
The Imbalance of Mitochondrial Fusion/Fission Drives High-Glucose-Induced Vascular Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Cell Culture and Experimental Designs
2.3. Western Blotting
2.4. Apoptosis Assay
2.5. Measurement of Mitochondrial Membrane Potential
2.6. Animal Models and Treatments
2.7. Plaque Area Quantitation
2.8. Immunohistochemistry Assay
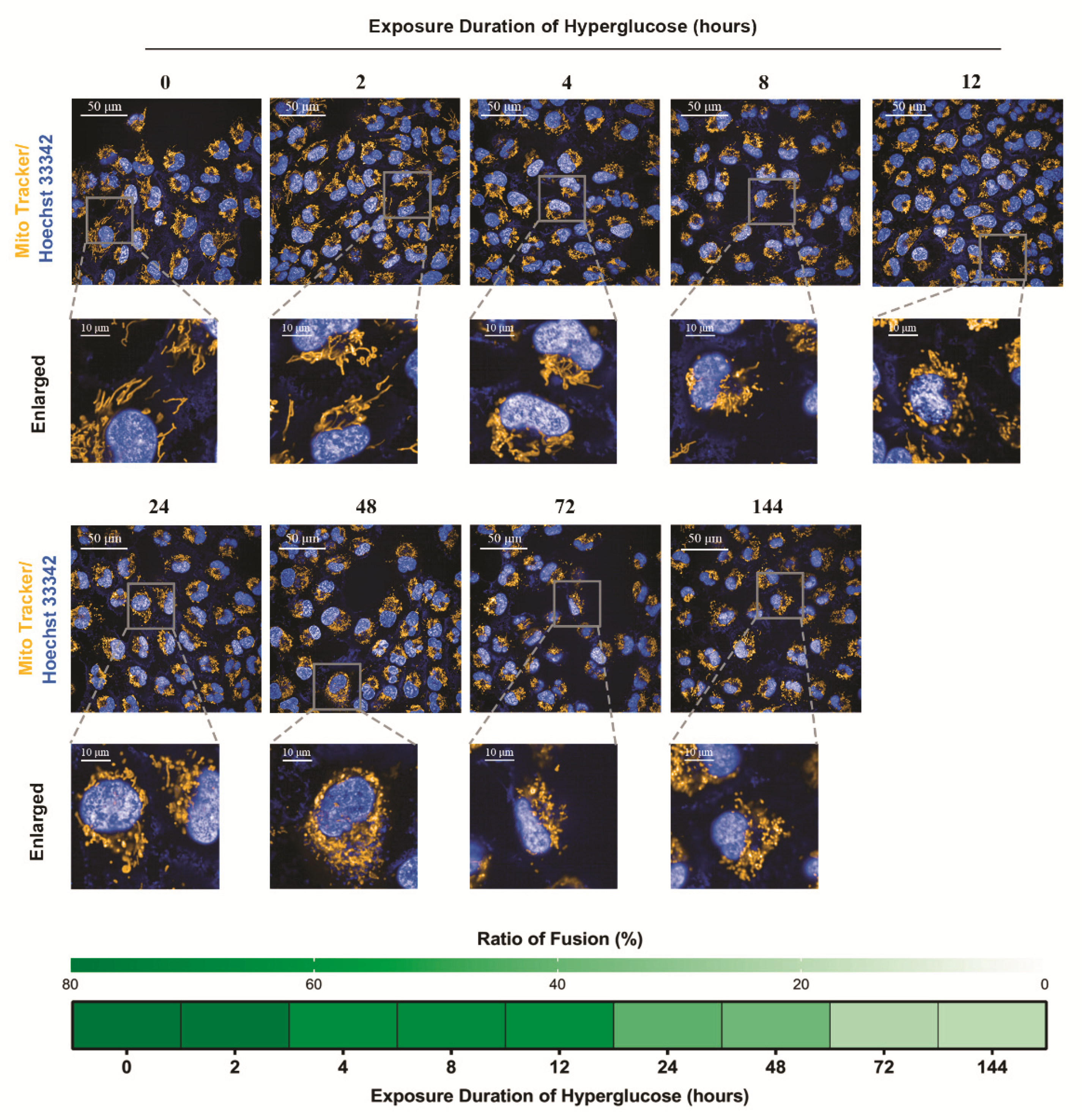
2.9. Mitochondrial Morphology Imaging
2.10. Statistical Analysis
3. Results
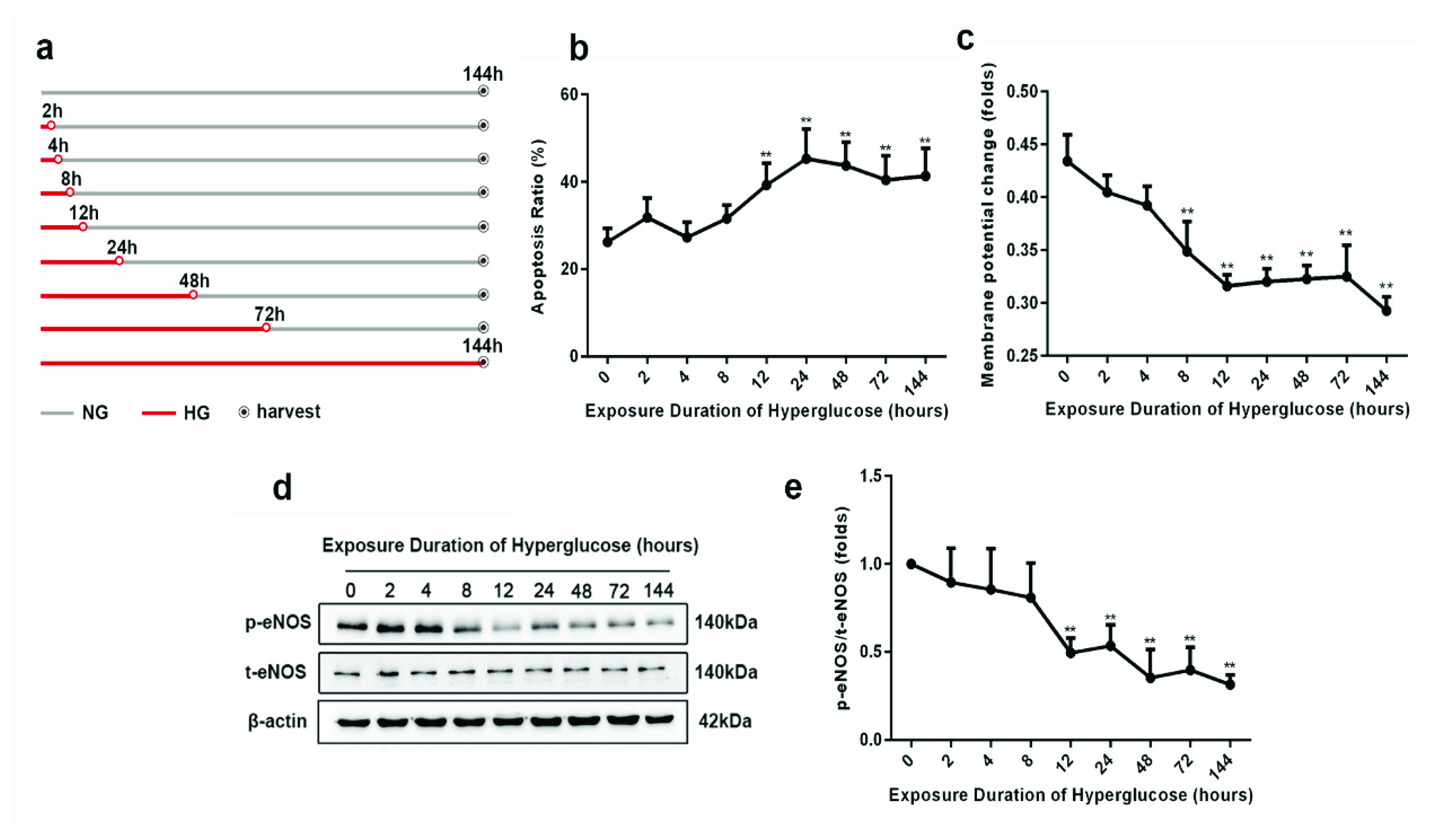
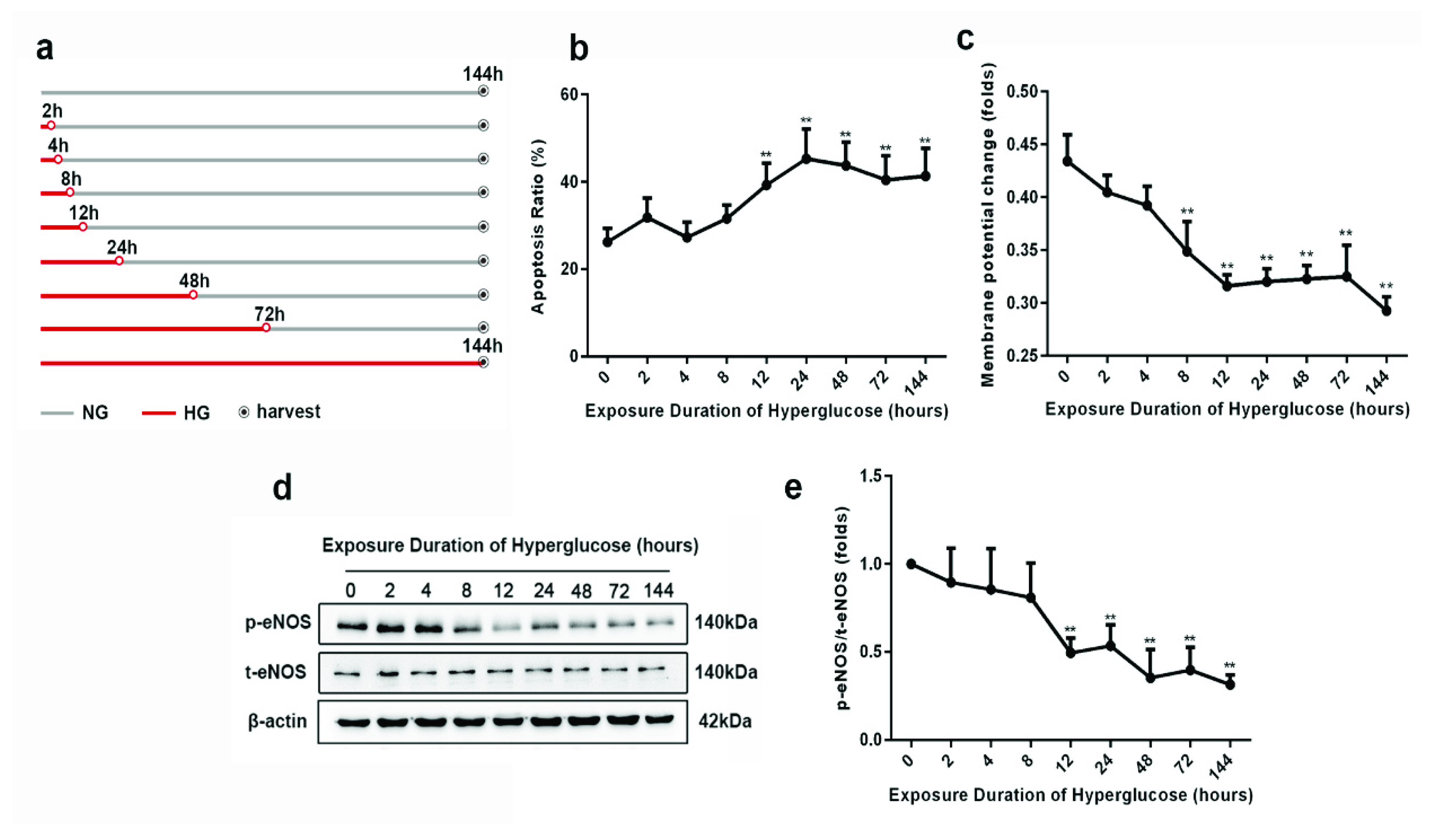
3.1. Progression of Metabolic Memory in High-Glucose-Exposed HUVECs
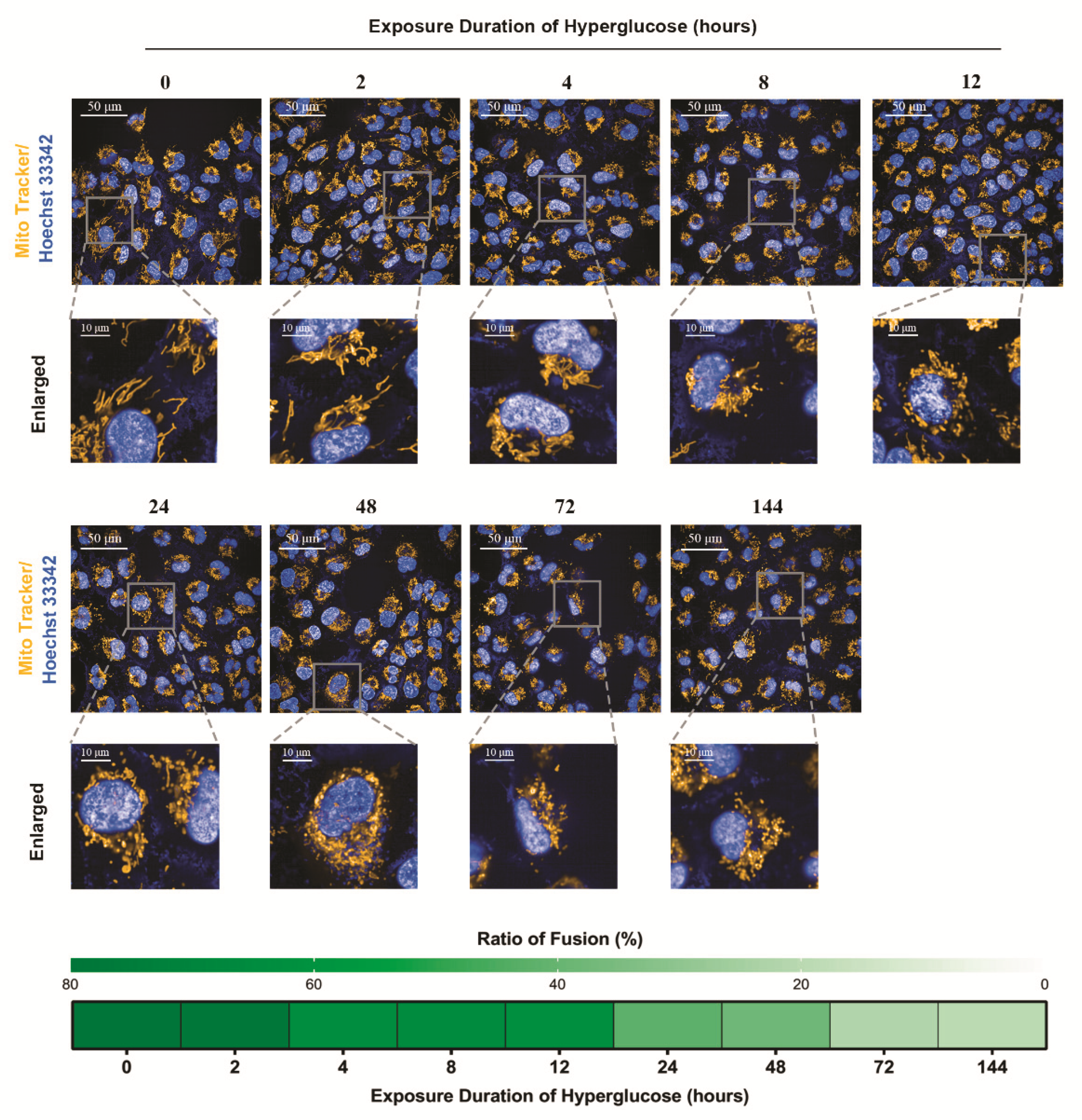
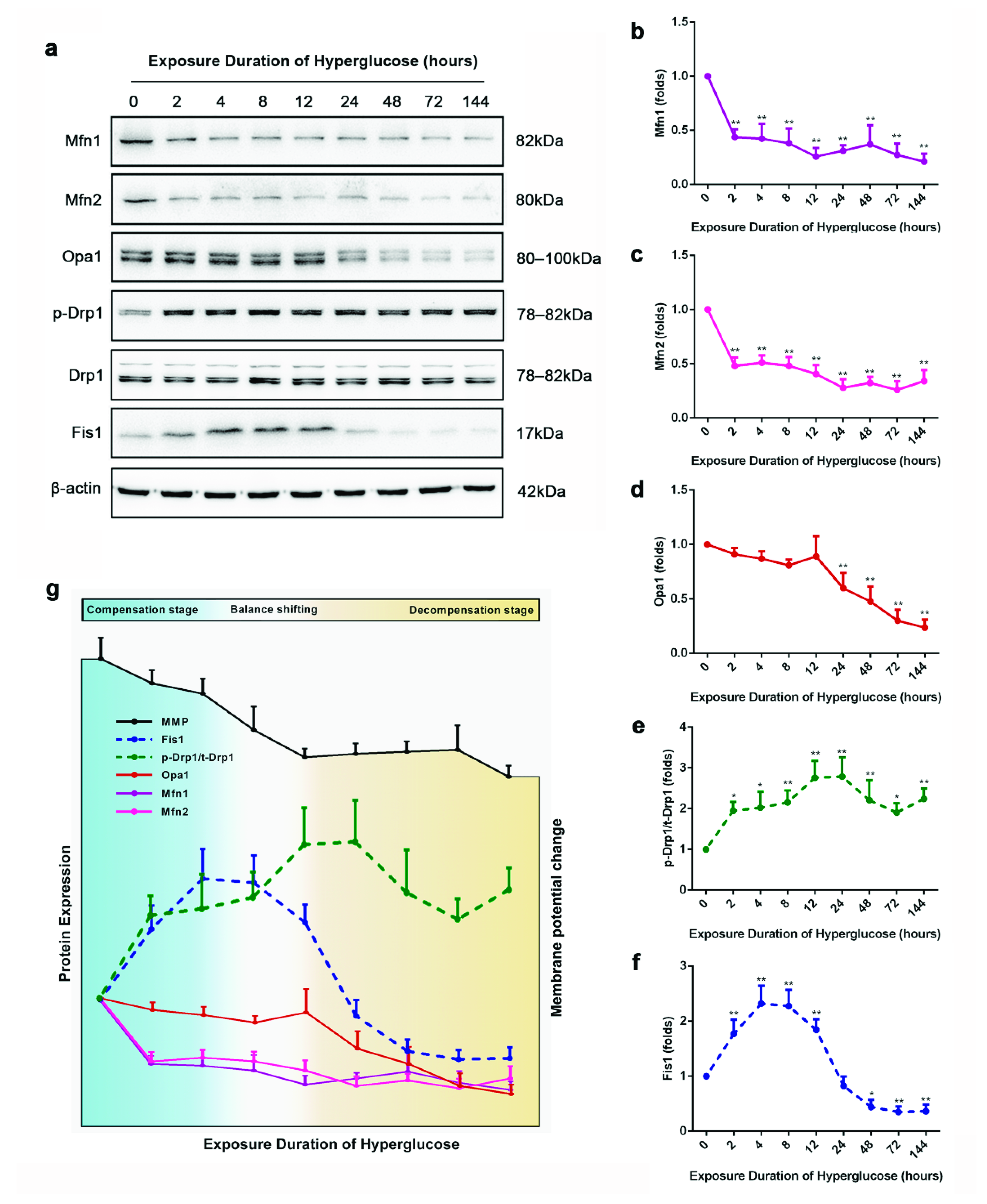
3.2. The Staged Mitochondrial Fusion/Fission Homeostasis during Metabolic Memory Formation
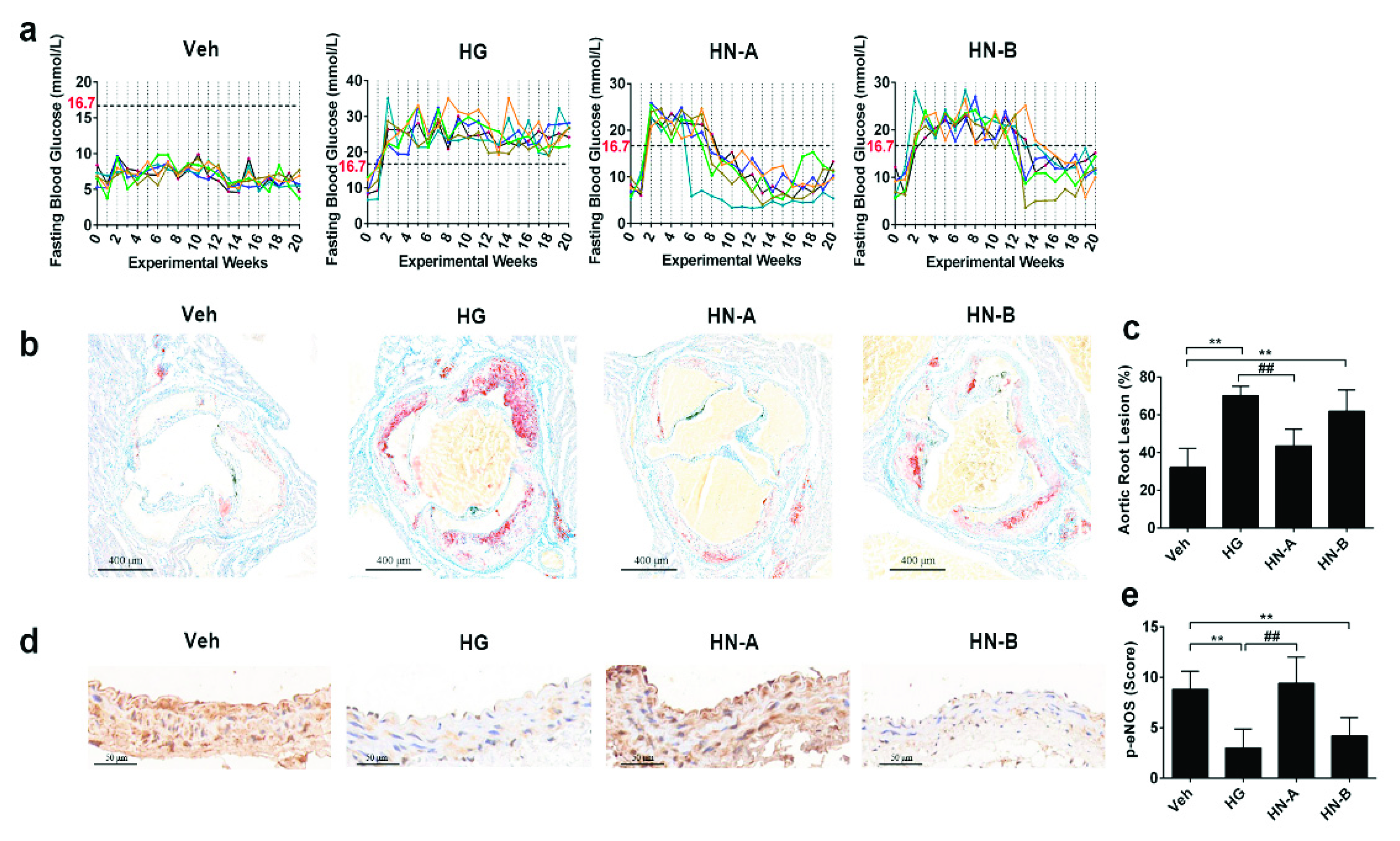
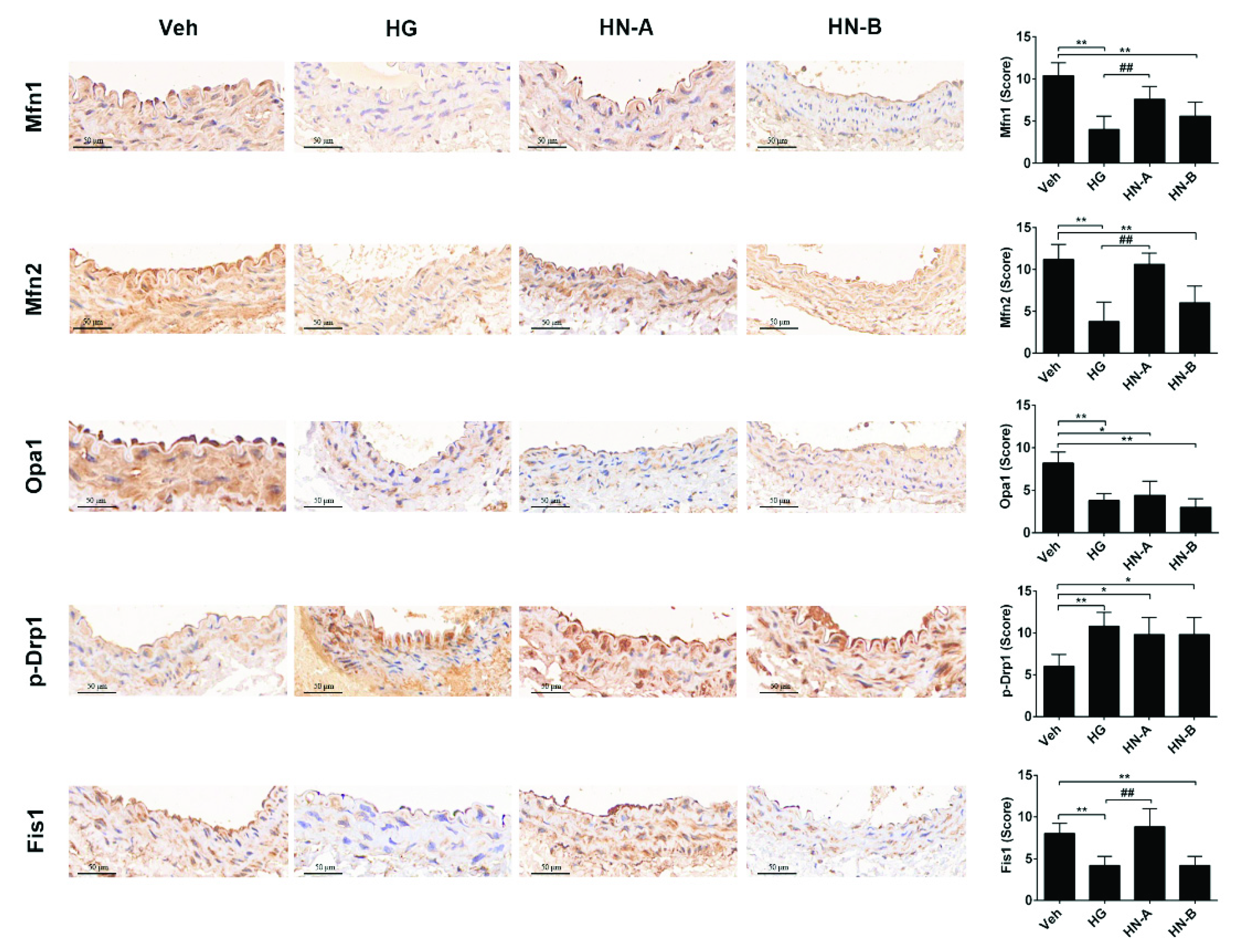
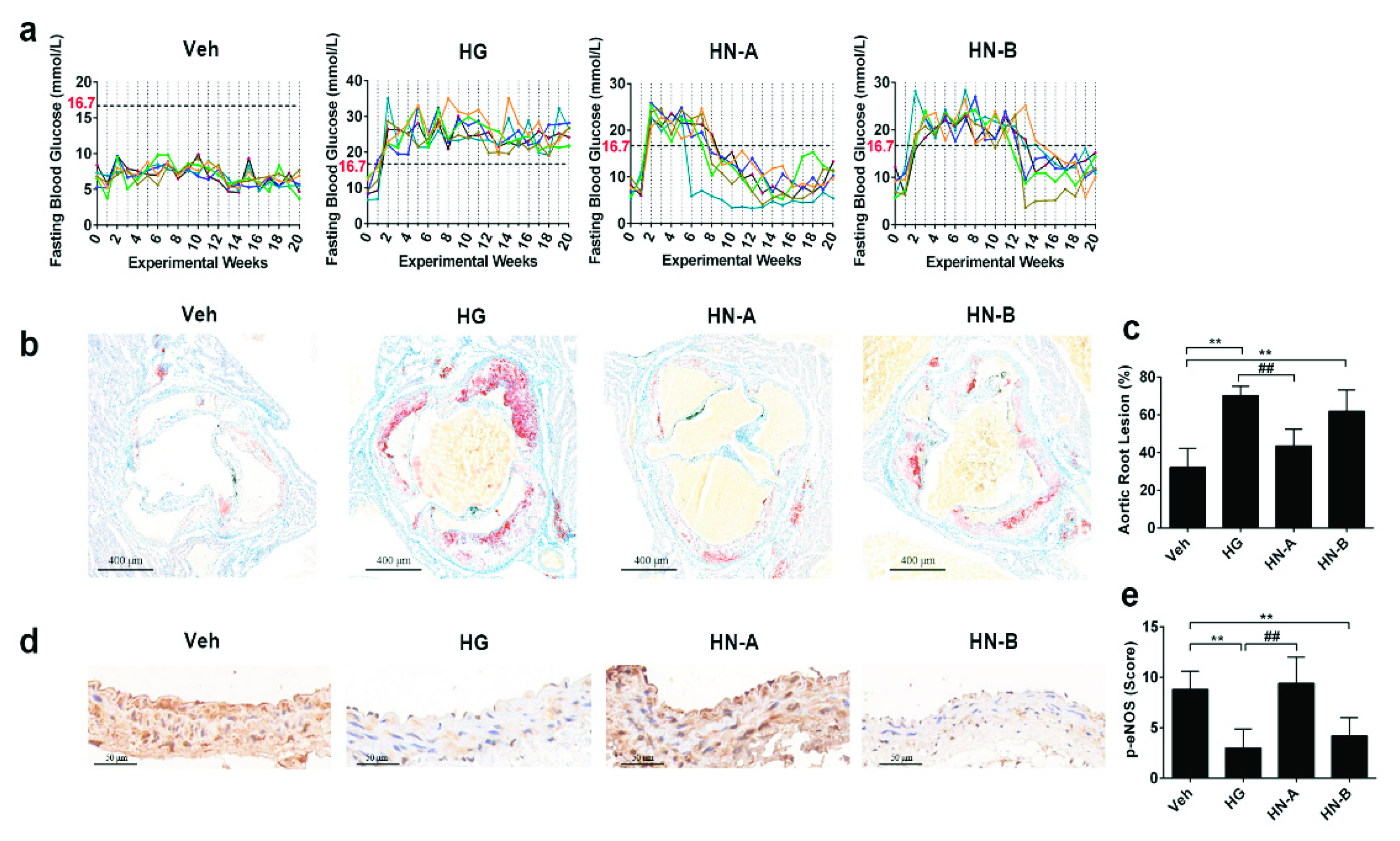
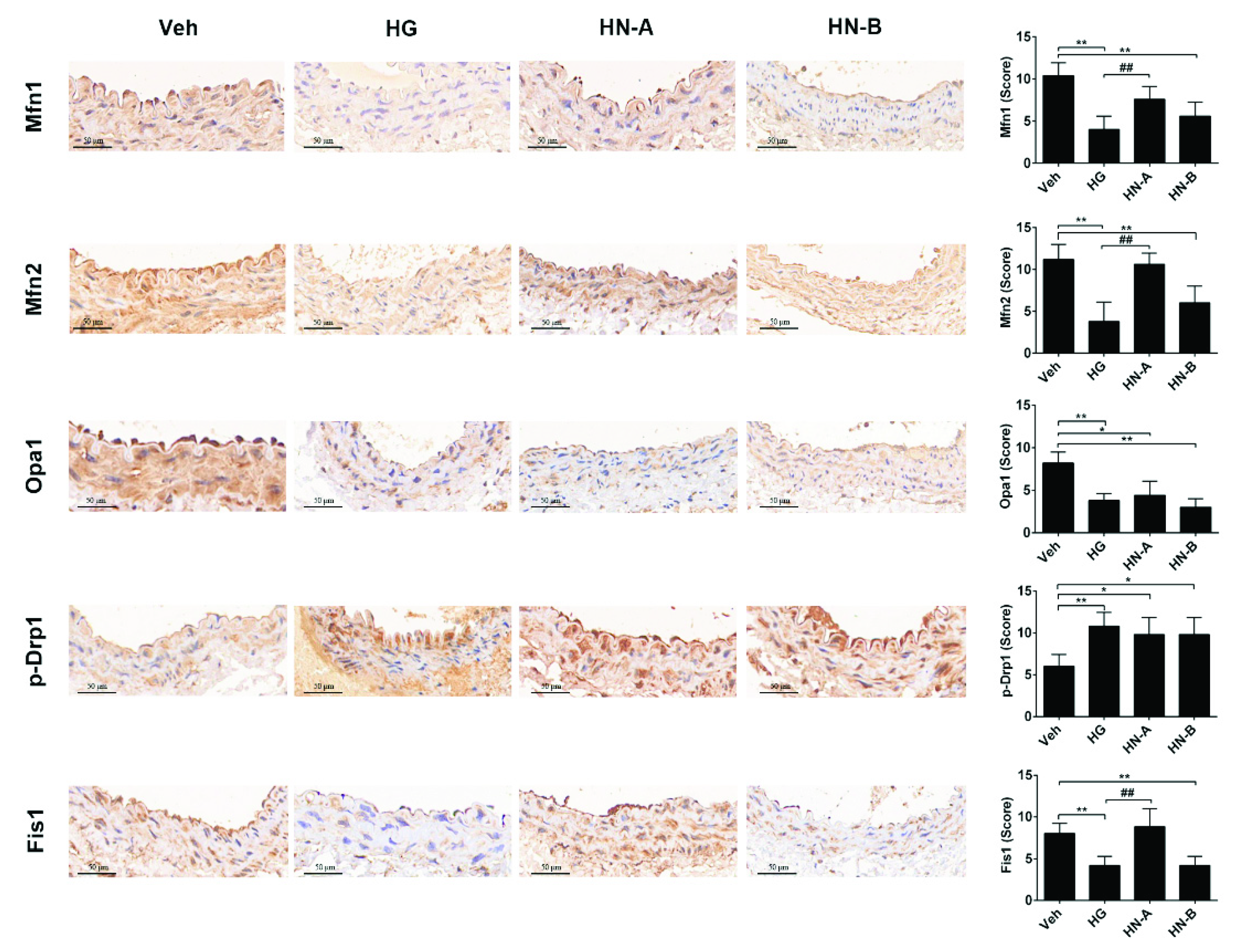
3.3. In Vivo Verification of the Staged Balancing of Mitochondrial Fusion/Fission
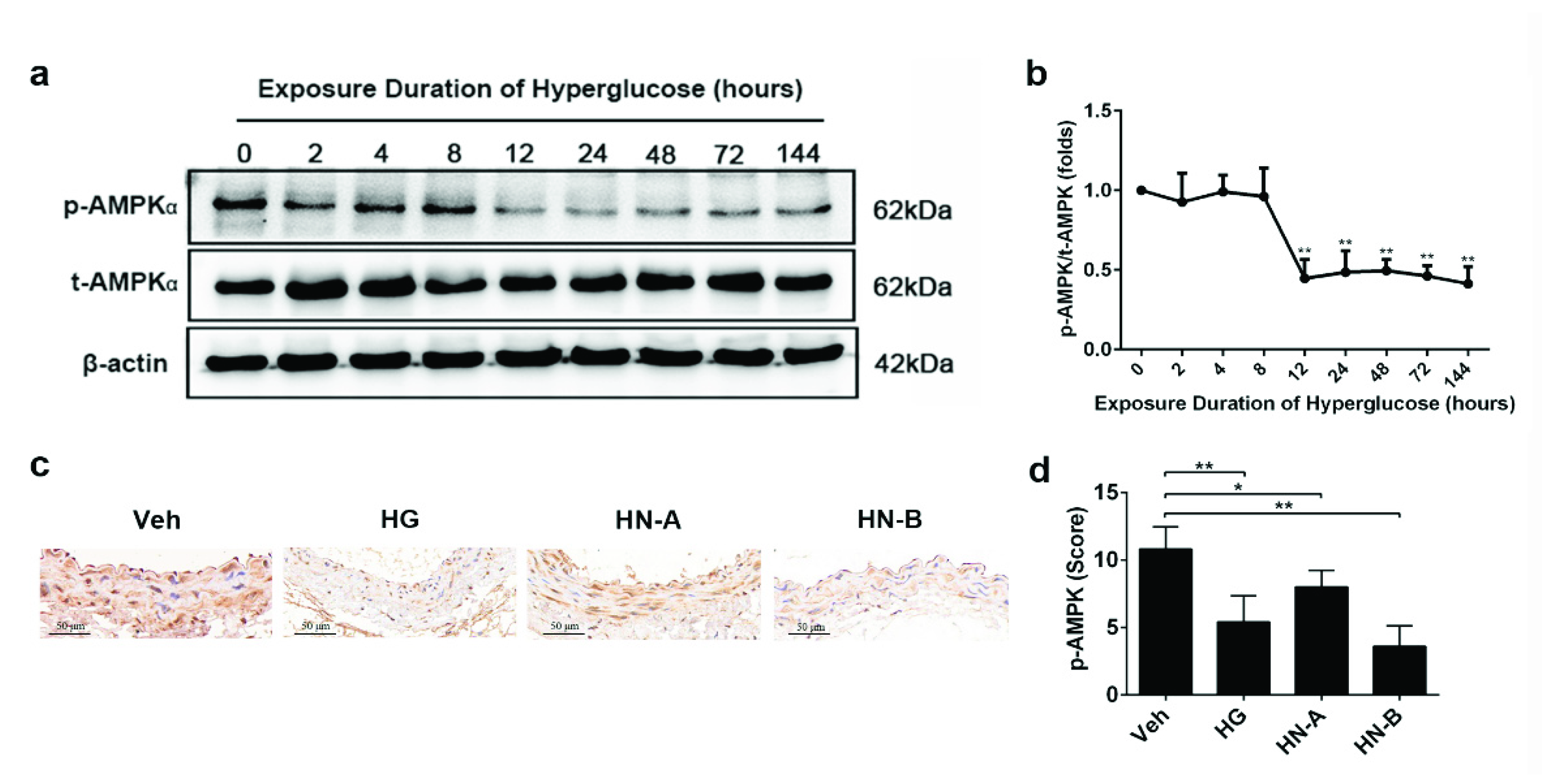
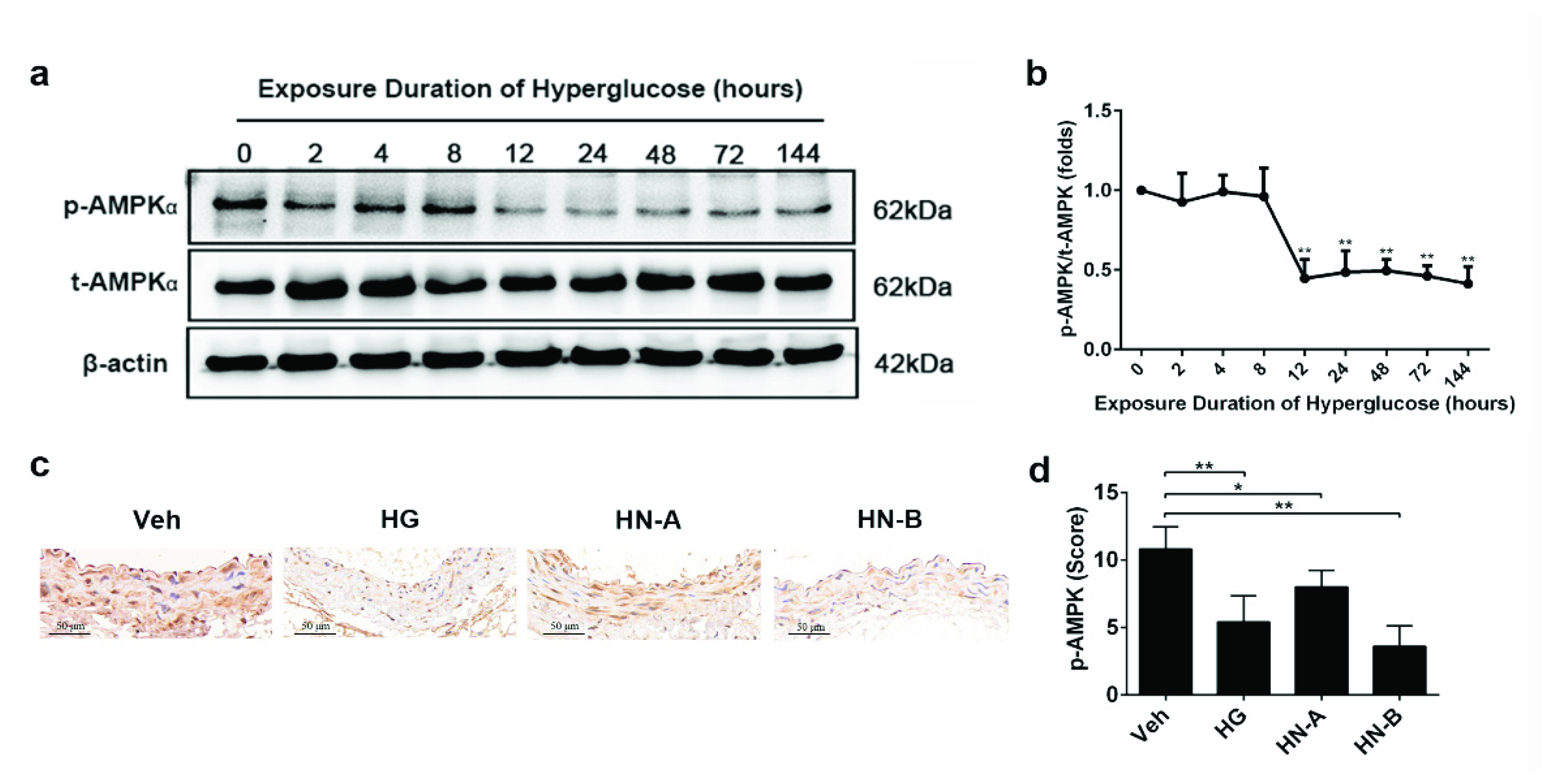
3.4. Involvement of AMPK in High-Glucose-Induced Mitochondrial Fusion/Fission Balance Shifting
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- O’Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E., Jr.; Chung, M.K.; de Lemos, J.A.; Ettinger, S.M.; Fang, J.C.; Fesmire, F.M.; Franklin, B.A.; et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 127, e362–e425. [Google Scholar] [CrossRef] [Green Version]
- Lind, M.; Svensson, A.-M.; Kosiborod, M.; Gudbjörnsdottir, S.; Pivodic, A.; Wedel, H.; Dahlqvist, S.; Clements, M.; Rosengren, A. Glycemic control and excess mortality in type 1 diabetes. N. Engl. J. Med. 2014, 371, 1972–1982. [Google Scholar] [CrossRef]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus: Atherosclerotic Cardiovascular Disease and Heart Failure in Type 2 Diabetes Mellitus-Mechanisms, Management, and Clinical Considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef]
- Engerman, R.L.; Kern, T.S. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 1987, 36, 808–812. [Google Scholar] [CrossRef]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A.W. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [Green Version]
- Diabetes, C.; Complications Trial /Epidemiology of Diabetes, I.; Complications Study Research, G. Intensive Diabetes Treatment and Cardiovascular Outcomes in Type 1 Diabetes: The DCCT/EDIC Study 30-Year Follow-up. Diabetes Care 2016, 39, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.E.; El-Osta, A.; Allen, T.J.; Watson, A.M.D.; Thomas, M.C.; Jandeleit-Dahm, K.A.M. Metabolic Karma-The Atherogenic Legacy of Diabetes: The 2017 Edwin Bierman Award Lecture. Diabetes 2018, 67, 785–790. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, C.A.; Yoon, Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid. Redox Signal. 2015, 22, 1545–1562. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.; Caino, M.C. Mitochondrial Dynamics in Type 2 Diabetes and Cancer. Front. Endocrinol. 2018, 9, 211. [Google Scholar] [CrossRef]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.W.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Lim, Y.C.; Kim, Y.M.; Ha, K.S. C-peptide activates AMPKalpha and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes 2013, 62, 3851–3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Núñez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L.; et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cell. Signal. 2015, 27, 978–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Chen, H.; Zhang, L.; Lin, X.; Li, X.; Zhuang, H.; Fan, H.; Meng, T.; He, Z.; Huang, H.; et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 2020, 17, 1142–1156. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. Off. J. Jpn. Circ. Soc. 2009, 73, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Shafique, E.; Torina, A.; Reichert, K.; Colantuono, B.; Nur, N.; Zeeshan, K.; Ravichandran, V.; Liu, Y.; Feng, J.; Zeeshan, K.; et al. Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium. Cardiovasc. Res. 2017, 113, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Oak, J.H.; Cai, H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 2007, 56, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Gou, L.; Chen, L.; Zhong, X.; Zhang, D.; Zhu, H.; Lu, X.; Zeng, T.; Deng, X.; Li, Y. NADPH oxidase 4 and endothelial nitric oxide synthase contribute to endothelial dysfunction mediated by histone methylations in metabolic memory. Free Radic. Biol. Med. 2018, 115, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Mocharla, P.; Akhmedov, A.; Costantino, S.; Osto, E.; Volpe, M.; Luscher, T.F.; Cosentino, F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ. Res. 2012, 111, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia Induces a Dynamic Cooperativity of Histone Methylase and Demethylase Enzymes Associated With Gene-Activating Epigenetic Marks That Coexist on the Lysine Tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Ebert, T.; Klöting, N.; Kolb, M.; Gericke, M.; Jeromin, F.; Jessnitzer, B.; Lössner, U.; Burkhardt, R.; Stumvoll, M.; et al. Leptin decreases circulating inflammatory IL-6 and MCP-1 in mice. Biofactors 2019, 45, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Candido, R.; Jandeleit-Dahm, K.A.; Cao, Z.; Nesteroff, S.P.; Burns, W.C.; Twigg, S.M.; Dilley, R.J.; Cooper, M.E.; Allen, T.J. Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 2002, 106, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Candido, R.; Allen, T.J.; Lassila, M.; Cao, Z.; Thallas, V.; Cooper, M.E.; Jandeleit-Dahm, K.A. Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 2004, 109, 1536–1542. [Google Scholar] [CrossRef]
- Soro-Paavonen, A.; Watson, A.M.; Li, J.; Paavonen, K.; Koitka, A.; Calkin, A.C.; Barit, D.; Coughlan, M.T.; Drew, B.G.; Lancaster, G.I.; et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 2008, 57, 2461–2469. [Google Scholar] [CrossRef] [Green Version]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Fealy, C.E.; Mulya, A.; Axelrod, C.L.; Kirwan, J.P. Mitochondrial dynamics in skeletal muscle insulin resistance and type 2 diabetes. Transl. Res. J. Lab. Clin. Med. 2018, 202, 69–82. [Google Scholar] [CrossRef]
- Schultz, J.; Waterstradt, R.; Kantowski, T.; Rickmann, A.; Reinhardt, F.; Sharoyko, V.; Mulder, H.; Tiedge, M.; Baltrusch, S. Precise expression of Fis1 is important for glucose responsiveness of beta cells. J. Endocrinol. 2016, 230, 81–91. [Google Scholar] [CrossRef]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef]
- Liu, H.; Xiang, H.; Zhao, S.; Sang, H.; Lv, F.; Chen, R.; Shu, Z.; Chen, A.F.; Chen, S.; Lu, H. Vildagliptin improves high glucose-induced endothelial mitochondrial dysfunction via inhibiting mitochondrial fission. J. Cell. Mol. Med. 2019, 23, 798–810. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, H.G.; Langer, T. The Good and the Bad of Mitochondrial Breakups. Trends Cell Biol. 2019, 29, 888–900. [Google Scholar] [CrossRef]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Liu, T.; Ning, C.; Tan, F.; Jin, S.-B.; Lendahl, U.; Zhao, J.; Nistér, M. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 2019, 294, 17262–17277. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Gaytan, N.; Garcia, I.; Herrera, A.; Ramos, M.; Agarwala, D.; Rana, M.; Innis-Whitehouse, W.; Schuenzel, E.; Gilkerson, R. A threshold of transmembrane potential is required for mitochondrial dynamic balance mediated by DRP1 and OMA1. Cell. Mol. Life Sci. CMLS 2017, 74, 1347–1363. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Jin, S.-B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Xian, H.; Yang, Q.; Xiao, L.; Shen, H.M.; Liou, Y.C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 2019, 10, 2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef]
- Seabright, A.P.; Lai, Y.C. Regulatory Roles of PINK1-Parkin and AMPK in Ubiquitin-Dependent Skeletal Muscle Mitophagy. Front. Physiol. 2020, 11, 608474. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Li, X.; Liu, J.; Sun, X.; Rousselle, T.; Ren, D.; Tong, N.; Li, J. AMPK is associated with the beneficial effects of antidiabetic agents on cardiovascular diseases. Biosci. Rep. 2019, 39, BSR20181995. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Luo, A.; Liu, X. The Imbalance of Mitochondrial Fusion/Fission Drives High-Glucose-Induced Vascular Injury. Biomolecules 2021, 11, 1779. https://doi.org/10.3390/biom11121779
Zheng Y, Luo A, Liu X. The Imbalance of Mitochondrial Fusion/Fission Drives High-Glucose-Induced Vascular Injury. Biomolecules. 2021; 11(12):1779. https://doi.org/10.3390/biom11121779
Chicago/Turabian StyleZheng, Yunsi, Anqi Luo, and Xiaoquan Liu. 2021. "The Imbalance of Mitochondrial Fusion/Fission Drives High-Glucose-Induced Vascular Injury" Biomolecules 11, no. 12: 1779. https://doi.org/10.3390/biom11121779
APA StyleZheng, Y., Luo, A., & Liu, X. (2021). The Imbalance of Mitochondrial Fusion/Fission Drives High-Glucose-Induced Vascular Injury. Biomolecules, 11(12), 1779. https://doi.org/10.3390/biom11121779