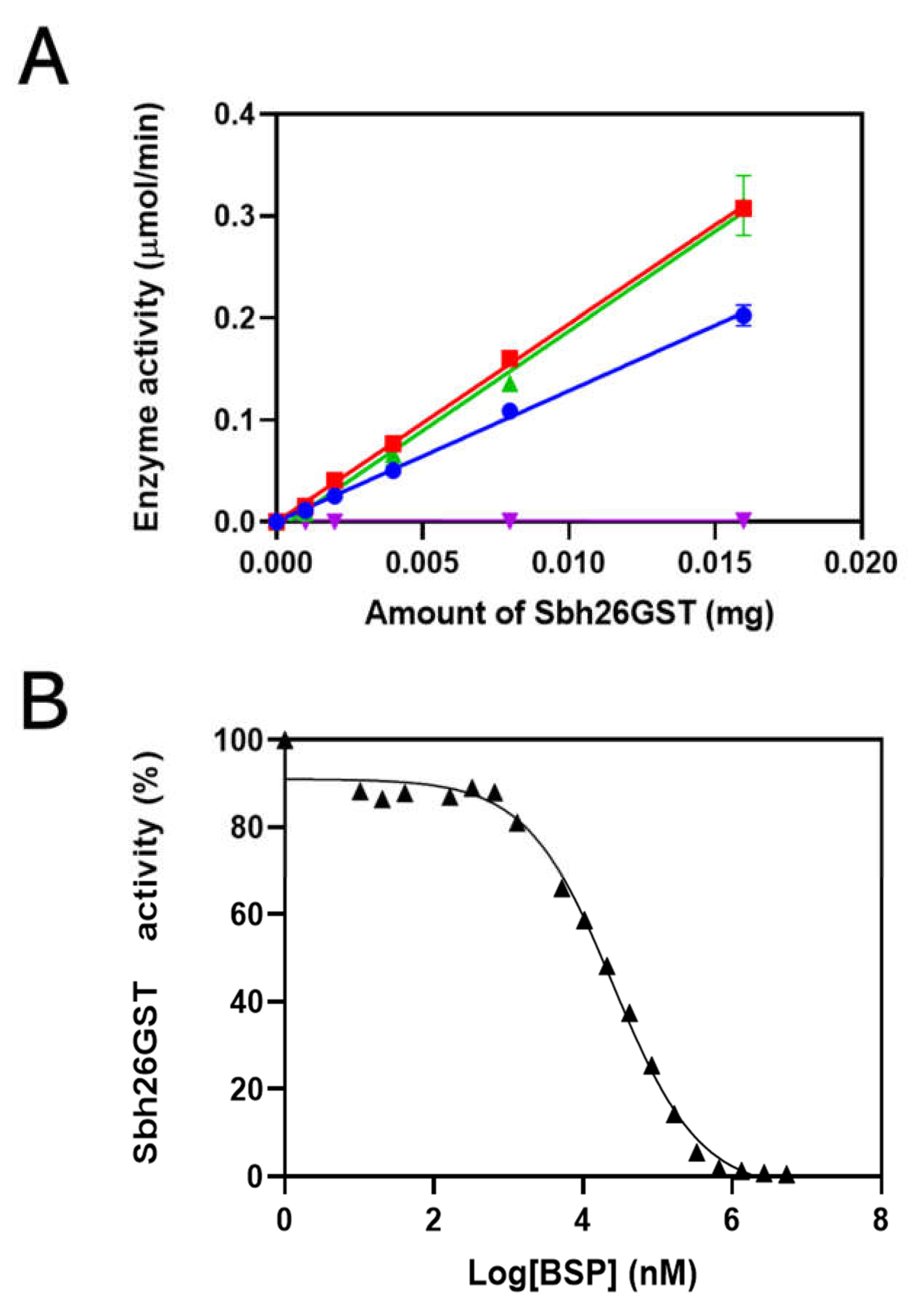
3.2. Characterization of Sbh26GST Using GSH–CDNB Conjugation Assay
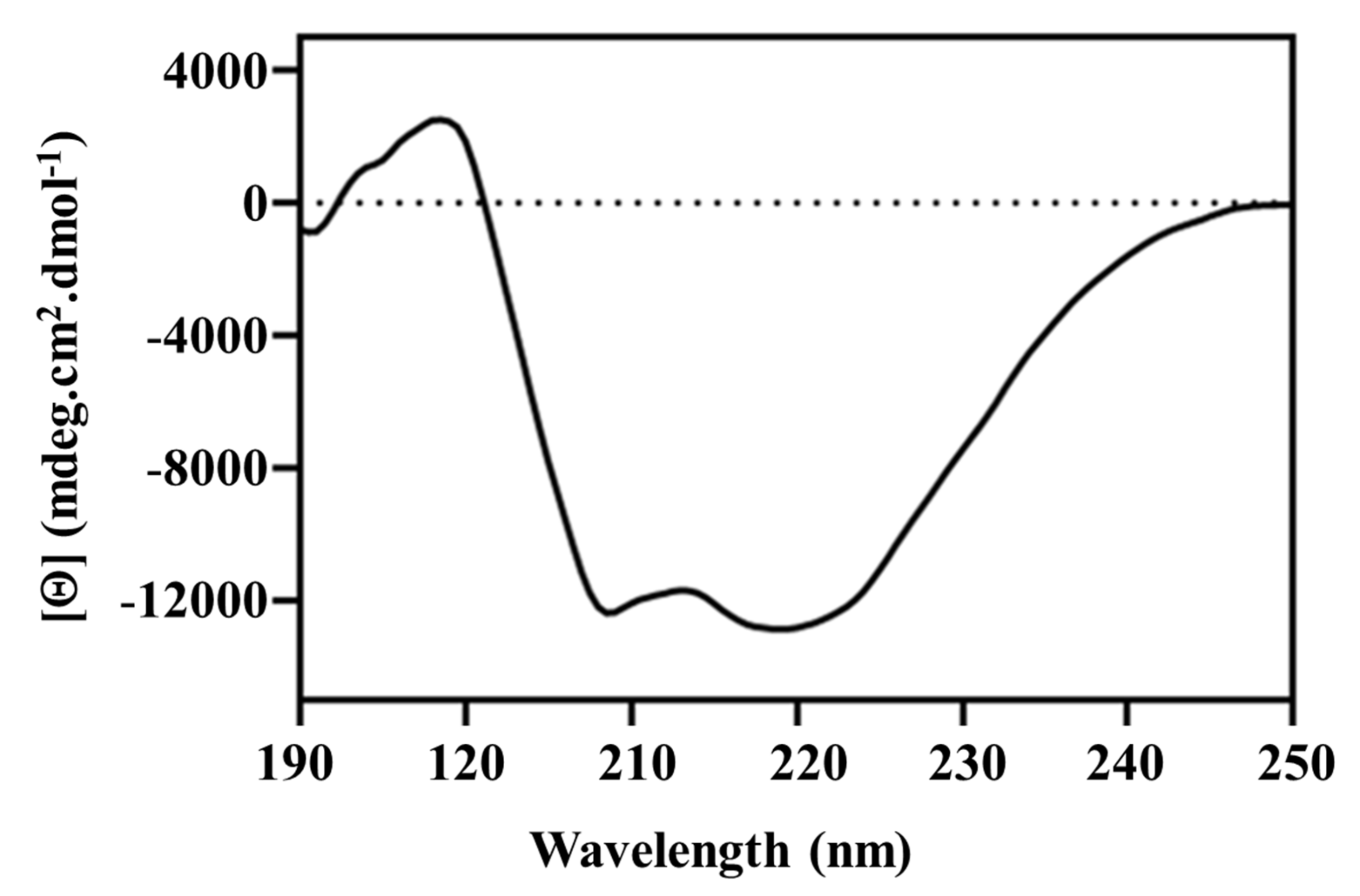
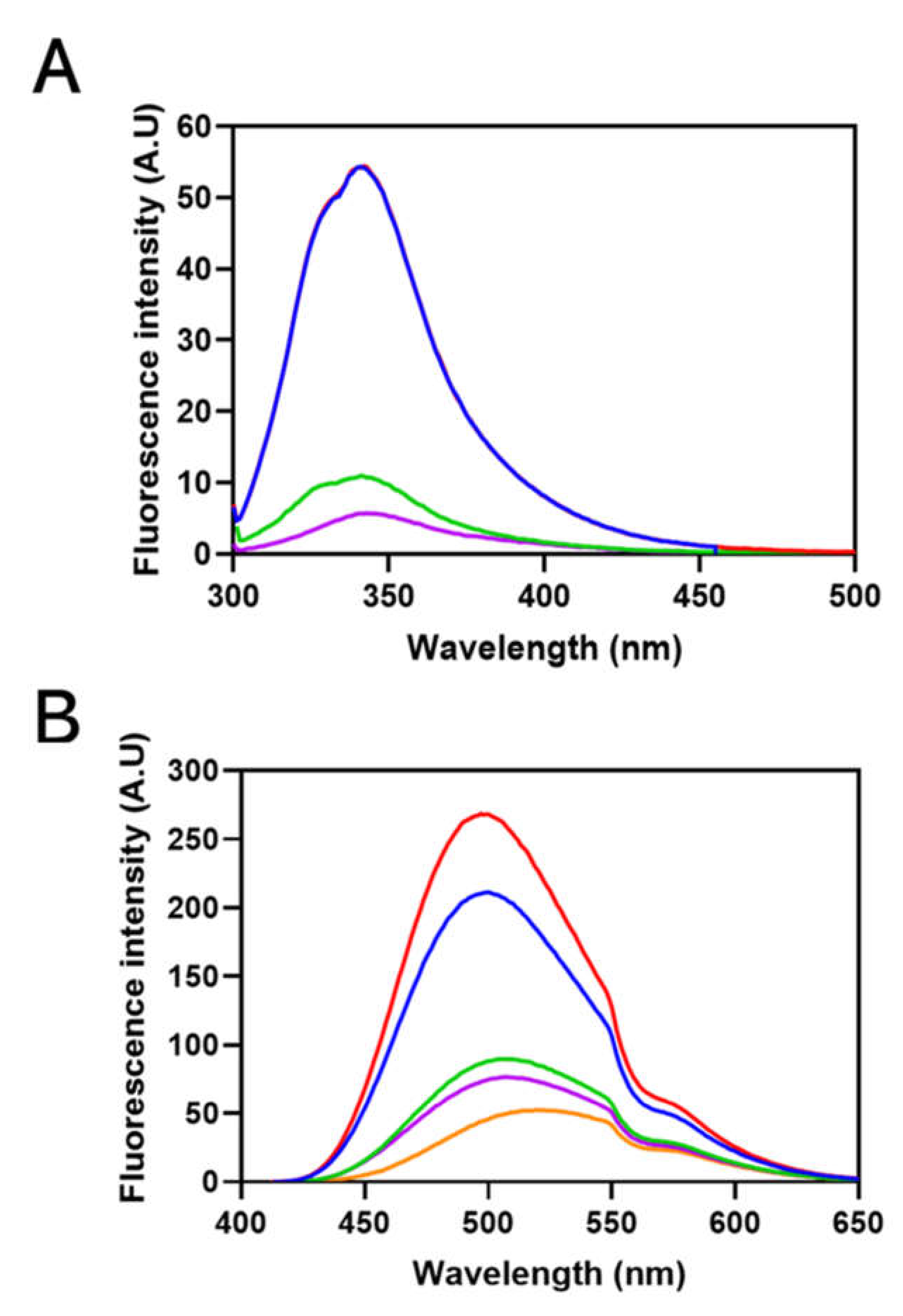
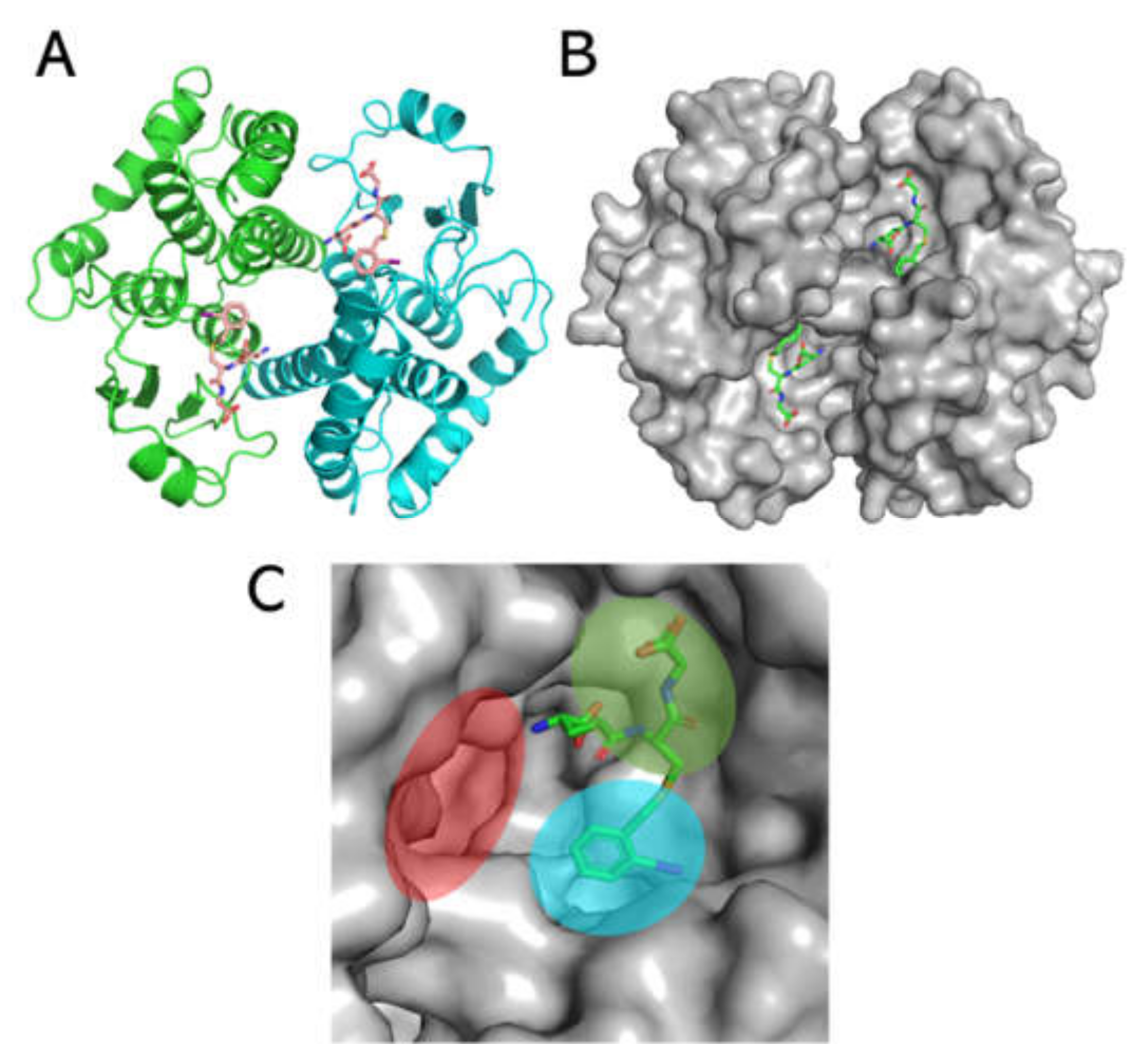
3.3. Structural Analysis of Sbh26GST
3.4. Thermodynamic Parameters of the Interactions between Sbh26GST and BSP
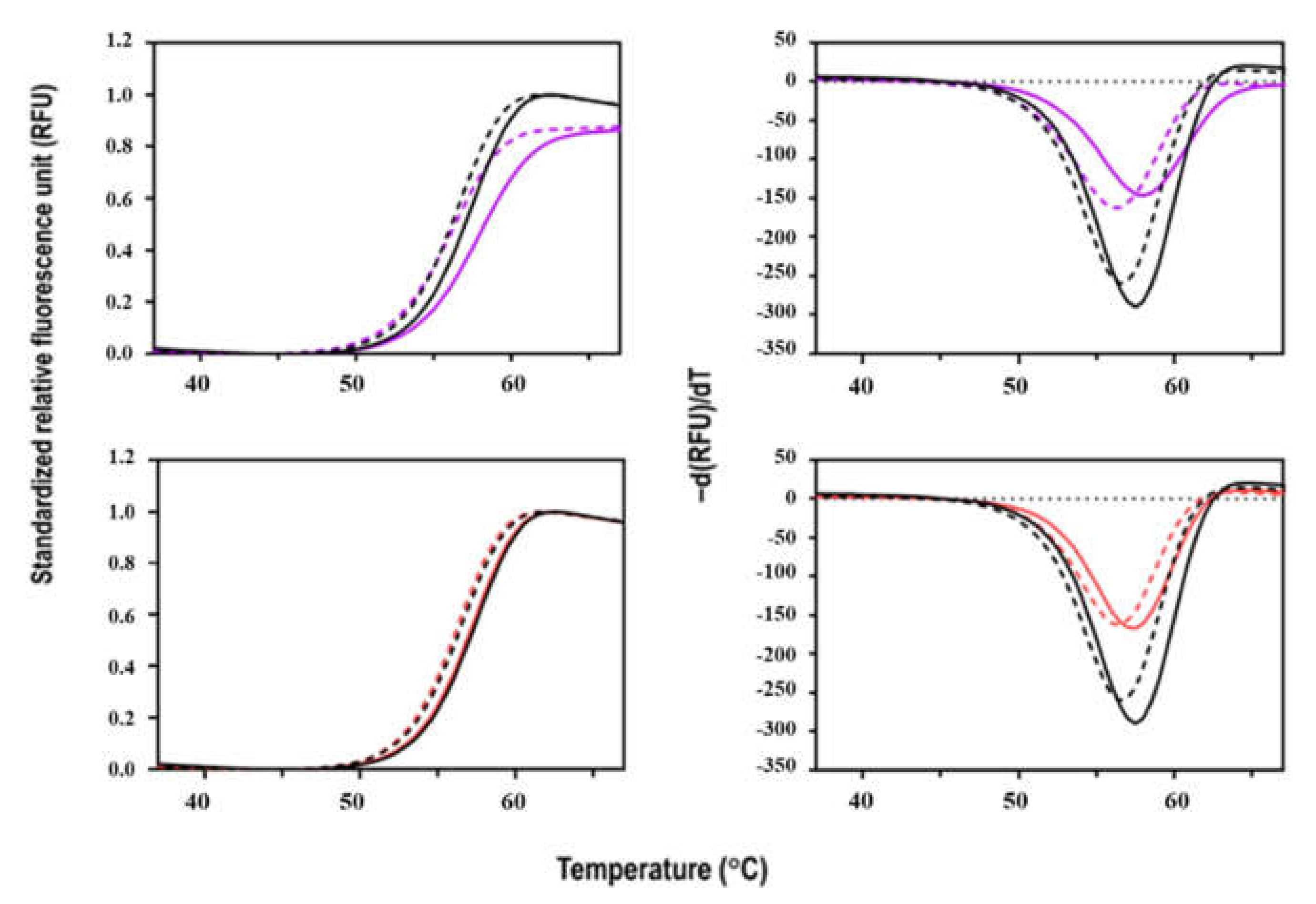
3.5. Thermal Stability of Sbh26GST
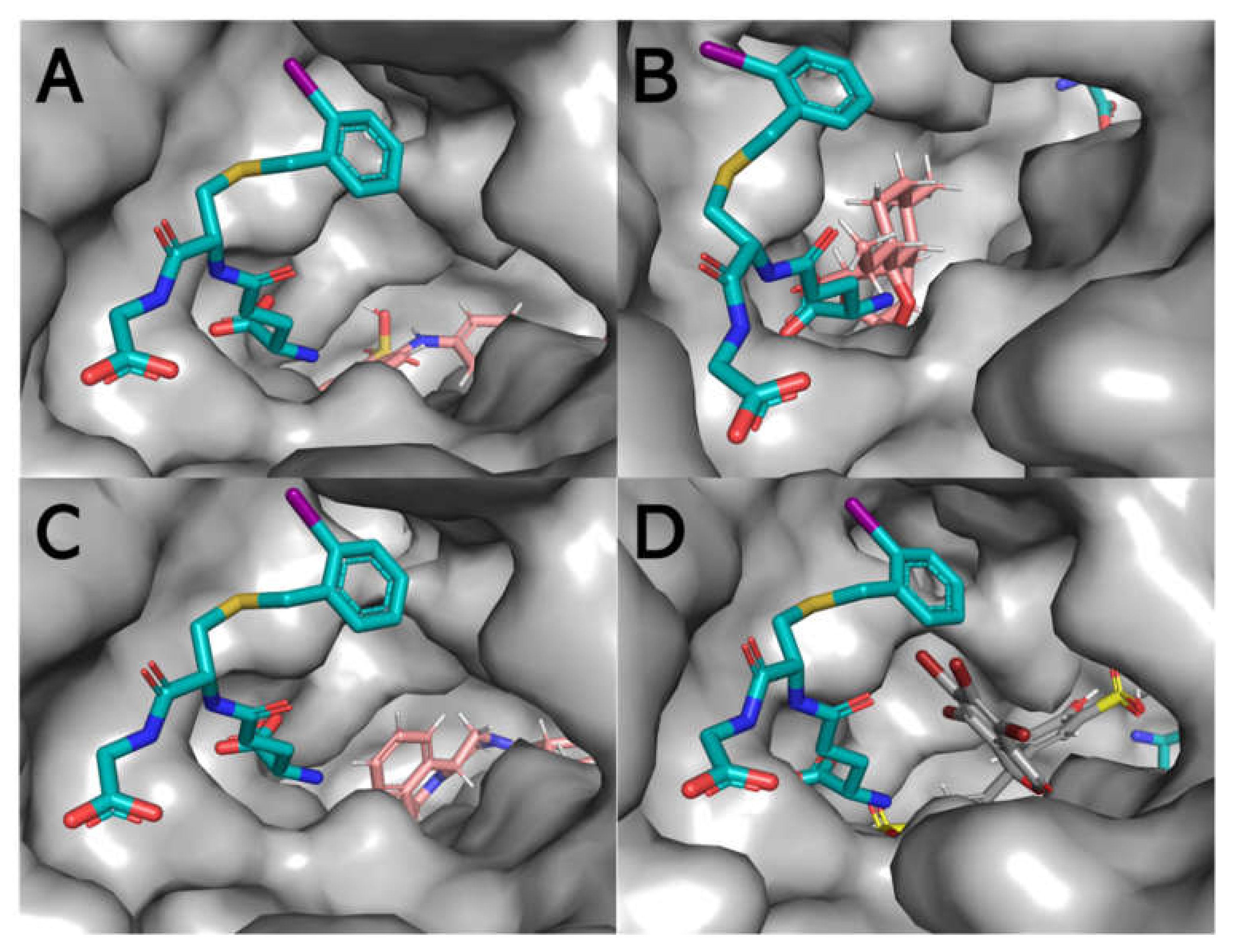
3.6. Theoretical Modelling of Sbh26GST Interacting with ANS, ART, PZQ, and BSP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PZQ | Praziquantel |
| ART | Artemisinin |
| ANS | 8-anilino-1-naphthalenesulfonate sulfonate |
| BSP | Bromosulfophthalein |
| GST | Glutathione transferase |
| IFD | Induced fit docking |
| CD | Circular dichroism |
| GSH | Glutathione |
| CDNB | 1-Chloro-2,4-dinitrobenzene |
| MBP | Maltose binding protein |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| ORF | Open reading frame |
| DTT | Dithiothreitol |
| EDTA | Ethylenediaminetetraacetic acid |
References
- Sangweme, D.T.; Midzi, N.; Zinyowera-Mutapuri, S.; Mduluza, T.; Diener-West, M.; Kumar, N. Impact of schistosome infection on Plasmodium falciparum Malariometric indices and immune correlates in school age children in Burma Valley, Zimbabwe. PLoS Negl. Trop. Dis. 2010, 4, e882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitsulo, L.; Engels, D.; Montresor, A.; Savioli, L. The global status of schistosomiasis and its control. Acta Trop. 2000, 77, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Cioli, D.; Pica-Mattoccia, L.; Basso, A.; Guidi, A. Schistosomiasis control: Praziquantel forever? Mol. Biochem. Parasitol. 2014, 195, 23–29. [Google Scholar] [CrossRef]
- Alger, H.M.; Williams, D.L. The disulfide redox system of Schistosoma mansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Mol. Biochem. Parasitol. 2002, 121, 129–139. [Google Scholar] [CrossRef]
- Huyse, T.; Webster, B.L.; Geldof, S.; Stothard, J.R.; Diaw, O.T.; Polman, K.; Rollinson, D. Bidirectional introgressive hybridization between a cattle and human schistosome species. PLoS Path. 2009, 5, e1000571. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.; Crowley, P.; Moreman, A.D.; Barrett, J. Biochemical properties of cloned glutathione S-transferases from Schistosoma mansoni and Schistosoma japonicum. Mol. Biochem. Parasitol. 1993, 61, 255–264. [Google Scholar] [CrossRef]
- Toh, S.Q.; Glanfield, A.; Gobert, G.N.; Jones, M.K. Heme and blood-feeding parasites: Friends or foes? Parasit. Vectors 2010, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhan, B.; Perally, S.; Brophy, P.M.; Xue, J.; Goud, G.; Liu, S.; Deumic, V.; de Oliveira, L.M.; Bethony, J.; Bottazzi, M.E. Molecular cloning, biochemical characterization, and partial protective immunity of the heme-binding glutathione S-transferases from the human hookworm Necator americanus. Infect. Immun. 2010, 78, 1552–1563. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M. Hybridisation experiments on five species of African schistosomes. J. Helminthol. 1970, 44, 253–314. [Google Scholar] [CrossRef]
- Agnew, A.; Murare, H.; Lucas, S.; Doenhoff, M. Schistosoma bovis as an immunological analogue of S. haematobium. Parasite Immunol. 1989, 11, 329–340. [Google Scholar] [CrossRef]
- Djuikwo-Teukeng, F.F.; Simo, A.K.; Allienne, J.-F.; Rey, O.; Ngapagna, A.N.; Tchuem-Tchuente, L.A.; Boissier, J. Population genetic structure of Schistosoma bovis in Cameroon. Parasit. Vectors 2019, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.; Li Hsu, H.; Chu, K.; Tsai, C.; Eveland, L. Immunization against Schistosoma haematobium in rhesus monkeys by administration of cercariae of Schistosoma bovis. Z. Fur Trop. Und Parasitol. 1966, 17, 407–412. [Google Scholar]
- McTigue, M.A.; Williams, D.R.; Tainer, J.A. Crystal Structures of a Schistosomal Drug and Vaccine Target: Glutathione S-Transferase from Schistosoma japonica and its Complex with the Leading Antischistomal Drug Praziquantel. J. Mol. Biol. 1995, 246, 21–27. [Google Scholar] [CrossRef]
- Pooe, K.; Worth, R.; Iwuchukwu, E.A.; Dirr, H.W.; Achilonu, I. An empirical and theoretical description of Schistosoma japonicum glutathione transferase inhibition by bromosulfophthalein and indanyloxyacetic acid 94. J. Mol. Struct. 2021, 1223, 128892. [Google Scholar] [CrossRef]
- Akumadu, B.O.; Pandian, R.; Olfsen, J.; Worth, R.; Thulo, M.; Mentor, T.; Fanucchi, S.; Sayed, Y.; Dirr, H.W.; Achilonu, I. Molecular basis of inhibition of Schistosoma japonicum glutathione transferase by ellagic acid: Insights into biophysical and structural studies. Mol. Biochem. Parasitol. 2020, 240, 111319. [Google Scholar] [CrossRef]
- Yassin, Z.; Ortiz-Salmerón, E.; García-Maroto, F.; Barón, C.; García-Fuentes, L. Implications of the ligandin binding site on the binding of non-substrate ligands to Schistosoma japonicum-glutathione transferase. Biochim. Biophys. Acta 2004, 1698, 227–237. [Google Scholar] [CrossRef]
- Schwenk, M.; Burr, R.; Schwarz, L.; Pfaff, E. Uptake of Bromosulfophthalein byIsolated Liver Cells. Eur. J. Biochem. 1976, 64, 189–197. [Google Scholar] [CrossRef]
- Cui, F.; Sequeira, S.B.; Huang, Z.; Shang, G.; Cui, Q.; Yang, X. Bromosulfophthalein suppresses inflammatory effects in lipopolysaccharide-stimulated RAW264. 7 macrophages. Immunopharmacol. Immunotoxicol. 2020, 42, 456–463. [Google Scholar] [CrossRef]
- Kolobe, D.; Sayed, Y.; Dirr, H.W. Characterization of bromosulphophthalein binding to human glutathione S-transferase A1-1: Thermodynamics and inhibition kinetics. Biochem. J. 2004, 382, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Bustamante, L.; Haynes, R.K.; Staines, H.M. Artemisinins: Their growing importance in medicine. Trends Pharmacol. Sci. 2008, 29, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Habig, W.H.; Jakoby, W.B. Assays for differentiation of glutathione S-transferases. Methods Enzymol. 1981, 77, 398–405. [Google Scholar]
- Whitmore, L.; Wallace, B. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [Green Version]
- Van Stokkum, I.H.; Spoelder, H.J.; Bloemendal, M.; Van Grondelle, R.; Groen, F.C. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990, 191, 110–118. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Introduction to fluorescence. In Principles of Fluorescence Spectroscopy; Springer Scinece & Business Media: Berlin, Germany, 1999; pp. 1–23. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Quesada-Soriano, I.; García-Maroto, F.; García-Fuentes, L.; Proteomics. Kinetic study on the irreversible thermal denaturation of Schistosoma japonicum glutathione S-transferase. Biochim. Biophys. Acta 2006, 1764, 979–984. [Google Scholar] [CrossRef]
- Jakoby, W.B. Reactions of glutathione transferases: A pattern for the enzymes of detoxication; Taylor and Francis: London, UK, 1990; Volume 1, p. 10. [Google Scholar]
- Armstrong, R.N. Glutathione S-transferases: Reaction mechanism, structure, and function. Chem. Res. Toxicol. 1991, 4, 131–140. [Google Scholar] [CrossRef]
- Dirr, H. Folding and assembly of glutathione transferases. Chem.-Biol. Interact. 2001, 133, 19–23. [Google Scholar]
- Dirr, H.; Reinemer, P.; Huber, R. X-ray crystal structures of cytosolic glutathione S-transferases: Implications for protein architecture, substrate recognition and catalytic function. Eur. J. Biochem. 1994, 220, 645–661. [Google Scholar] [CrossRef]
- Fanucchi, S.; Adamson, R.J.; Dirr, H.W. Formation of an unfolding intermediate state of soluble chloride intracellular channel protein CLIC1 at acidic pH. Biochemistry 2008, 47, 11674–11681. [Google Scholar] [CrossRef]
- Gildenhuys, S.; Dobreva, M.; Kinsley, N.; Sayed, Y.; Burke, J.; Pelly, S.; Gordon, G.P.; Sayed, M.; Sewell, T.; Dirr, H.W. Arginine 15 stabilizes an SNAr reaction transition state and the binding of anionic ligands at the active site of human glutathione transferase A1-1. Biophys. Chem. 2010, 146, 118–125. [Google Scholar] [CrossRef]
- Masino, L.; Kelly, G.; Leonard, K.; Trottier, Y.; Pastore, A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002, 513, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Sayed, Y.; Hornby, J.A.; Lopez, M.; Dirr, H. Thermodynamics of the ligandin function of human class Alpha glutathione transferase A1-1: Energetics of organic anion ligand binding. Biochem. J. 2002, 363, 341–346. [Google Scholar] [CrossRef]
- Walmsley, A.R.; Martin, G.; Henderson, P. 8-Anilino-1-naphthalenesulfonate is a fluorescent probe of conformational changes in the D-galactose-H+ symport protein of Escherichia coli. J. Biol. Chem. 1994, 269, 17009–17019. [Google Scholar] [CrossRef]
- Lo, M.-C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Parameter | Value ± SD | ||
|---|---|---|---|
| No Ligand | ART | BSP | |
| Vmax (μmol/min) | 0.19 ± 0.020 | 0.32 ± 0.080 | 0.03 ± 0.001 |
| Km (μM) | 120.00 ± 0.030 | 250.00 ± 0.090 | 36.50 ± 0.006 |
| kcat (S−1) | 9.80 ± 0.056 | 39.00 ± 0.011 | 1.20 ± 0.130 |
| kcat/Km (M/s) | 0.08 ± 0.041 | 150.00 ± 0.012 | 0.03 ± 0.003 |
| Thermodynamic Parameter | Independent 1 Binding Model (Value ± SD) | Multiple Site Binding Model (Value ± SD) |
|---|---|---|
| Stoichiometry (n1) | 1.13 ± 0.02 | 1.12 ± 0.05 |
| Stoichiometry (n2) | 4.83 ± 0.70 | |
| ∆H°1 (kJ/mol) | −23.35 ± 0.28 | −24.20 ± 0.33 |
| ∆H°2 (kJ/mol) | 0.53 ± 1.20 | |
| ∆S°1 (J/mol·K) | 26.12 ± 0.02 | 91.10 ± 0.06 |
| ∆S°2 (J/mol·K) | 140.90 ± 0.90 | |
| ∆G°1 (kJ/mol) | −31.14 ± 0.02 | −51.37 ± 0.07 |
| ∆G°2 (kJ/mol) | −41.48 ± 0.87 | |
| Kd1 (nM) | 35.07 ± 0.01 | 1.00 ± 0.01 |
| Kd2 (nM) | 53.83 ± 0.81 |
| Ramachandran Plot Region | Number of Residues | Percentage (%) |
|---|---|---|
| Most favored | 352 | 94.60 |
| Additional allowed | 18 | 4.80 |
| Generously allowed | 2 | 0.50 |
| Disallowed | 0 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padi, N.; Akumadu, B.O.; Faerch, O.; Aloke, C.; Meyer, V.; Achilonu, I. Engineering a Pseudo-26-kDa Schistosoma Glutathione Transferase from bovis/haematobium for Structure, Kinetics, and Ligandin Studies. Biomolecules 2021, 11, 1844. https://doi.org/10.3390/biom11121844
Padi N, Akumadu BO, Faerch O, Aloke C, Meyer V, Achilonu I. Engineering a Pseudo-26-kDa Schistosoma Glutathione Transferase from bovis/haematobium for Structure, Kinetics, and Ligandin Studies. Biomolecules. 2021; 11(12):1844. https://doi.org/10.3390/biom11121844
Chicago/Turabian StylePadi, Neo, Blessing Oluebube Akumadu, Olga Faerch, Chinyere Aloke, Vanessa Meyer, and Ikechukwu Achilonu. 2021. "Engineering a Pseudo-26-kDa Schistosoma Glutathione Transferase from bovis/haematobium for Structure, Kinetics, and Ligandin Studies" Biomolecules 11, no. 12: 1844. https://doi.org/10.3390/biom11121844
APA StylePadi, N., Akumadu, B. O., Faerch, O., Aloke, C., Meyer, V., & Achilonu, I. (2021). Engineering a Pseudo-26-kDa Schistosoma Glutathione Transferase from bovis/haematobium for Structure, Kinetics, and Ligandin Studies. Biomolecules, 11(12), 1844. https://doi.org/10.3390/biom11121844