
Towards the De Novo Design of HIV-1 Protease Inhibitors Based on Natural Products


 , and
, and 
Abstract
:1. Introduction
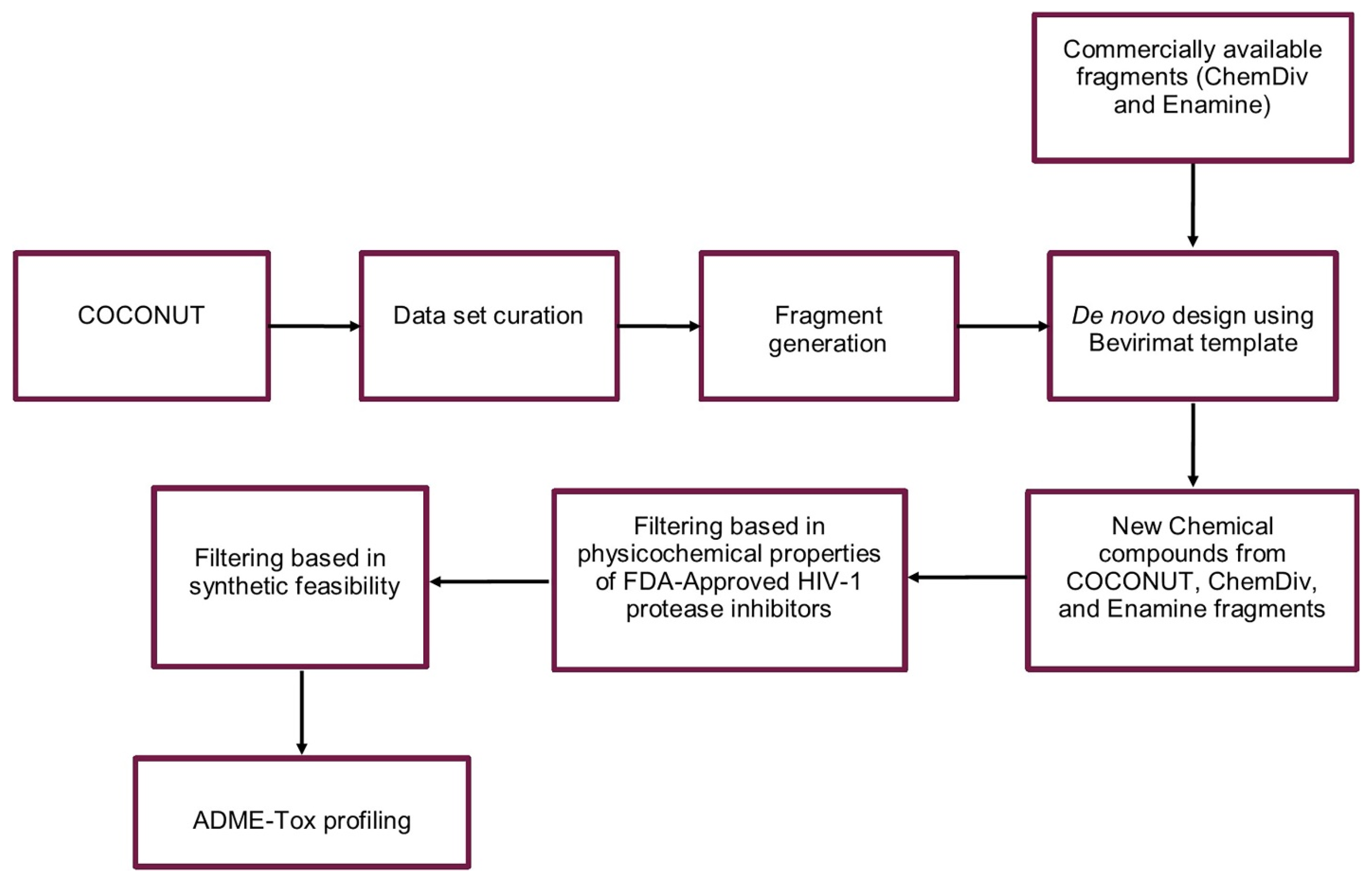
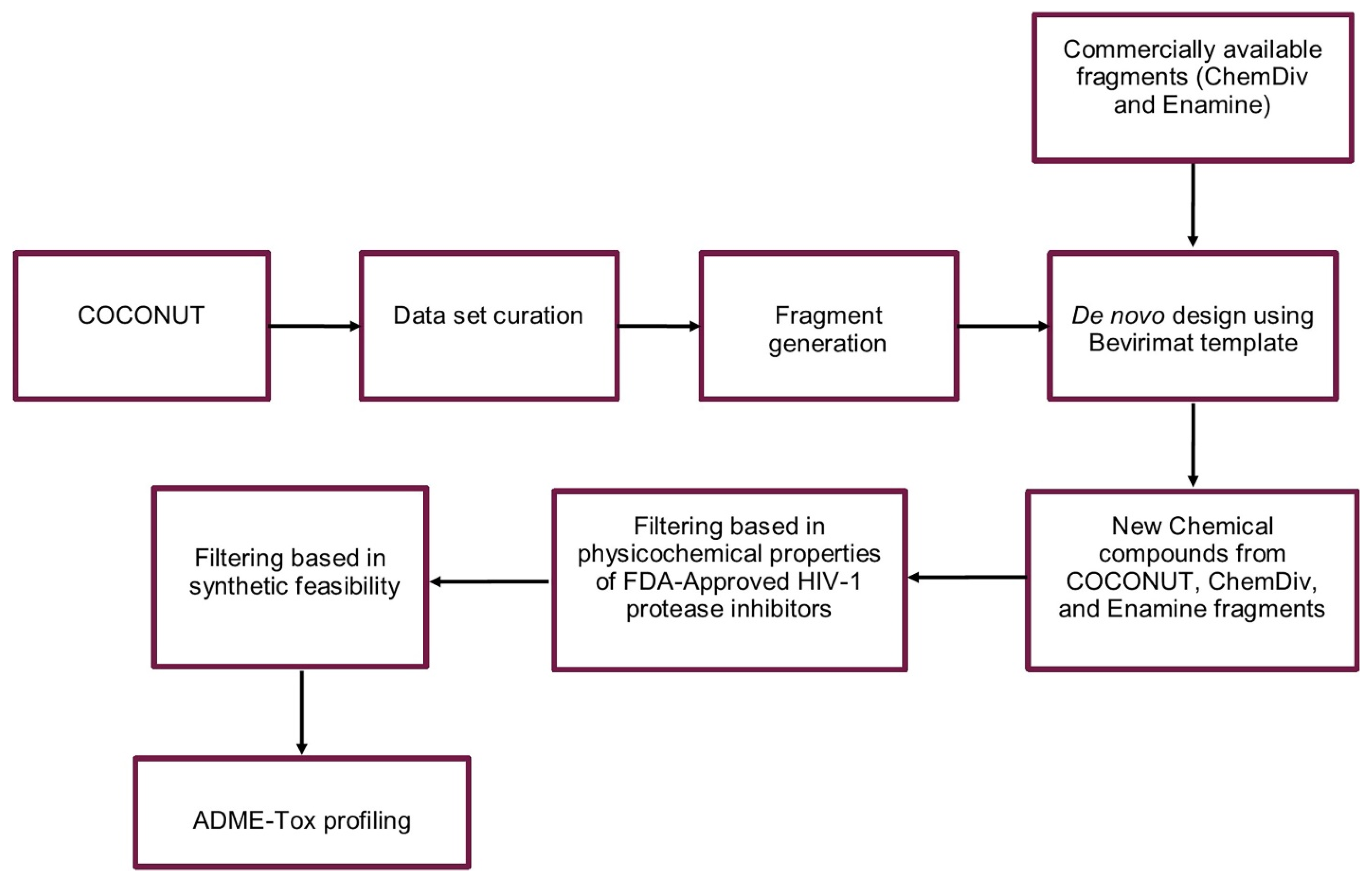
2. Materials and Methods
2.1. Dataset Curation
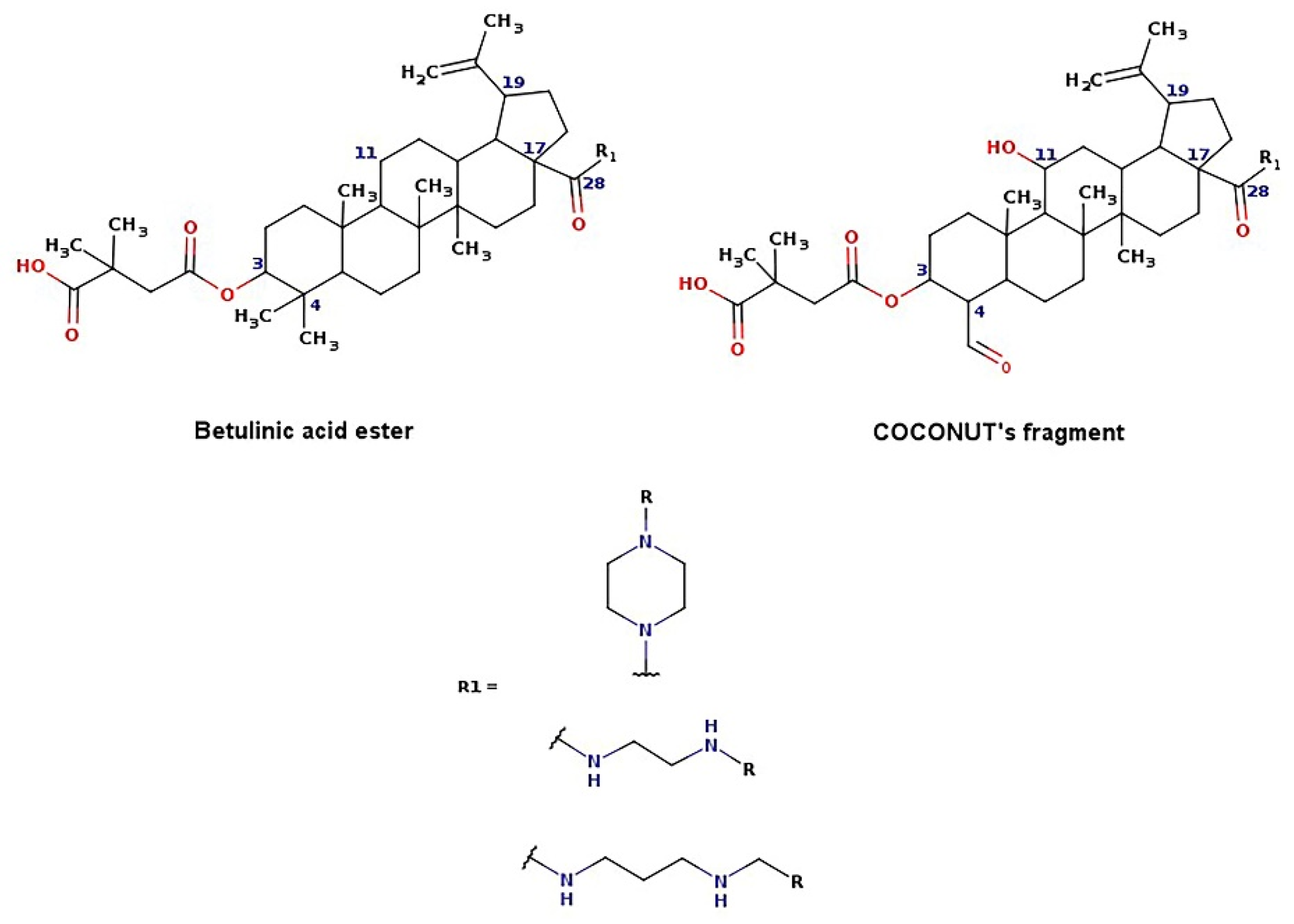
2.2. Generation of Unique Fragments Using Retrosynthetic Rules
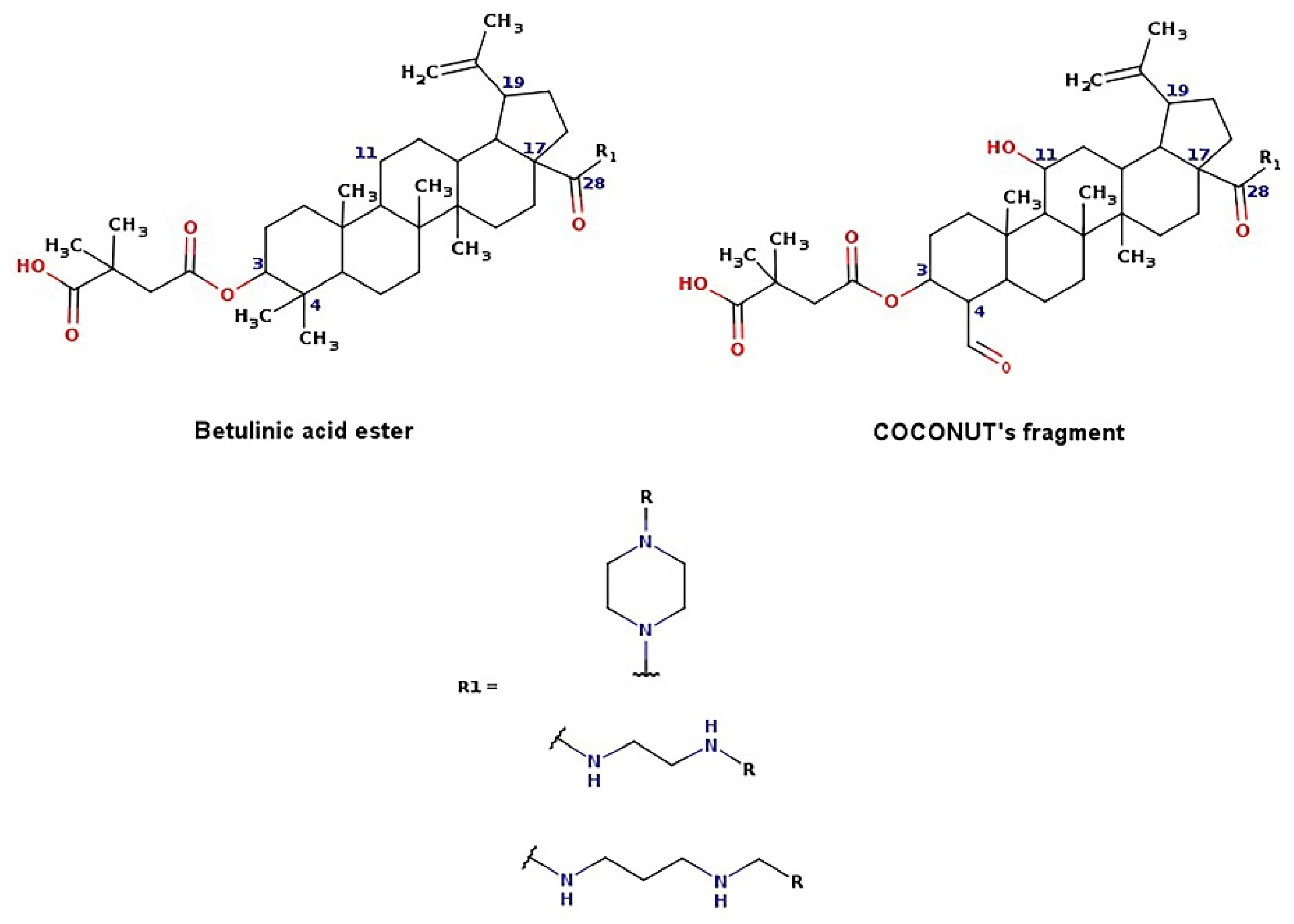
2.3. De Novo Design
2.4. Structural Diversity and Complexity
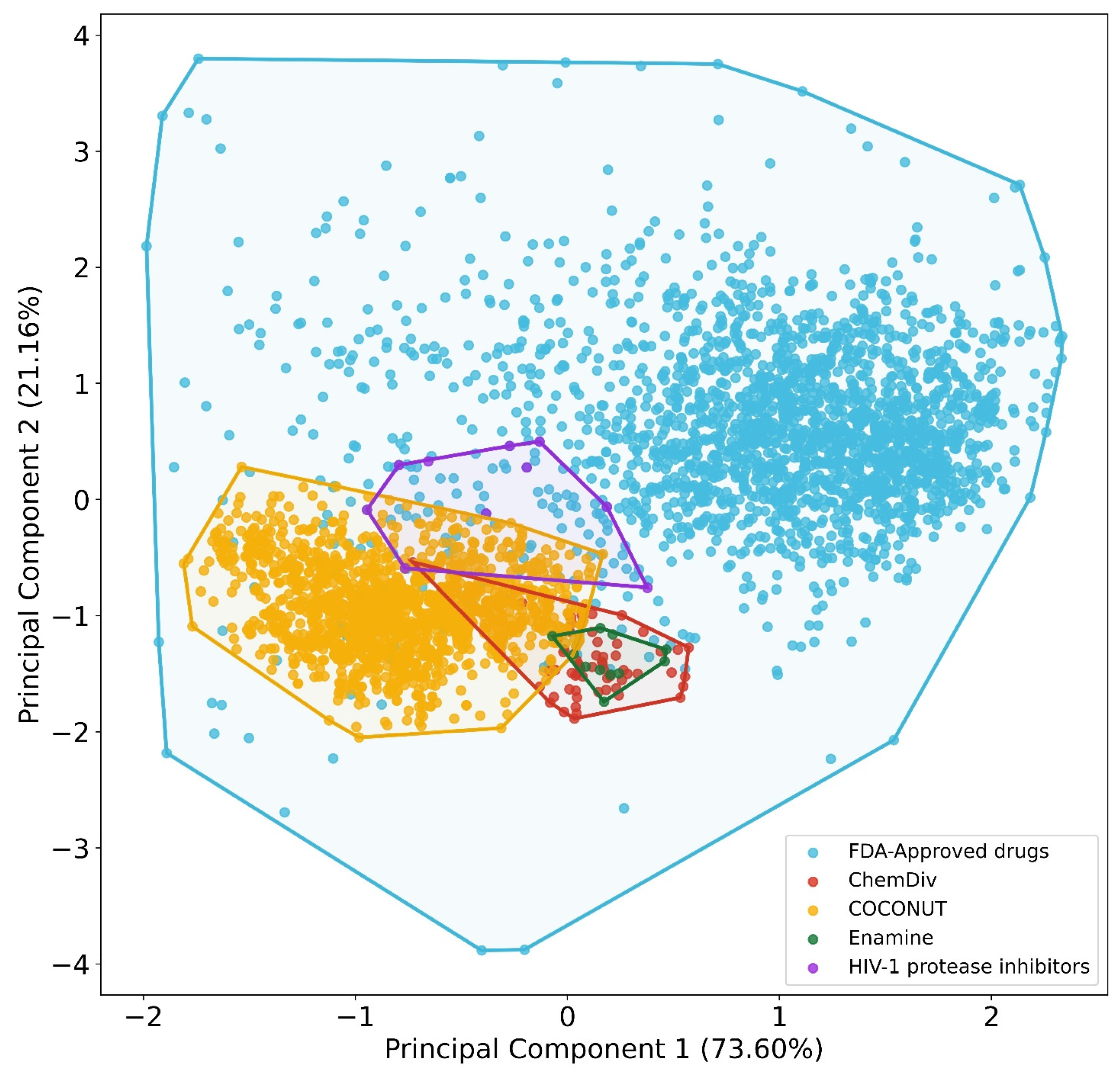
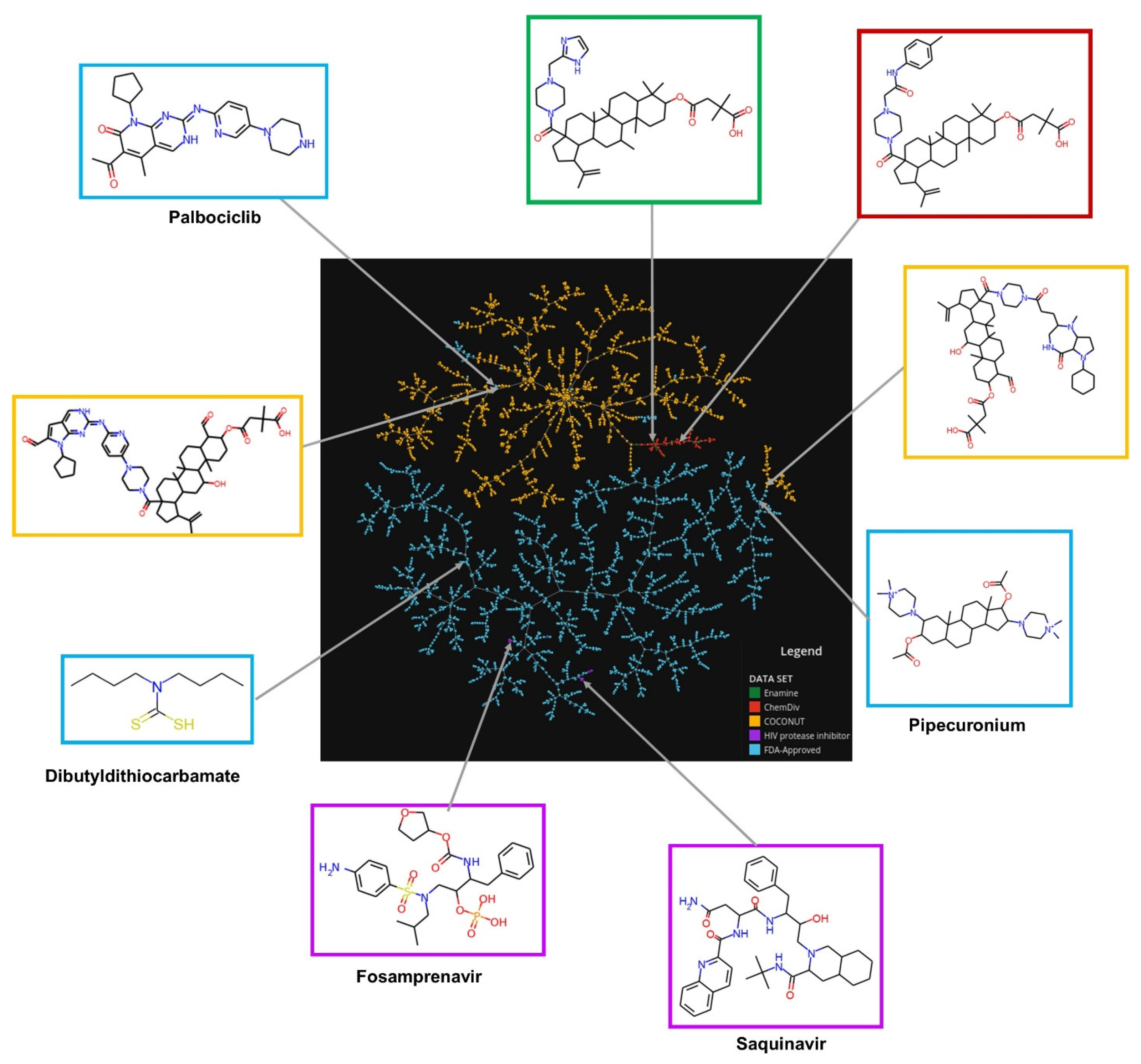
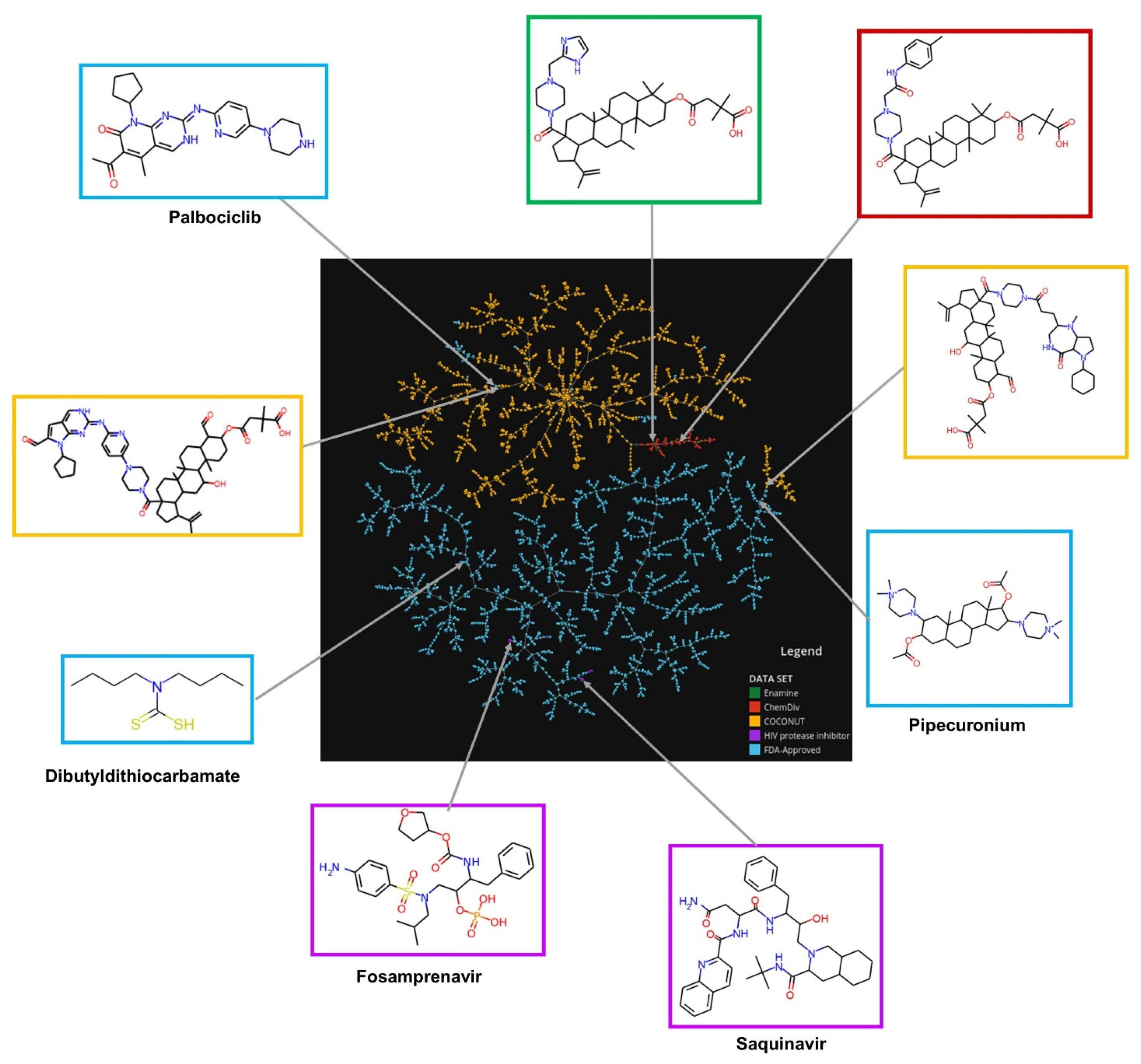
2.5. Chemical Space Visualization
2.6. Filtering of the New Chemical Compounds Generated
2.7. Synthetic Feasibility
2.8. ADME-Tox Profiling
3. Results and Discussion
3.1. Structural Diversity
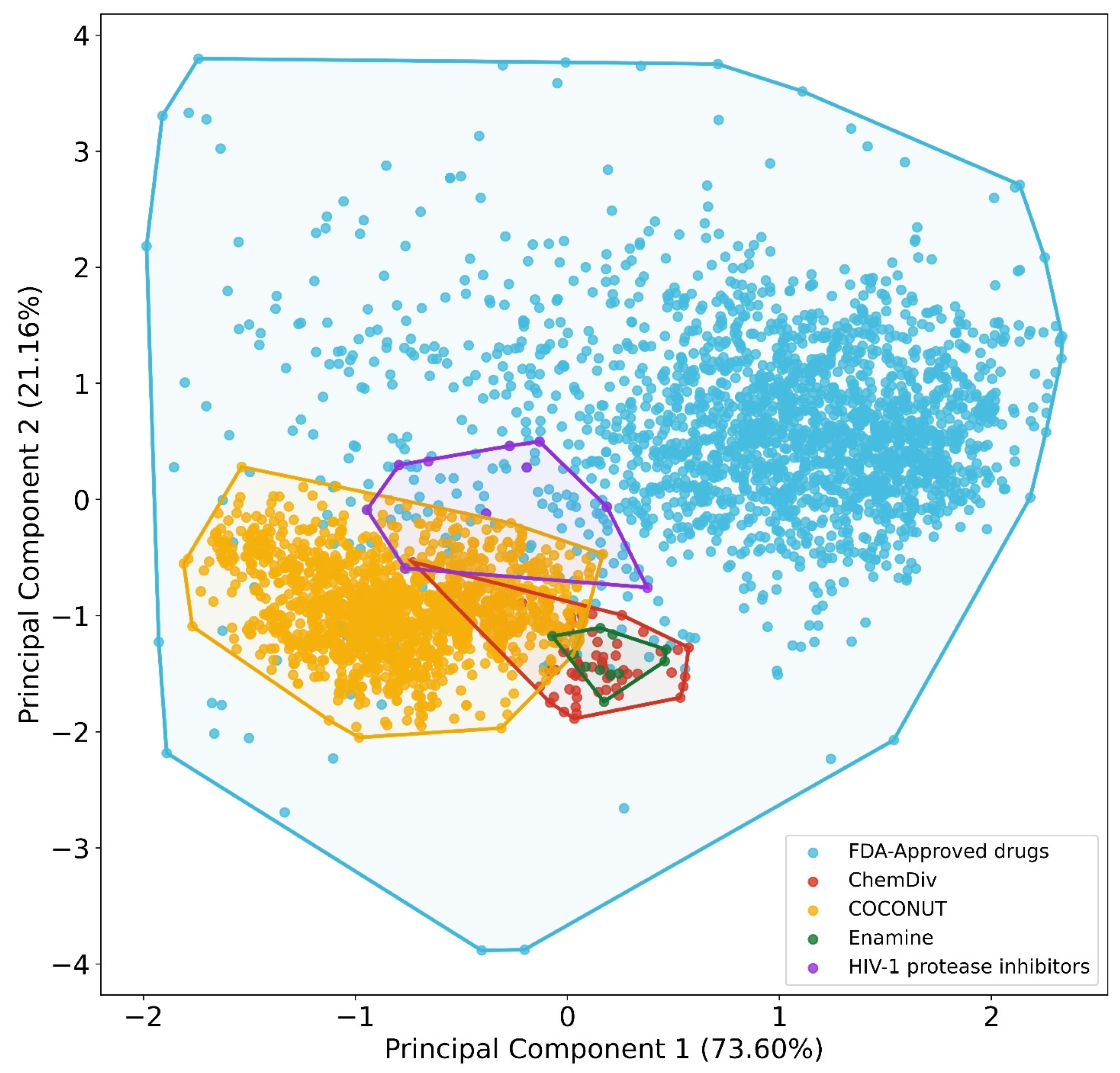
3.2. Chemical Space Visualization
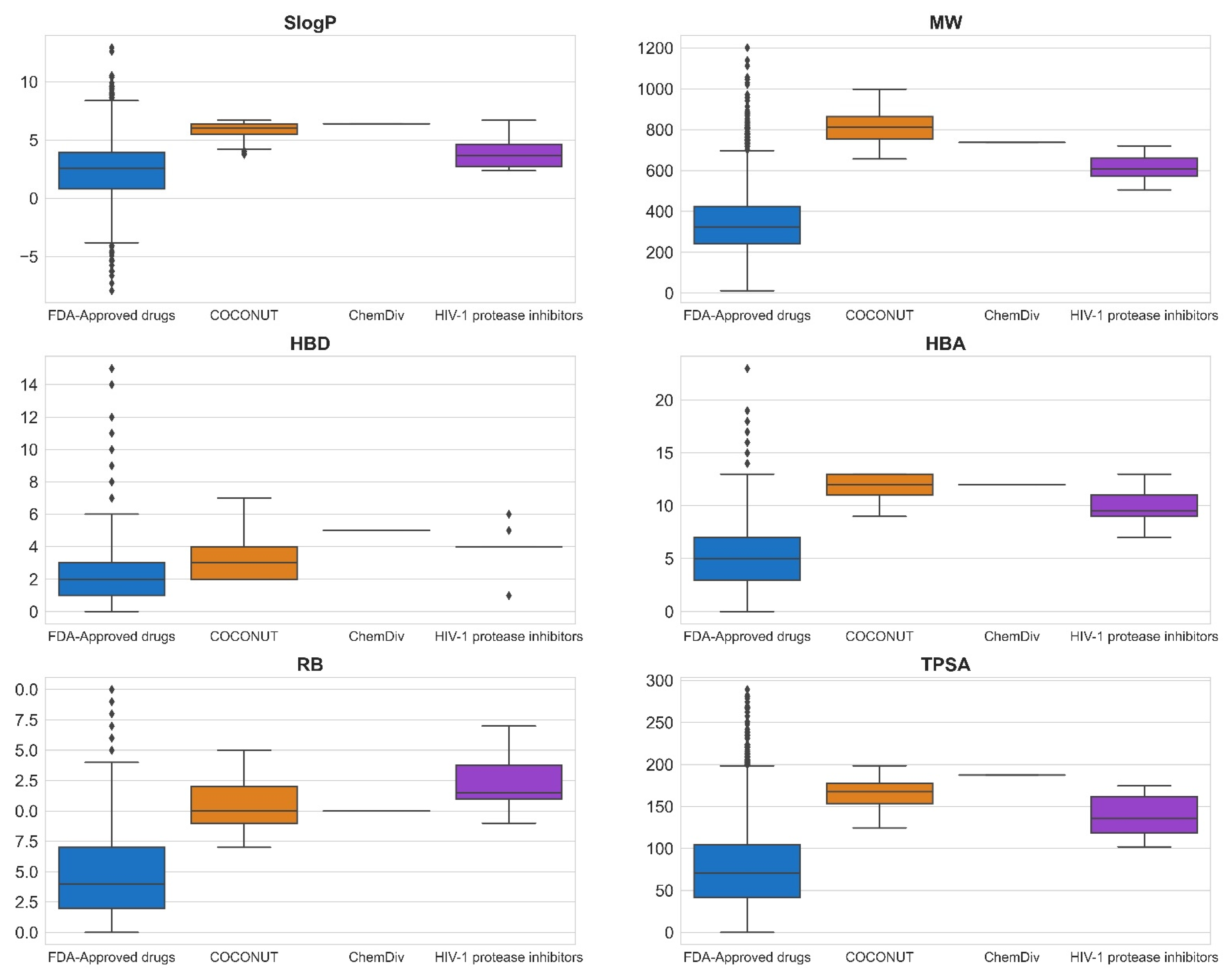
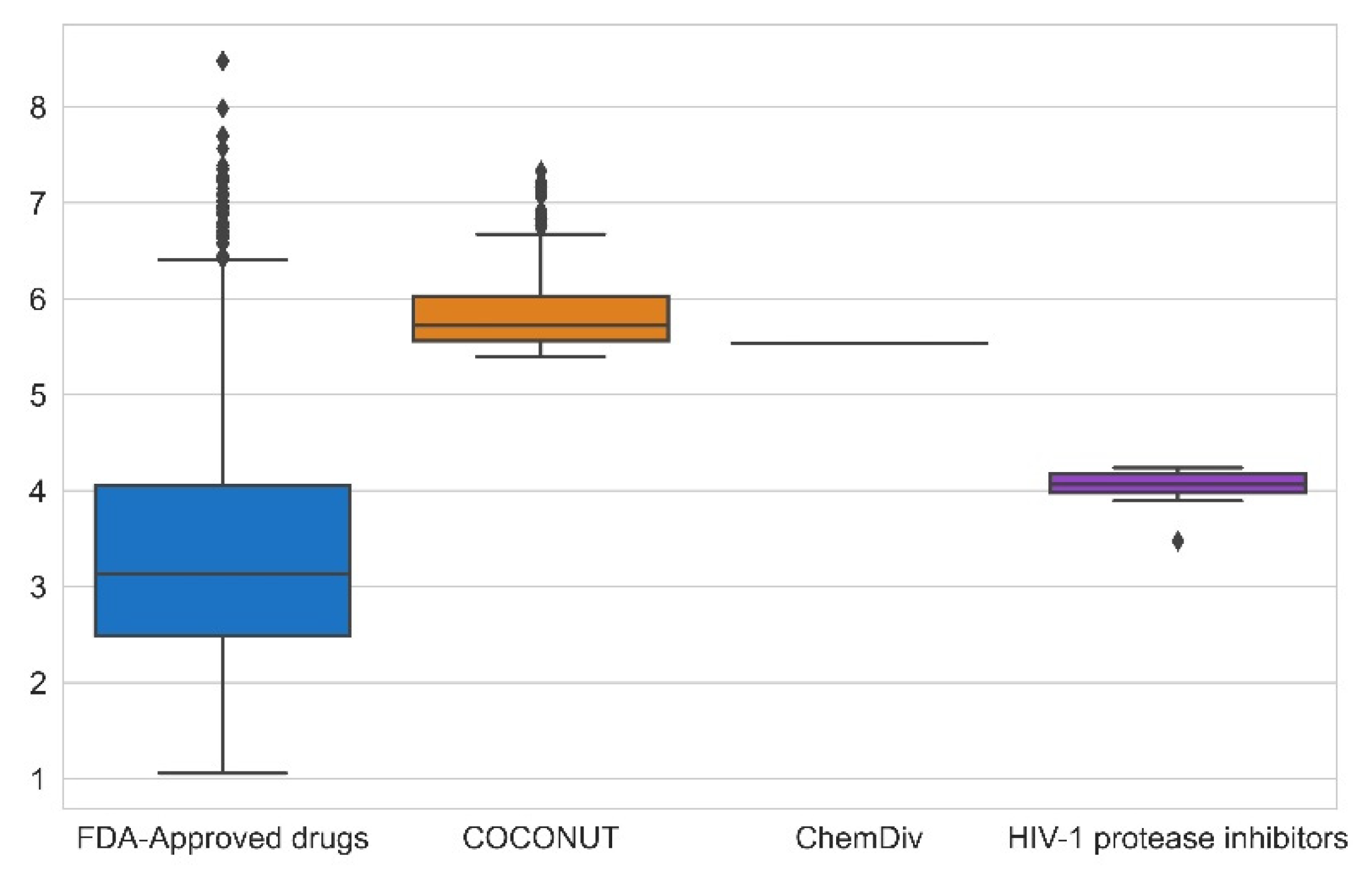
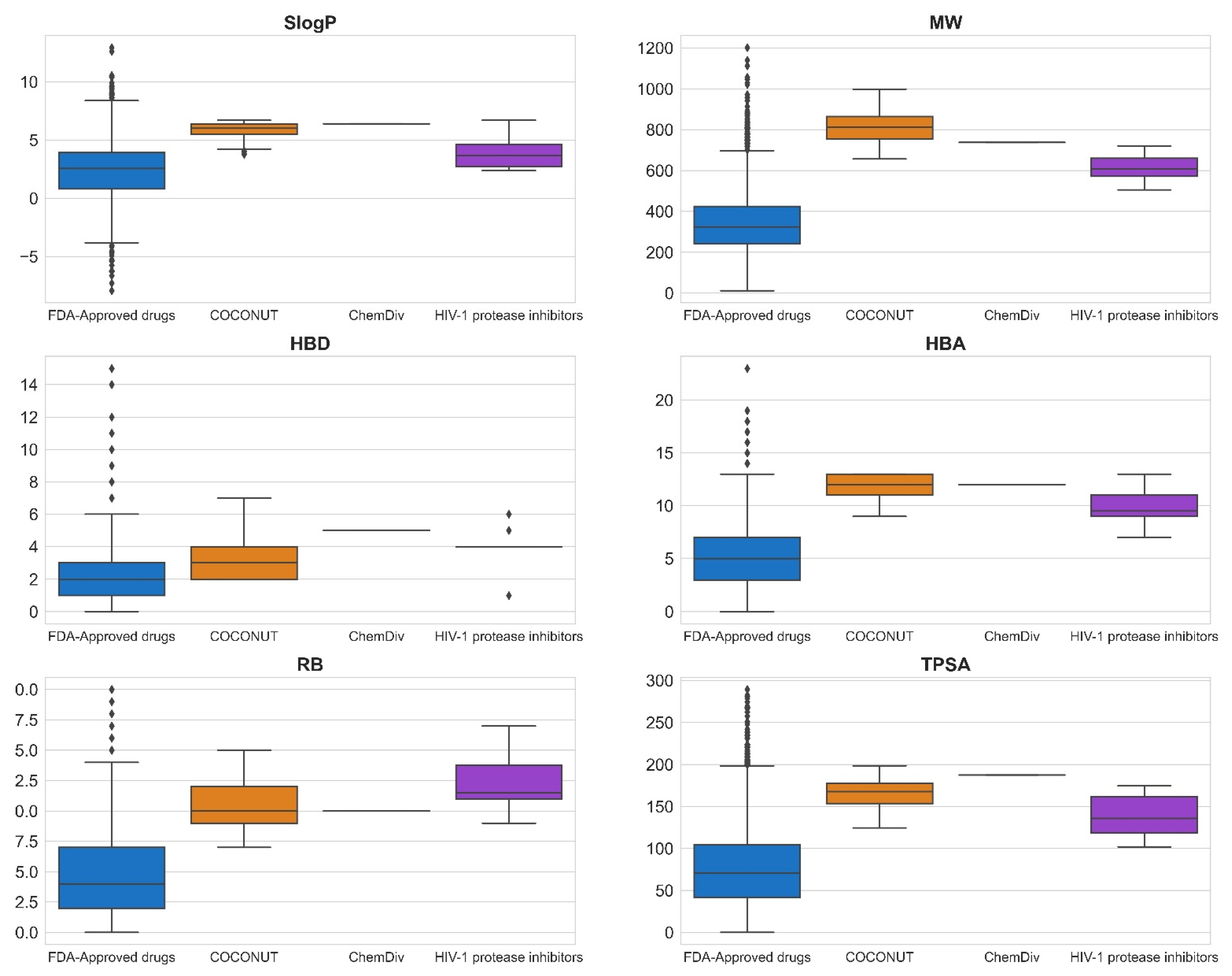
3.3. Compound Filtering Based on Physicochemical Properties
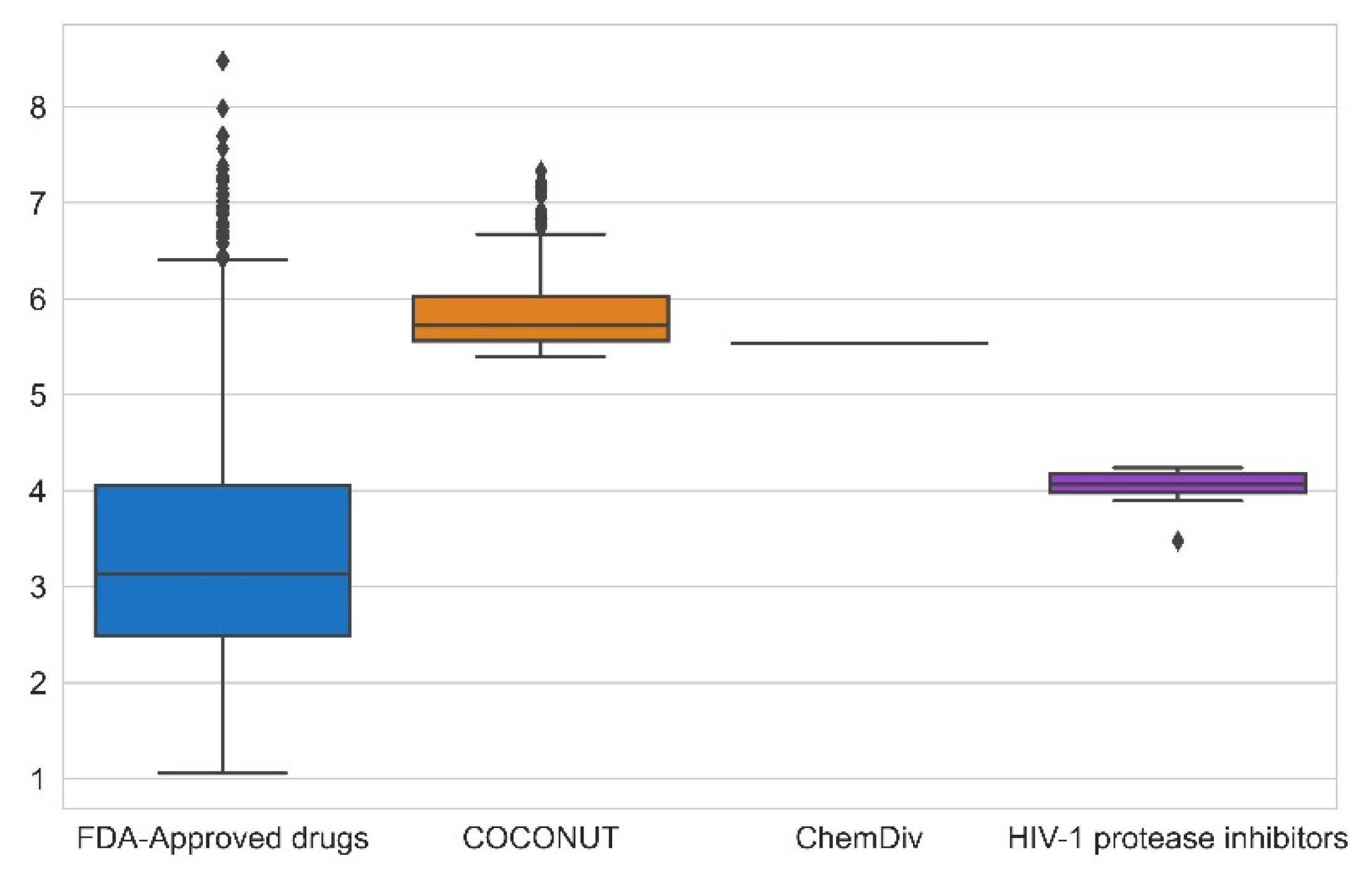
3.4. Filtering Based on Synthetic Feasibility
3.5. ADME-Tox Profiling
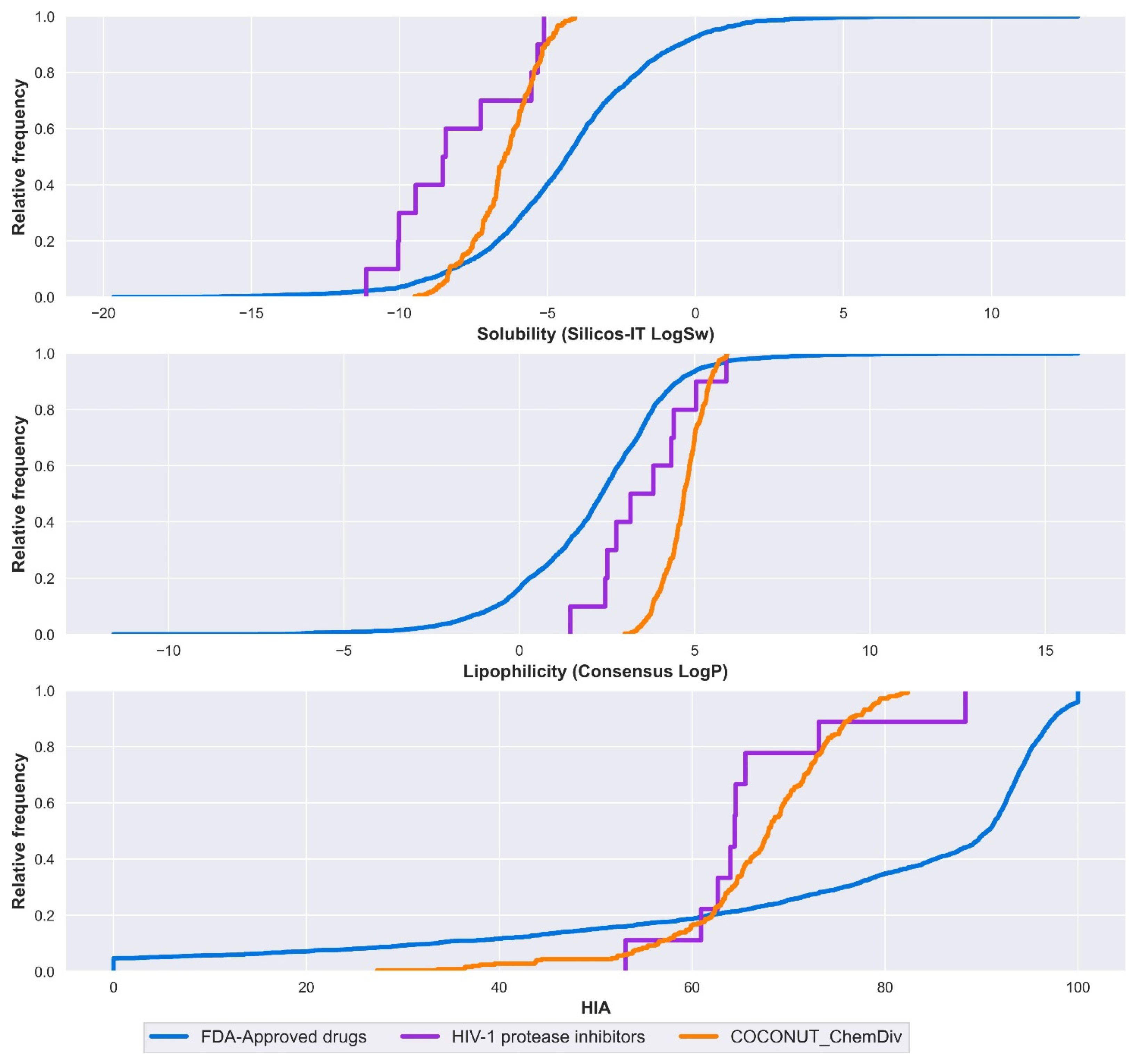
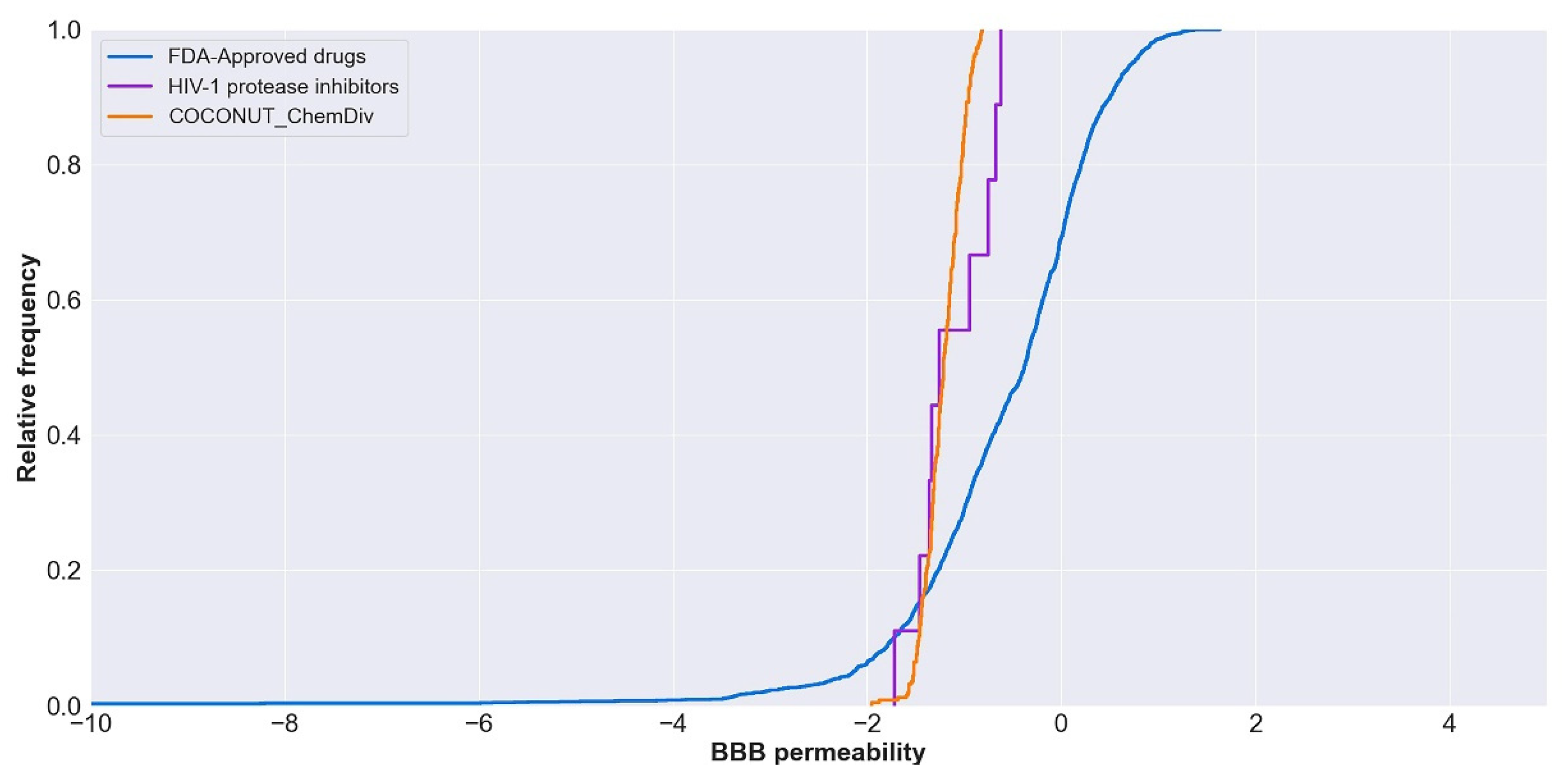
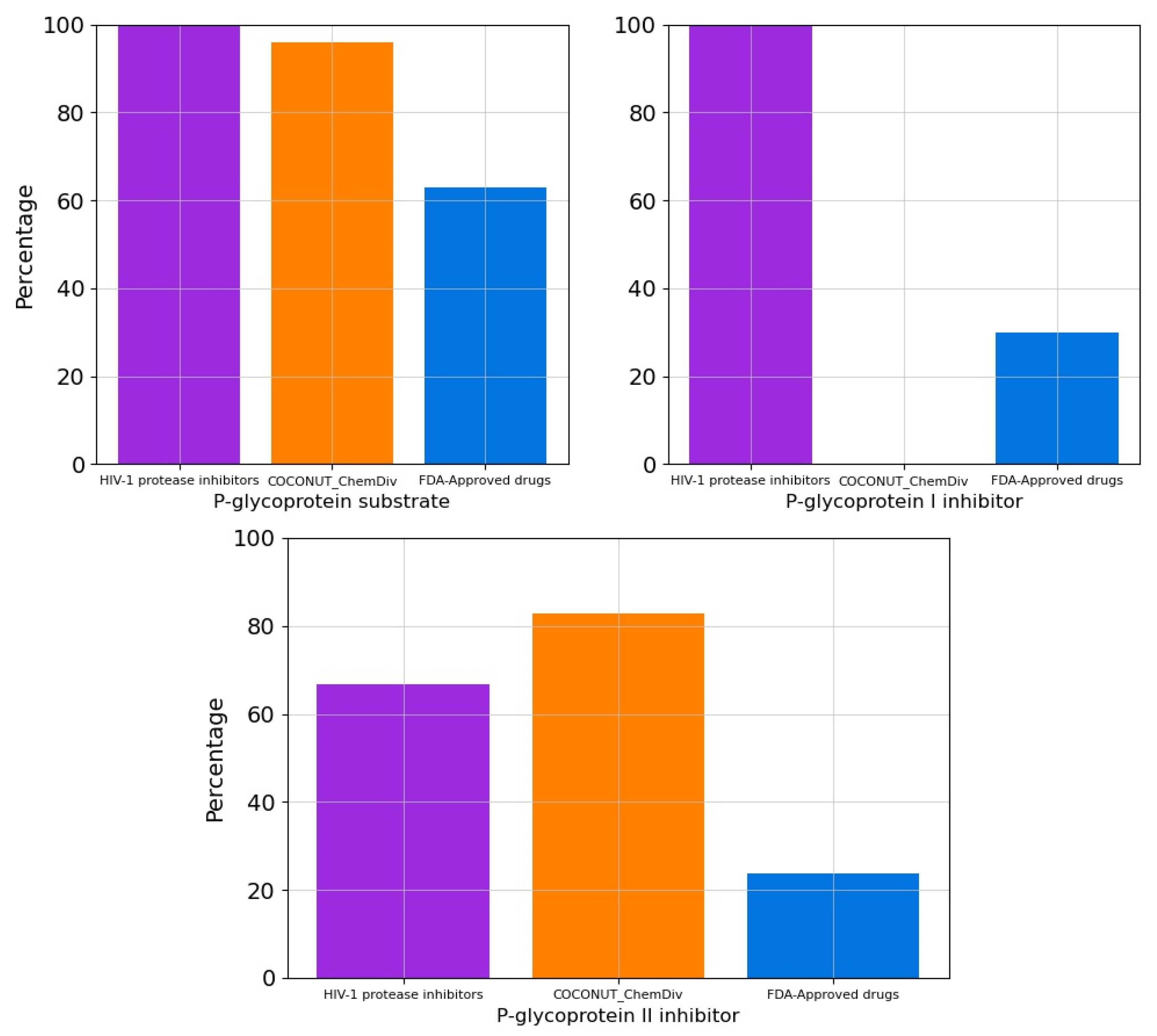
3.5.1. Absorption
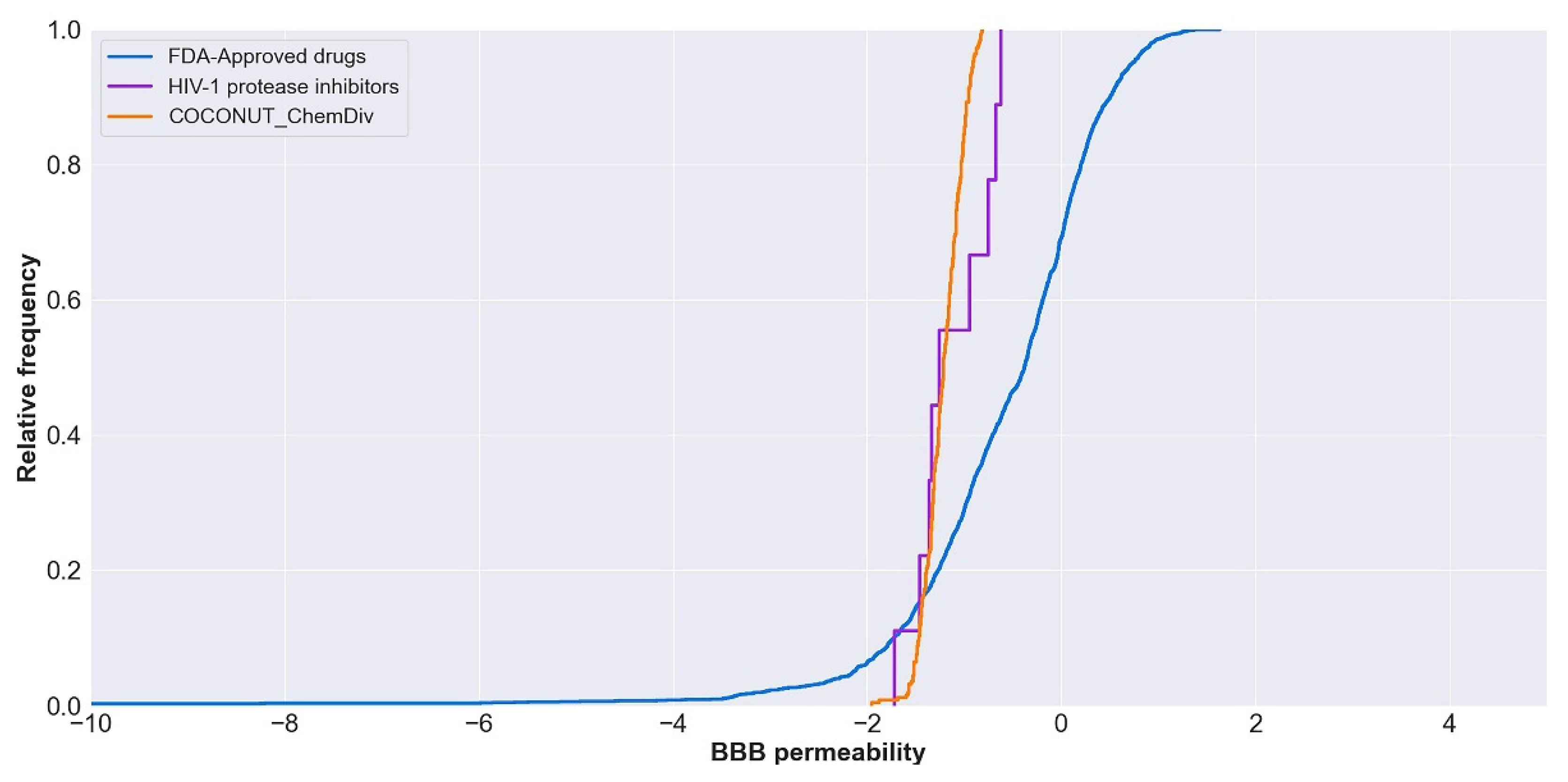
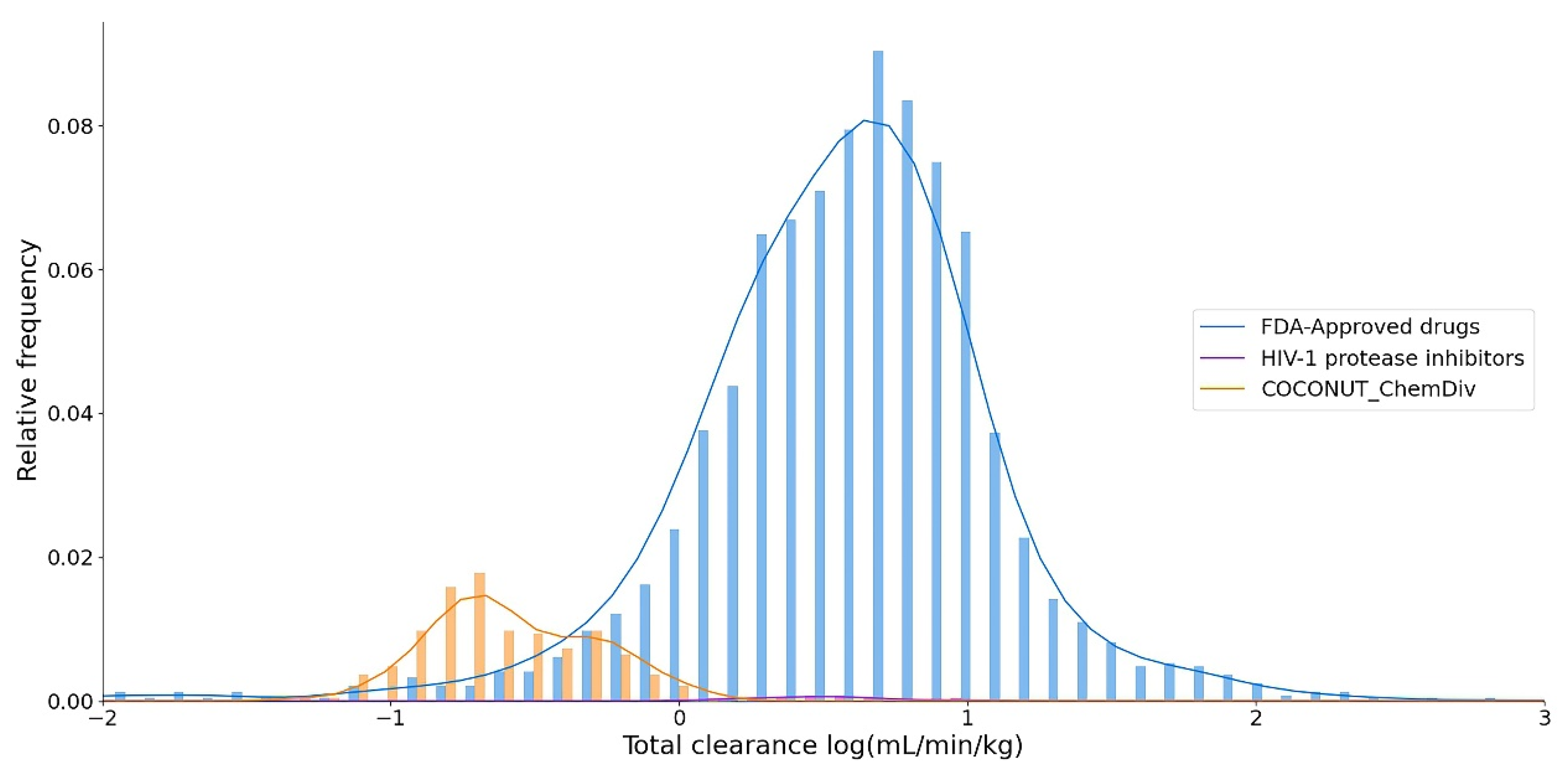
3.5.2. Distribution
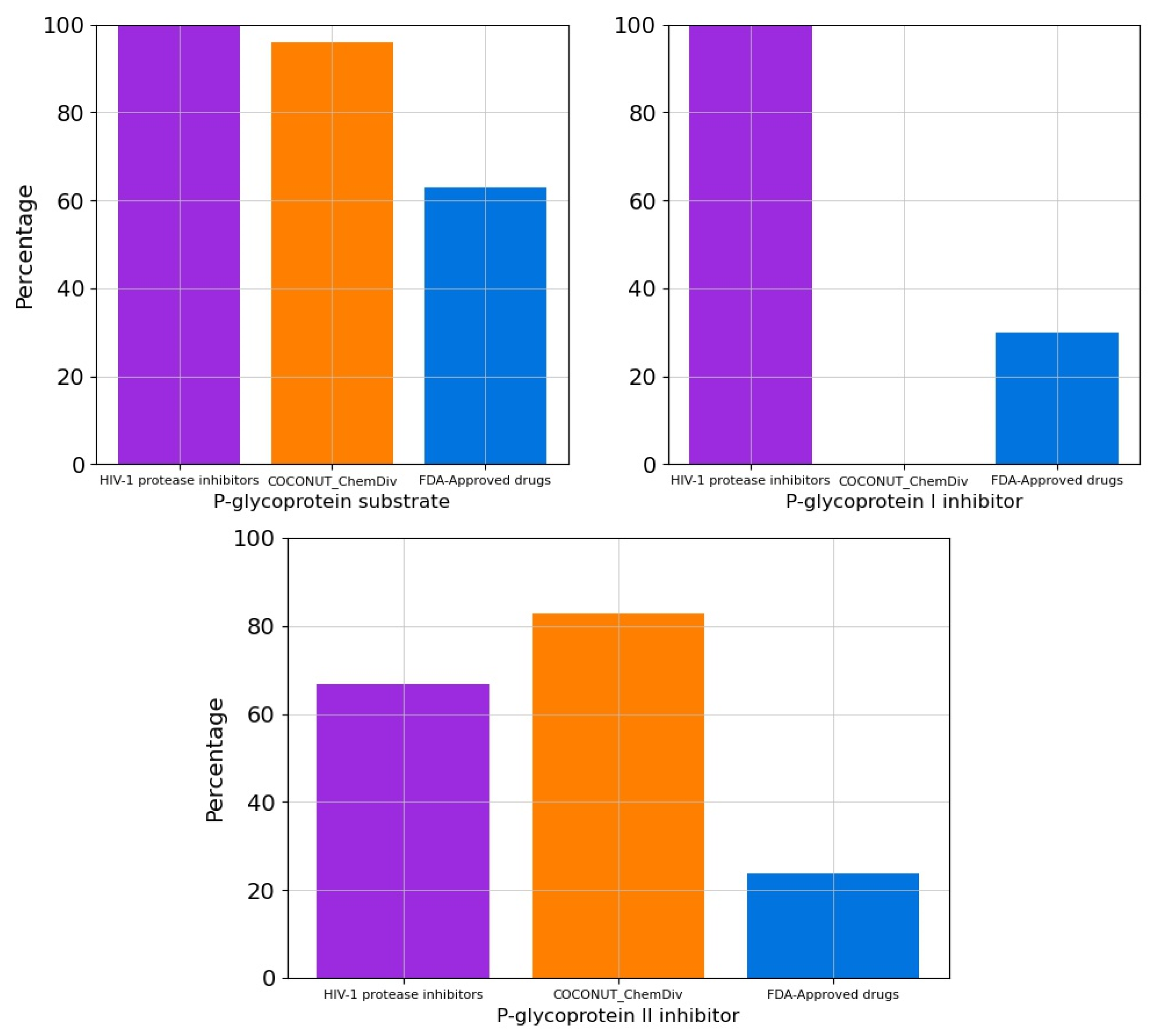
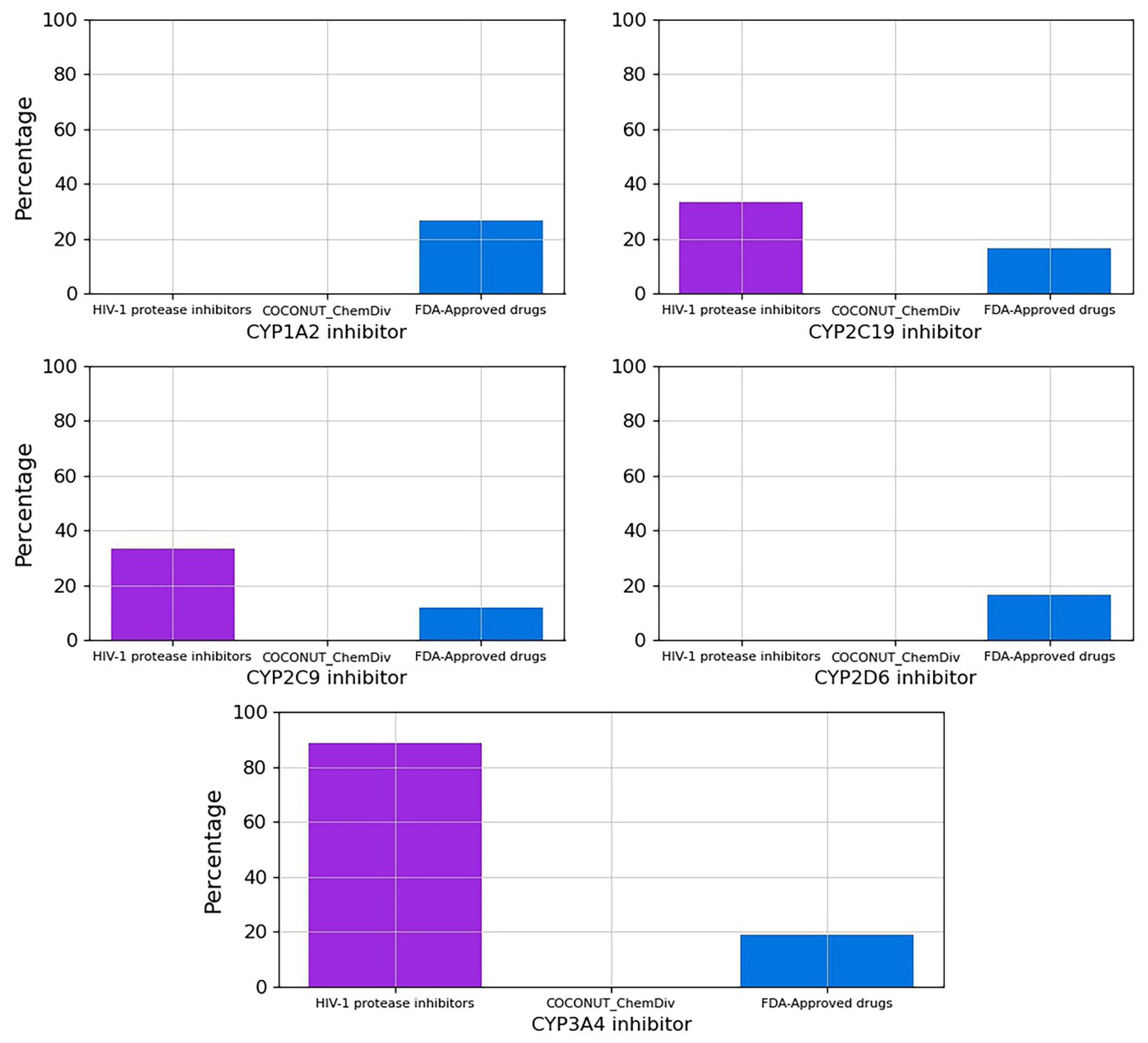
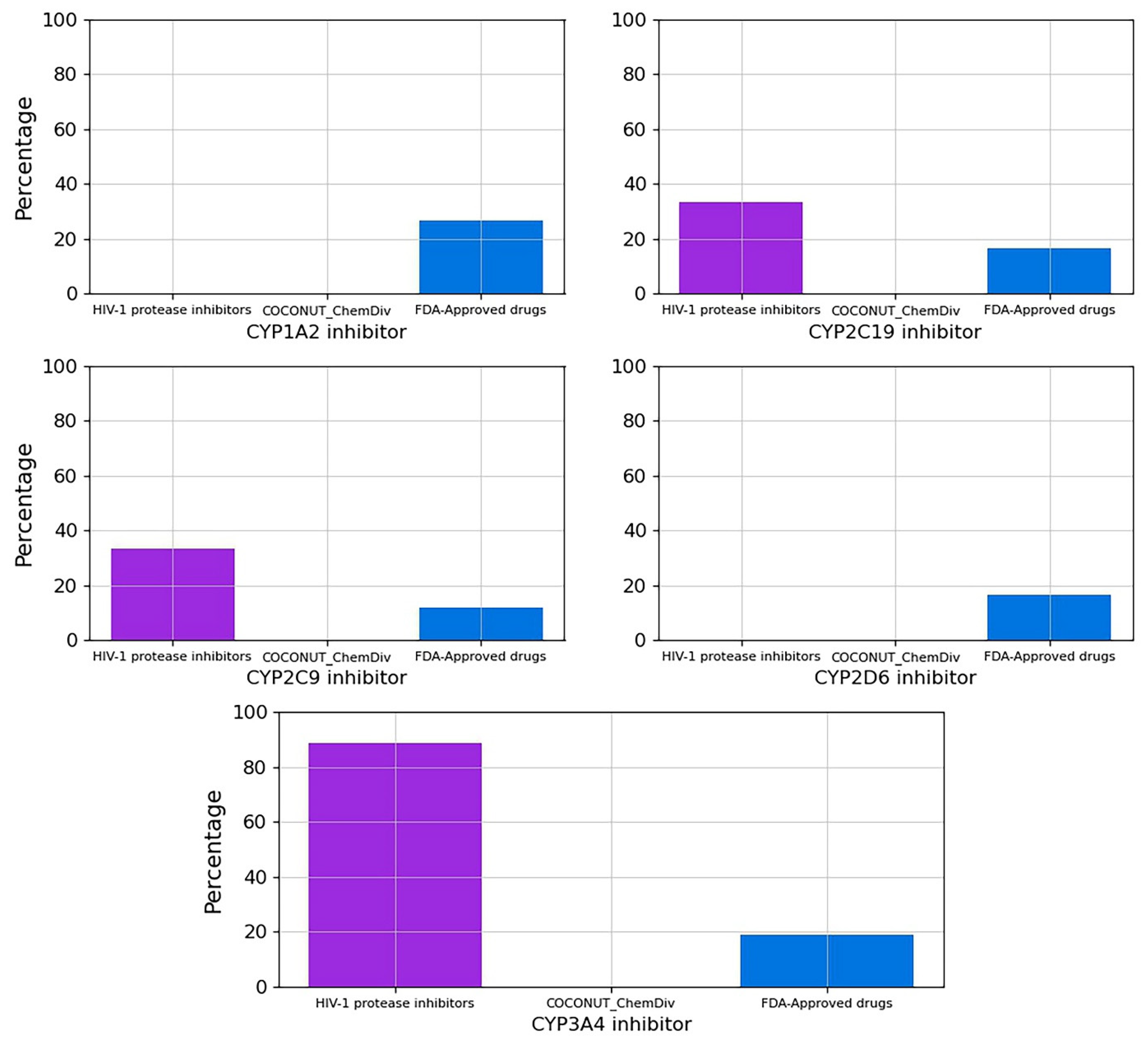
3.5.3. Metabolism
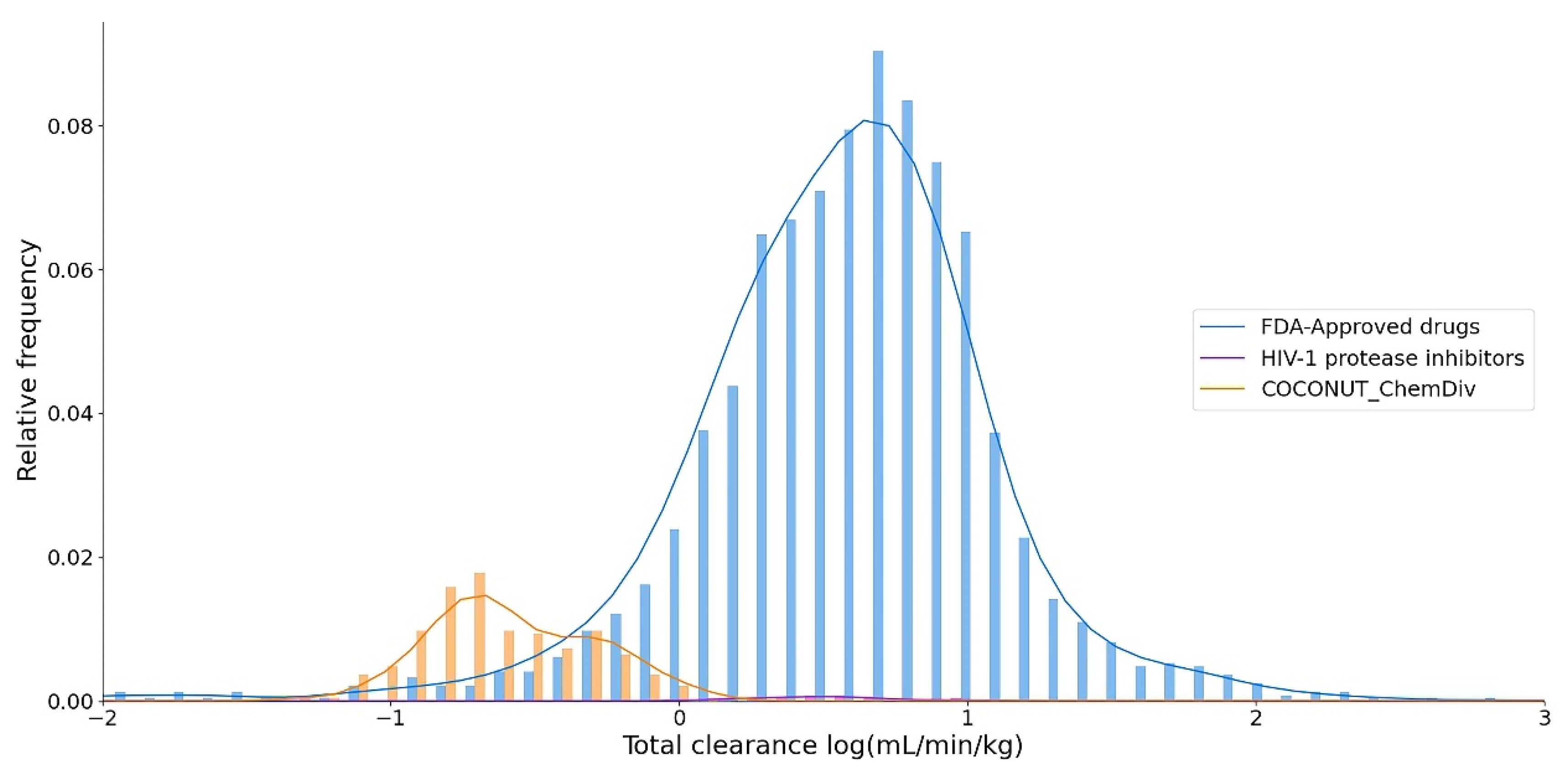
3.5.4. Excretion
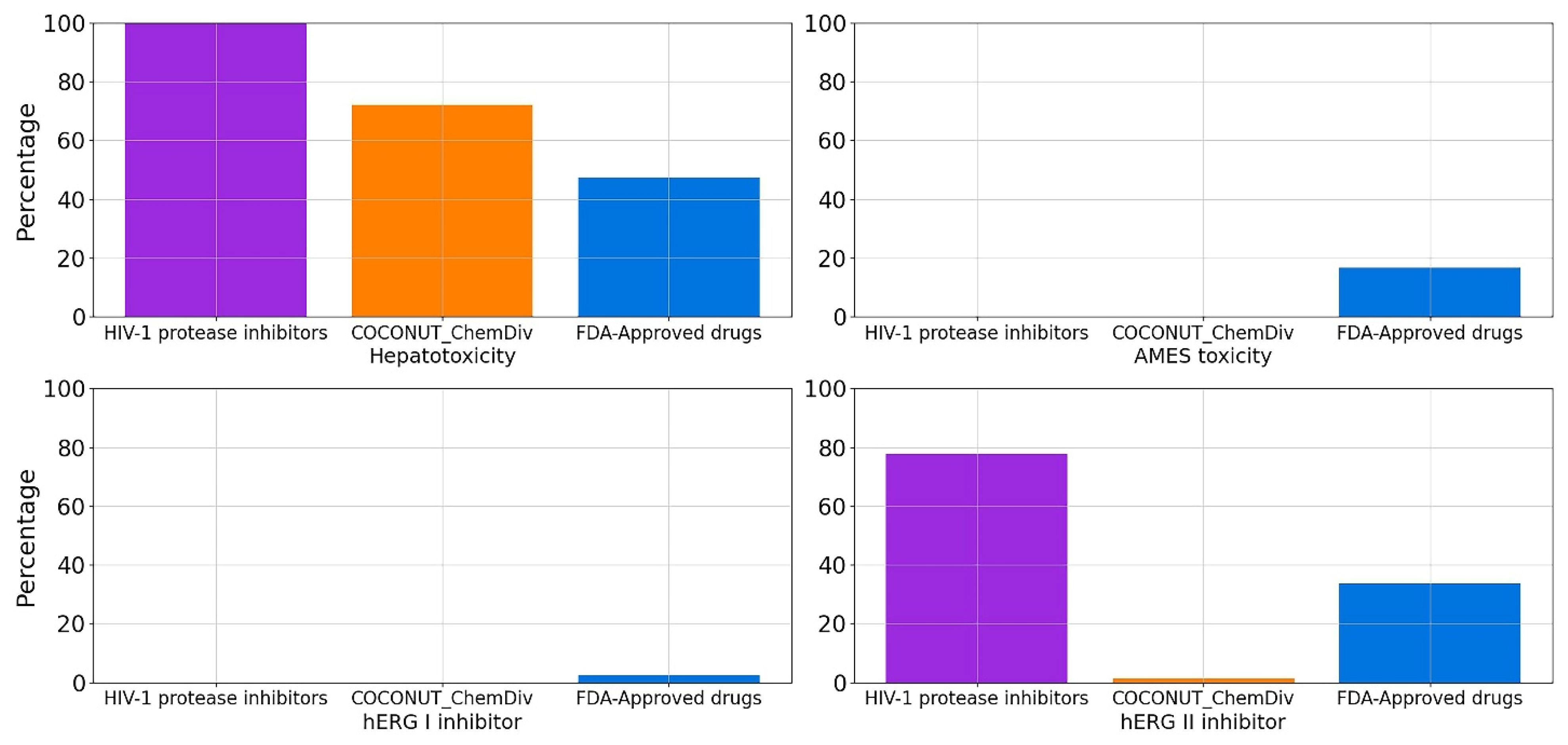
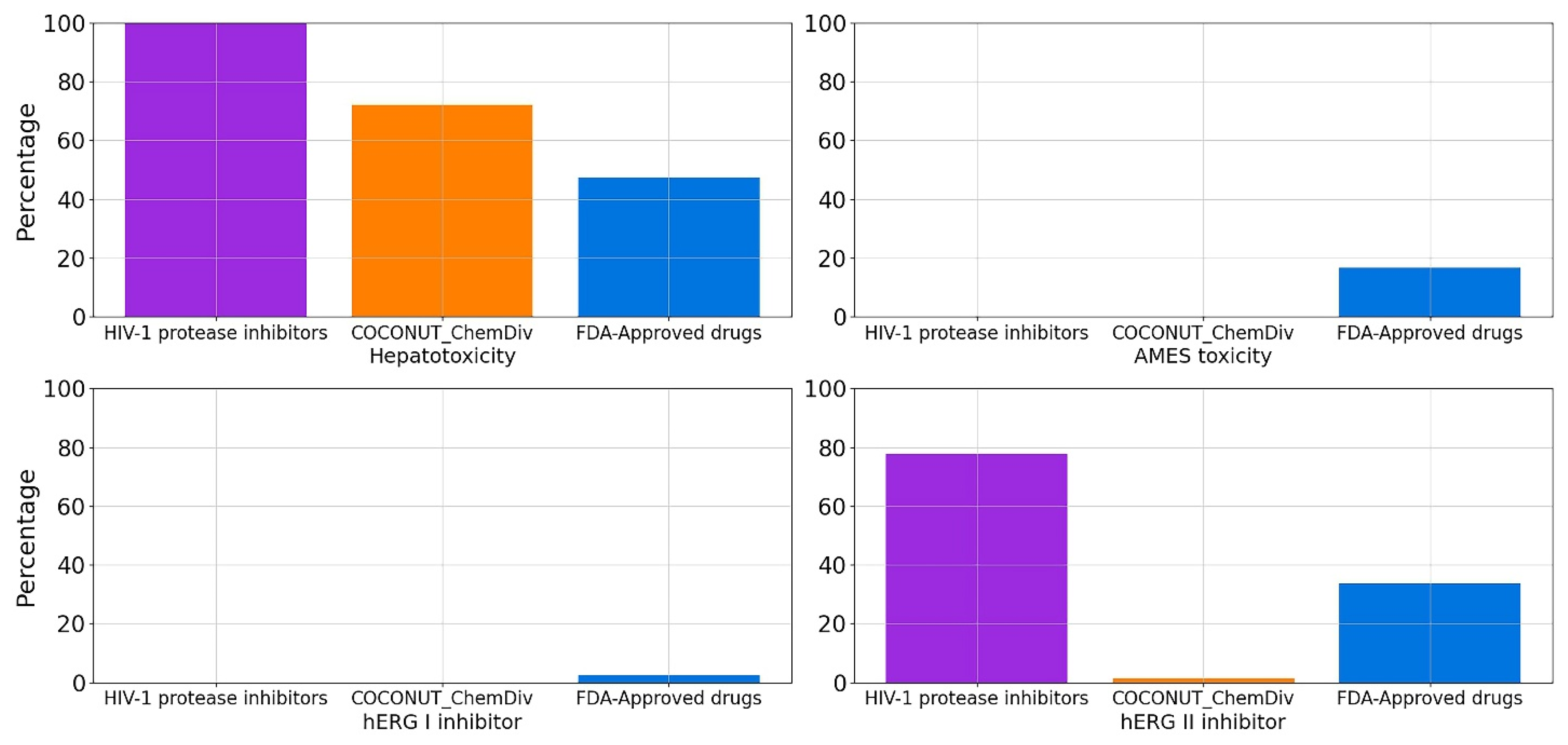
3.5.5. Toxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 15 July 2021).
- Zulfiqar, H.F.; Javed, A.; Sumbal; Afroze, B.; Ali, Q.; Akbar, K.; Nadeem, T.; Rana, M.A.; Nazar, Z.A.; Nasir, I.A.; et al. HIV Diagnosis and Treatment through Advanced Technologies. Front. Public Health 2017, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS 2015, 7, 95–104. [Google Scholar] [CrossRef] [Green Version]
- FDA. Available online: https://www.fda.gov/consumers/free-publications-women/hiv-and-aids-medicines-help-you (accessed on 28 April 2021).
- Schneider, G.; Clark, D.E. Automated de novo drug design: Are we nearly there yet? Angew. Chem. Int. Ed. 2019, 58, 10792–10803. [Google Scholar] [CrossRef]
- Torjesen, I. Drug Development: The Journey of a Medicine from Lab to Shelf. Available online: https://pharmaceutical-journal.com/article/feature/drug-development-the-journey-of-a-medicine-from-lab-to-shelf (accessed on 29 May 2021).
- Medina-Franco, J.L. Grand challenges of computer-aided drug design: The road ahead. Front. Drug Discov. 2021, 1, 728551. [Google Scholar] [CrossRef]
- Bung, N.; Krishnan, S.R.; Bulusu, G.; Roy, A. De novo design of new chemical entities for SARS-CoV-2 using artificial intelligence. Future Med. Chem. 2021, 13, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; IJzerman, A.P.; van Westen, G.J.P. Computational approaches for de novo drug design: Past, present, and future. In Artificial Neural Networks; Cartwright, H., Ed.; Springer: New York, NY, USA, 2021; pp. 139–165. ISBN 978-1-0716-0826-5. [Google Scholar]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Meyers, J.; Fabian, B.; Brown, N. De novo molecular design and generative models. Drug Discov. Today 2021, 26, 2707–2715. [Google Scholar] [CrossRef]
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Hartenfeller, M.; Schneider, G. De Novo Drug Design. In Chemoinformatics and Computational Chemical Biology; Bajorath, J., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 299–323. ISBN 978-1-60761-839-3. [Google Scholar]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Osborne, J.; Panova, S.; Rapti, M.; Urushima, T.; Jhoti, H. Fragments: Where are we now? Biochem. Soc. Trans. 2020, 48, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Shinde, P.B.; Bhowmick, S.; Alfantoukh, E.; Patil, P.C.; Wabaidur, S.M.; Chikhale, R.V.; Islam, M.A. De novo design based identification of potential HIV-1 integrase inhibitors: A pharmacoinformatics study. Comput. Biol. Chem. 2020, 88, 107319. [Google Scholar] [CrossRef]
- Ghiandoni, G.M.; Bodkin, M.J.; Chen, B.; Hristozov, D.; Wallace, J.E.A.; Webster, J.; Gillet, V.J. Enhancing reaction-based de novo design using a multi-label reaction class recommender. J. Comput. Aided Mol. Des. 2020, 34, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Saldívar-González, F.I.; Huerta-García, C.S.; Medina-Franco, J.L. Chemoinformatics-based enumeration of chemical libraries: A tutorial. J. Cheminform. 2020, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Prado-Romero, D.L.; Medina-Franco, J.L. Advances in the exploration of the epigenetic televant chemical space. ACS Omega 2021, 6, 22478–22486. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.C.; Kumar, R.; Davis, R.A. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep. 2016, 33, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Hernández, A.L.; Sánchez-Cruz, N.; Medina-Franco, J.L. A Fragment library of natural products and its comparative chemoinformatic characterization. Mol. Inform. 2020, 39, 2000050. [Google Scholar] [CrossRef] [PubMed]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [Google Scholar] [CrossRef]
- ChemDiv. Available online: https://store.chemdiv.com/ (accessed on 19 July 2021).
- Enamine. Available online: https://enamine.net/compound-collections/fragment-collection (accessed on 16 July 2021).
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminform. 2020, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- toolkit RDKit. Available online: http://rdkit.org (accessed on 21 May 2021).
- MolVS. Available online: https://molvs.readthedocs.io/en/latest/ (accessed on 21 May 2021).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
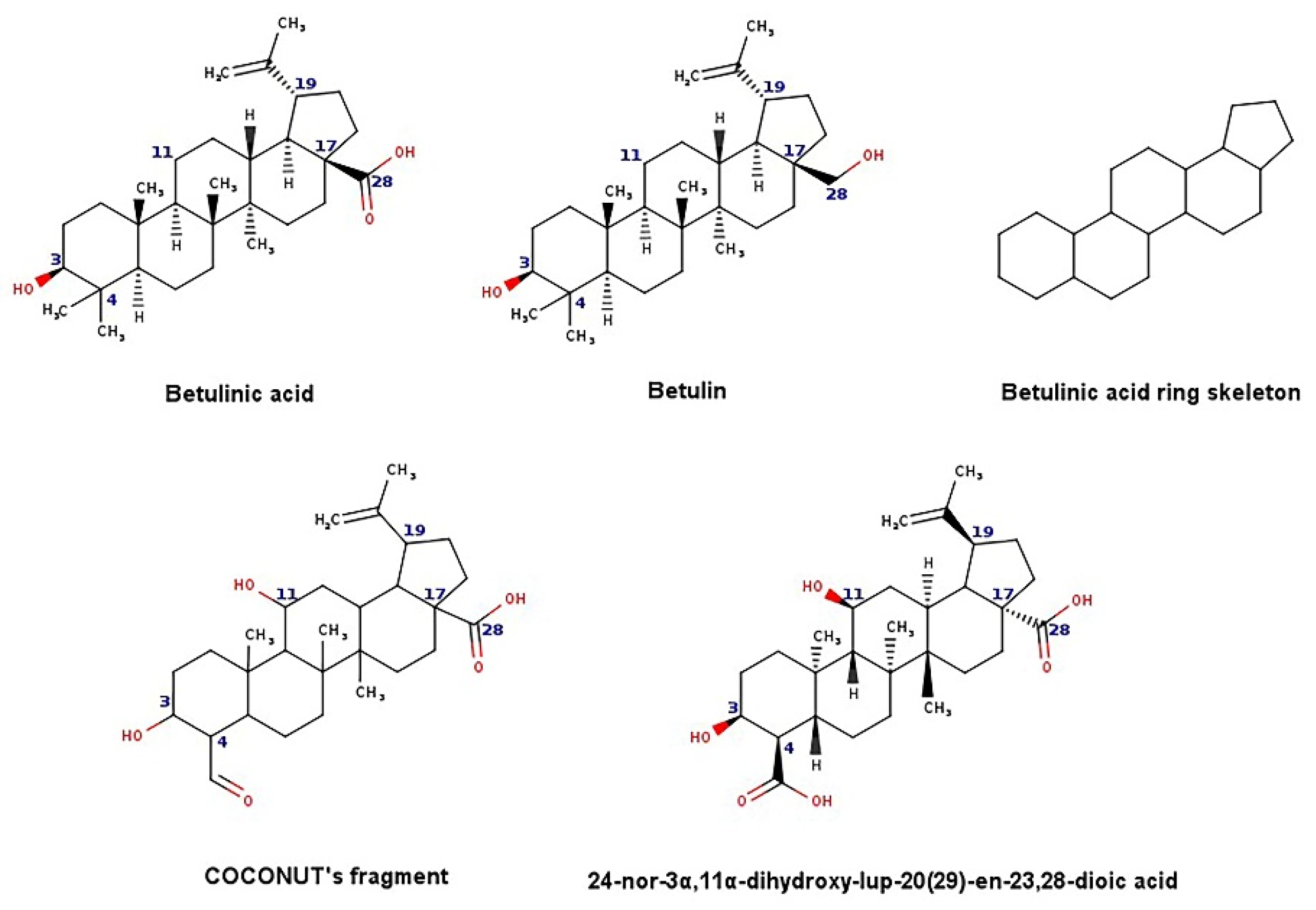
- Lewell, X.Q.; Judd, D.B.; Watson, S.P.; Hann, M.M. RECAPRetrosynthetic combinatorial analysis procedure: A powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J. Chem. Inf. Comput. Sci. 1998, 38, 511–522. [Google Scholar] [CrossRef] [PubMed]
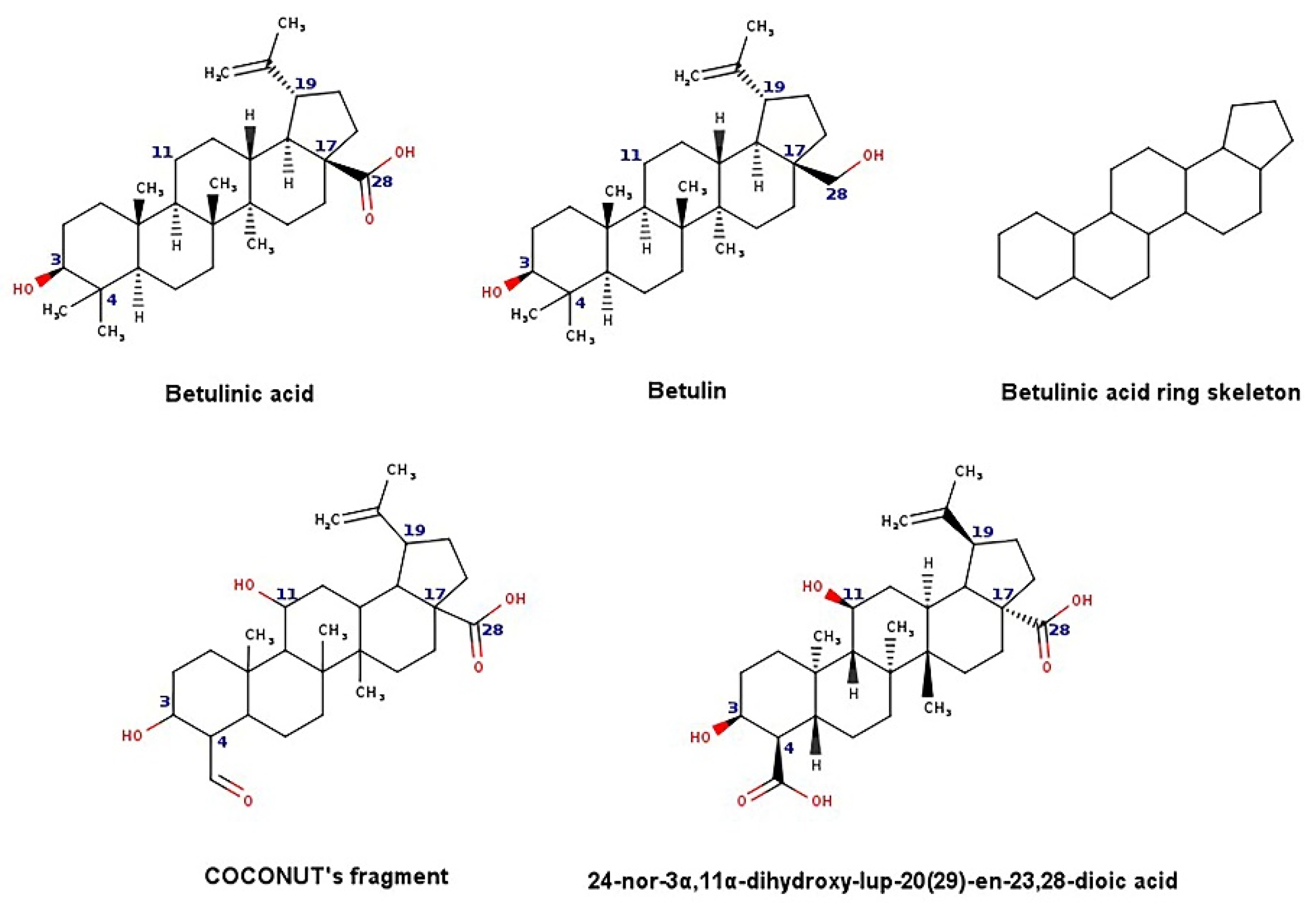
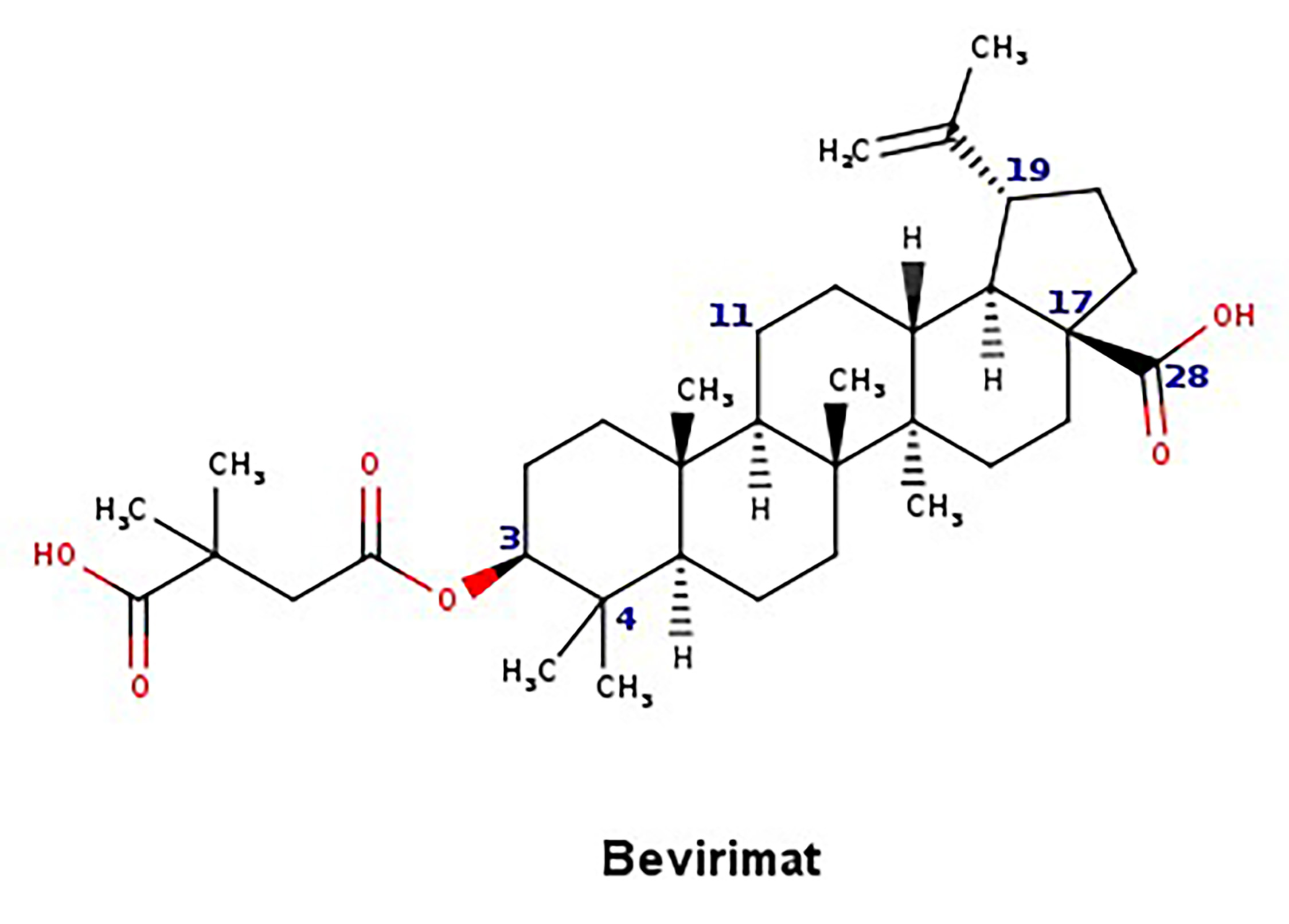
- Zhao, Y.; Chen, C.-H.; Morris-Natschke, S.L.; Lee, K.-H. Design, synthesis, and structure activity relationship analysis of new betulinic acid derivatives as potent HIV inhibitors. Eur. J. Med. Chem. 2021, 215, 113287. [Google Scholar] [CrossRef] [PubMed]
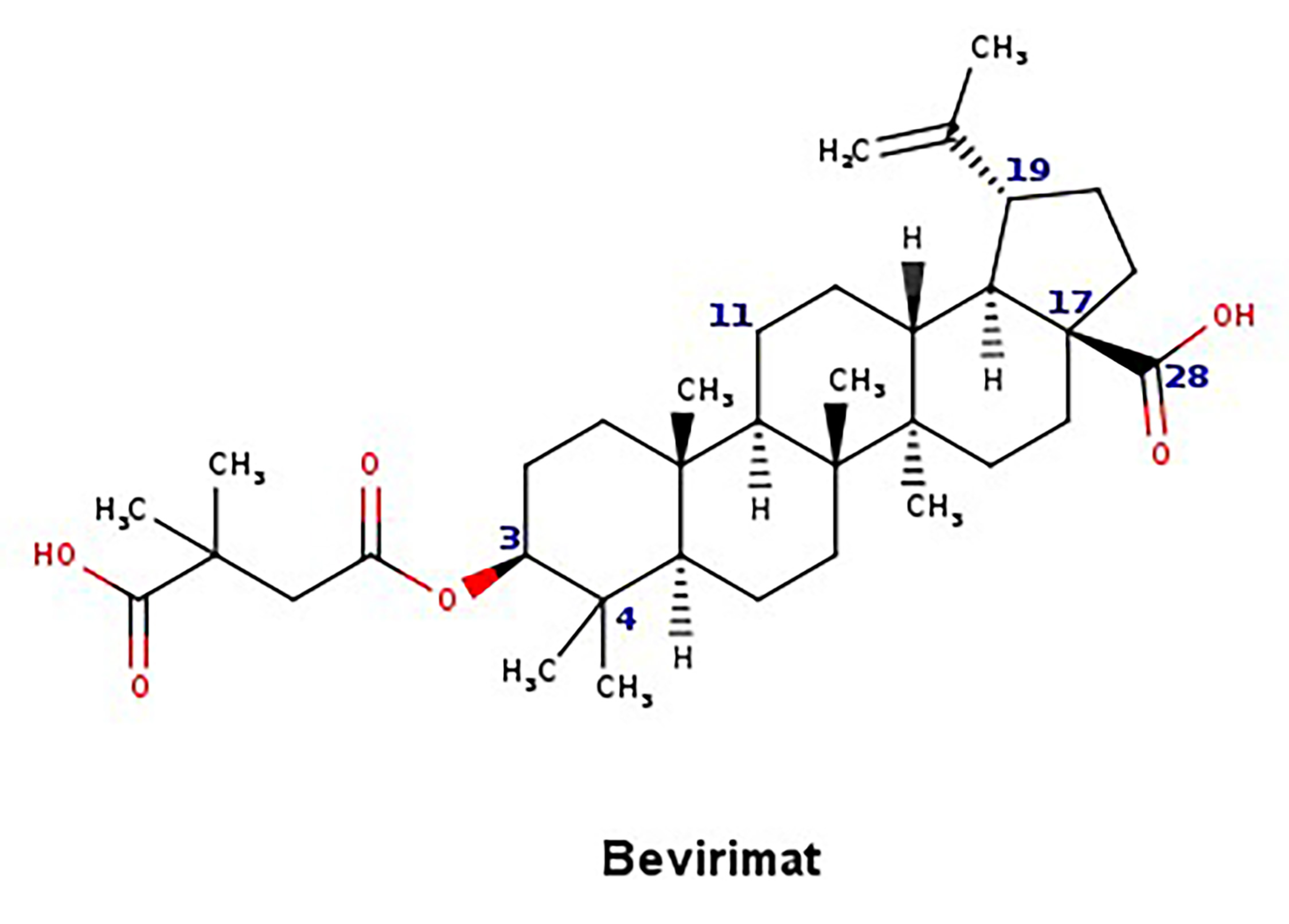
- Martin, D.E.; Salzwedel, K.; Allaway, G.P. Bevirimat: A novel maturation inhibitor for the treatment of hiv-1 infection. Antivir. Chem. Chemother. 2008, 19, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Lazerwith, S.E.; Siegel, D.; McFadden, R.M.; Mish, M.R.; Tse, W.C. 5.19—New Antiretrovirals for HIV and Antivirals for HBV; Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 628–664. ISBN 978-0-12-803201-5. [Google Scholar]
- Qian, K.; Kuo, R.-Y.; Chen, C.-H.; Huang, L.; Morris-Natschke, S.L.; Lee, K.-H. Anti-AIDS Agents 81. Design, synthesis, and structure−activity relationship study of betulinic acid and moronic acid derivatives as potent HIV maturation inhibitors. J. Med. Chem. 2010, 53, 3133–3141. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Chen, H.; Luo, X.; Zhang, Y.; Yao, X.; Zheng, X. Structure and anti-HIV activity of betulinic acid analogues. Curr. Med. Sci. 2018, 38, 387–397. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Probst, D.; Reymond, J.-L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminform. 2020, 12, 12. [Google Scholar] [CrossRef] [Green Version]
- TMAP. Available online: https://tmap.gdb.tools/ (accessed on 14 September 2021).
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef]
- Sánchez-Cruz, N.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Functional group and diversity analysis of BIOFACQUIM: A Mexican natural product database. F1000Research 2020, 8, 2071. [Google Scholar] [CrossRef]
- Chávez-Hernández, A.L.; Sánchez-Cruz, N.; Medina-Franco, J.L. Fragment library of natural products and compound databases for drug discovery. Biomolecules 2020, 10, 1518. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Durán-Iturbide, N.A.; Díaz-Eufracio, B.I.; Medina-Franco, J.L. In silico ADME/Tox profiling of natural products: A focus on BIOFACQUIM. ACS Omega 2020, 5, 16076–16084. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Lenci, E.; Calugi, L.; Medina-Franco, J.L.; Trabocchi, A. Computational-aided design of a library of lactams through a diversity-oriented synthesis strategy. Bioorg. Med. Chem. 2020, 28, 115539. [Google Scholar] [CrossRef]
- Laurini, R. 5—Geographic Relations; Laurini, R.B.T.-G.K.I., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 83–109. ISBN 978-1-78548-243-4. [Google Scholar]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Tinworth, C.P.; Young, R.J. Facts, Patterns, and principles in drug discovery: Appraising the Rule of 5 with measured physicochemical data. J. Med. Chem. 2020, 63, 10091–10108. [Google Scholar] [CrossRef] [PubMed]
- Bergström, C.A.S.; Yazdanian, M. Lipophilicity in drug development: Too much or not enough? AAPS J. 2016, 18, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Clearance in drug design. J. Med. Chem. 2019, 62, 2245–2255. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
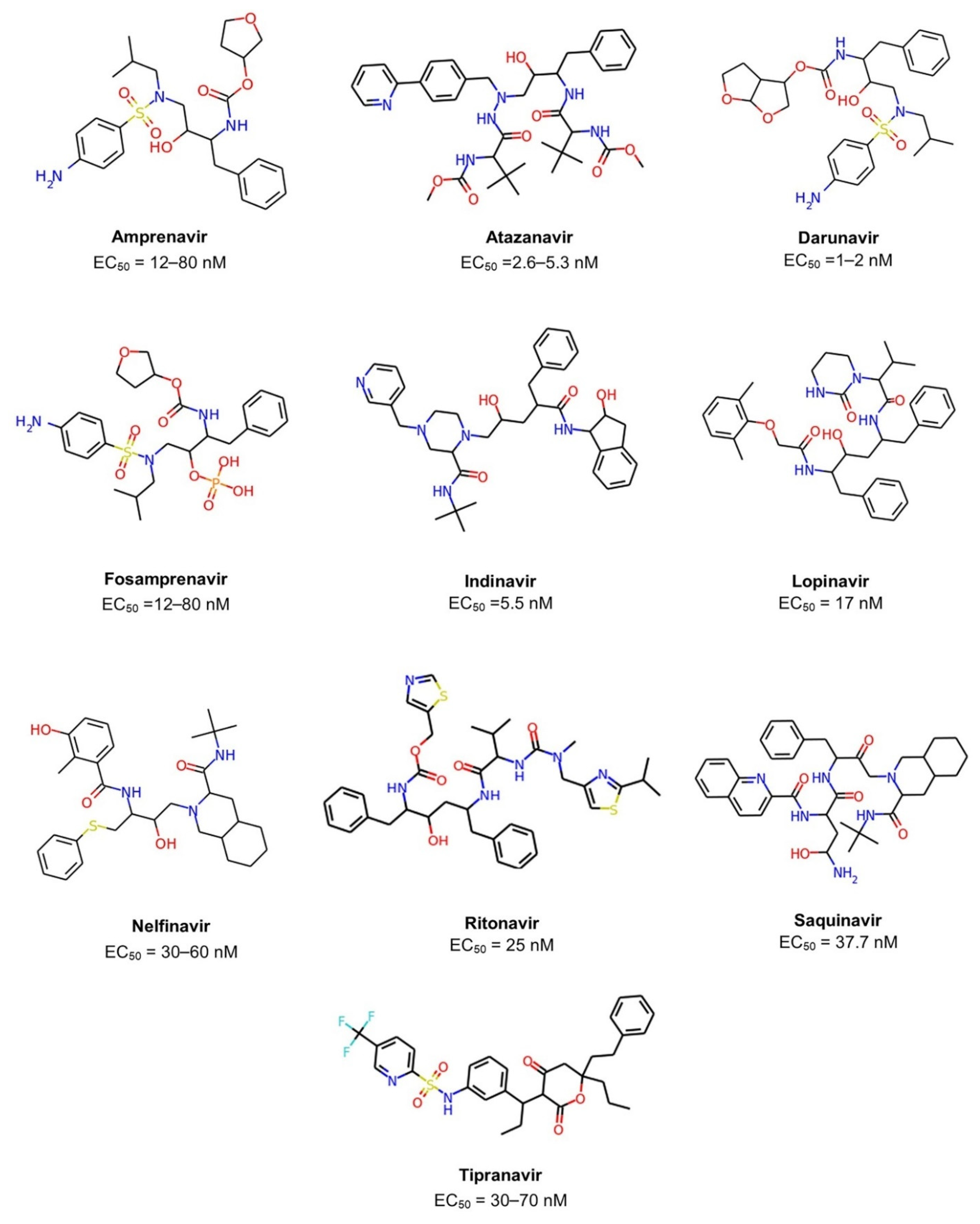
| Generic Name | Brand Name | EC50 [3] | FDA Approval |
|---|---|---|---|
| Amprenavir | Agenerase | 12–80 nM | 1999 |
| Atazanavir | Reyataz | 2.6–5.3 nM | 2003 |
| Darunavir | Prezista | 1–2 nM | 2006 |
| Fosamprenavir a | Lexiva | 12–80 nM | 2003 |
| Indinavir | Crixivan | 5.5 nM | 1996 |
| Lopinavir | Kaletra | 17 nM | 2000 |
| Nelfinavir | Viracept | 30–60 nM | 1997 |
| Ritonavir | Norvir | 25 nM | 1996 |
| Saquinavir | Invirase | 37.7 nM | 1995 |
| Tipranavir | Aptivus | 30–70 nM | 2005 |
| Description | Scheme |
|---|---|
| Reaction 1 | 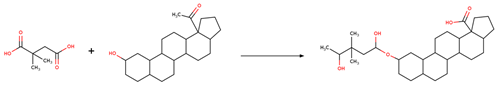 |
| SMIRKS 1 | 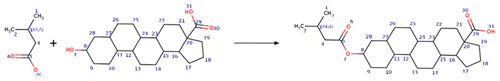 [#6:1][#6;A;X4:3]([#6:2])[#6:4]-[#6:5]([#8;A])=[O:6].[#8:7]-[#6:8]-1-[#6:9]-[#6:10]-[#6:11]-2-[#6:27](-[#6:26]-[#6:25]-[#6:24]-3-[#6:23]-4-[#6:22]-[#6:21][C:20]5([#6:19]-[#6:18]-[#6:17]-[#6:16]5-[#6:15]-4-[#6:14]-[#6:13]-[#6:12]-2-3)[#6:29](-[#8:31])=[O:30])-[#6:28]-1>>[#6:2][#6;A;X4:3]([#6:1])[#6:4]-[#6:5](=[O:6])-[#8:7]-[#6:8]-1-[#6:9]-[#6:10]-[#6:11]-2-[#6:27](-[#6:26]-[#6:25]-[#6:24]-3-[#6:23]-4-[#6:22]-[#6:21][C:20]5([#6:19]-[#6:18]-[#6:17]-[#6:16]5-[#6:15]-4-[#6:14]-[#6:13]-[#6:12]-2-3)[#6:29](-[#8:31])=[O:30])-[#6:28]-1 |
| Reaction 2.1 | 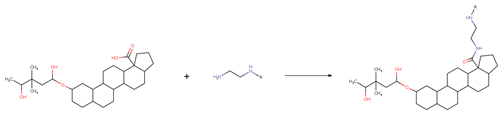 |
| SMIRKS 2.1 | 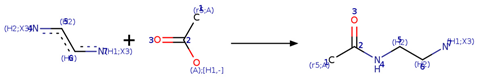 [#7;H1;X3:7][#6H2:6][#6;H2:5][#7;H2;X3:4].[#6;A;r5:1][#6:2]([#8;A;H1,-])=[O:3]>>[#6;A;r5:1][#6:2](=[O:3])-[#7:4]-[#6;H2:5]-[#6;H2:6]-[#7;H1;X3:7] |
| Reaction 2.2 | 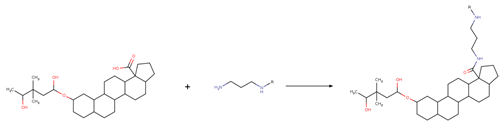 |
| SMIRKS 2.2 | 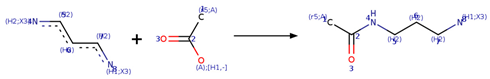 [#7;H1X3:8][#6H2:7][#6H2:6][#6H2:5][#7;H2X3:4].[#6;A;r5:1][#6:2]([#8;A;H1,-])=[O:3]>>[#6;A;r5:1][#6:2](=[O:3])-[#7:4]-[#6H2:5]-[#6H2:6]-[#6H2:7]-[#7;H1X3:8] |
| Reaction 2.3 | 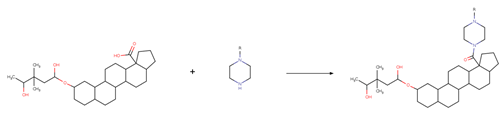 |
| SMIRKS 2.3 | 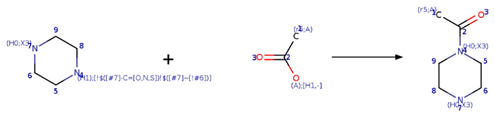 [#6:9]-1-[#6:8]-[#7H1;!$([#7]-C=[O,N,S])!$([#7]~[!#6]):4]-[#6:5]-[#6:6]-[#7;H0X3:7]-1.[#6;A;r5:1][#6:2]([#8;A;H1,-])=[O:3]>>[#6;A;r5:1][#6:2](=[O:3])-[#7;H0X3:4]-1-[#6:5]-[#6:6]-[#7;H0X3:7]-[#6:8]-[#6:9]-1 |
| Functional Groups | SMARTS |
|---|---|
| Aliphatic alcohol (cyclohexanol) | [#8;H1]-[#6]-1-[#6]-[#6]-[#6]-2-[#6](-[#6]-[#6]-[#6]-3-[#6]-4-[#6]-[#6]C5([#6]-[#6]-[#6]-[#6]5-[#6]-4-[#6]-[#6]-[#6]-2-3)[#6]([#8;H1])=O)-[#6]-1 |
| 2,2-dimethyl succinic acid | [#6]C([#6])([#6]-[#6](-[#8])=O)[#6](-[#8])=O |
| piperazine | [#6;H2;X4]1-[#6;H2;X4][#7;X3;!H1][#6;H2;X4]-[#6;H2;X4][#7;H1;X3]1 |
| 1,2-diaminoethane | [#7;H1;X3][#6;H2;X4][#6;H2;X4][#7;H2;X3] |
| 1,3-diaminopropane | [#7;H1;X3][#6;H2;X4][#6;H2;X4][#6;H2;X4][#7;H2;X3] |
| Cyclic system skeleton derived from betulinic acid | [#6]1-[#6]-[#6]-[#6]2-[#6](-[#6]-1)-[#6]-[#6]-[#6]1-[#6]-2-[#6]-[#6]-[#6]2-[#6]3-[#6]-[#6]-[#6]-[#6]-3-[#6]-[#6]-[#6]-1-2 |
| Parent Molecule | SlogP | MW | HBD | HBA | TPSA | RB |
|---|---|---|---|---|---|---|
| Amprenavir | 2.40 | 505.22 | 4 | 9 | 131.19 | 11 |
| Atazanavir | 4.21 | 704.39 | 5 | 13 | 171.22 | 14 |
| Darunavir | 2.38 | 547.24 | 4 | 10 | 140.42 | 11 |
| Fosamprenavir a | 2.69 | 585.19 | 4 | 12 | 174.56 | 13 |
| Indinavir | 2.87 | 613.36 | 4 | 9 | 118.03 | 11 |
| Lopinavir | 4.33 | 628.36 | 4 | 9 | 120.00 | 15 |
| Nelfinavir | 4.75 | 567.31 | 4 | 7 | 101.90 | 9 |
| Ritonavir | 5.91 | 720.31 | 4 | 11 | 145.78 | 17 |
| Saquinavir | 3.09 | 670.38 | 6 | 11 | 166.75 | 12 |
| Tipranavir | 6.70 | 602.21 | 1 | 7 | 102.43 | 11 |
| Minimum a | 2.40 | 505.20 | 1 | 7 | 101.90 | 9 |
| Maximum a | 6.70 | 720.30 | 6 | 13 | 174.60 | 17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chávez-Hernández, A.L.; Juárez-Mercado, K.E.; Saldívar-González, F.I.; Medina-Franco, J.L. Towards the De Novo Design of HIV-1 Protease Inhibitors Based on Natural Products. Biomolecules 2021, 11, 1805. https://doi.org/10.3390/biom11121805
Chávez-Hernández AL, Juárez-Mercado KE, Saldívar-González FI, Medina-Franco JL. Towards the De Novo Design of HIV-1 Protease Inhibitors Based on Natural Products. Biomolecules. 2021; 11(12):1805. https://doi.org/10.3390/biom11121805
Chicago/Turabian StyleChávez-Hernández, Ana L., K. Eurídice Juárez-Mercado, Fernanda I. Saldívar-González, and José L. Medina-Franco. 2021. "Towards the De Novo Design of HIV-1 Protease Inhibitors Based on Natural Products" Biomolecules 11, no. 12: 1805. https://doi.org/10.3390/biom11121805
APA StyleChávez-Hernández, A. L., Juárez-Mercado, K. E., Saldívar-González, F. I., & Medina-Franco, J. L. (2021). Towards the De Novo Design of HIV-1 Protease Inhibitors Based on Natural Products. Biomolecules, 11(12), 1805. https://doi.org/10.3390/biom11121805