
Vitis OneGenE: A Causality-Based Approach to Generate Gene Networks in Vitis vinifera Sheds Light on the Laccase and Dirigent Gene Families

 ,
,  , ,
, , 
 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computation of OneGenE Expansion Lists
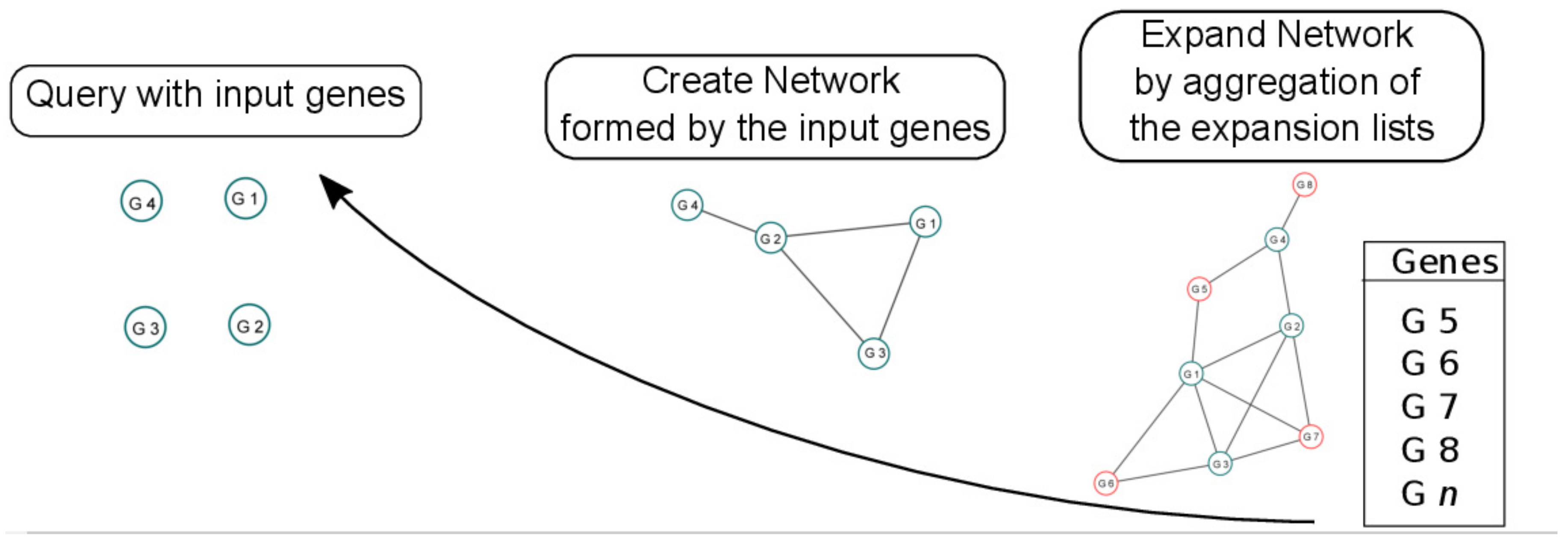
2.2. Programs Developed to Reconstruct Gene Networks Using OneGenE Output
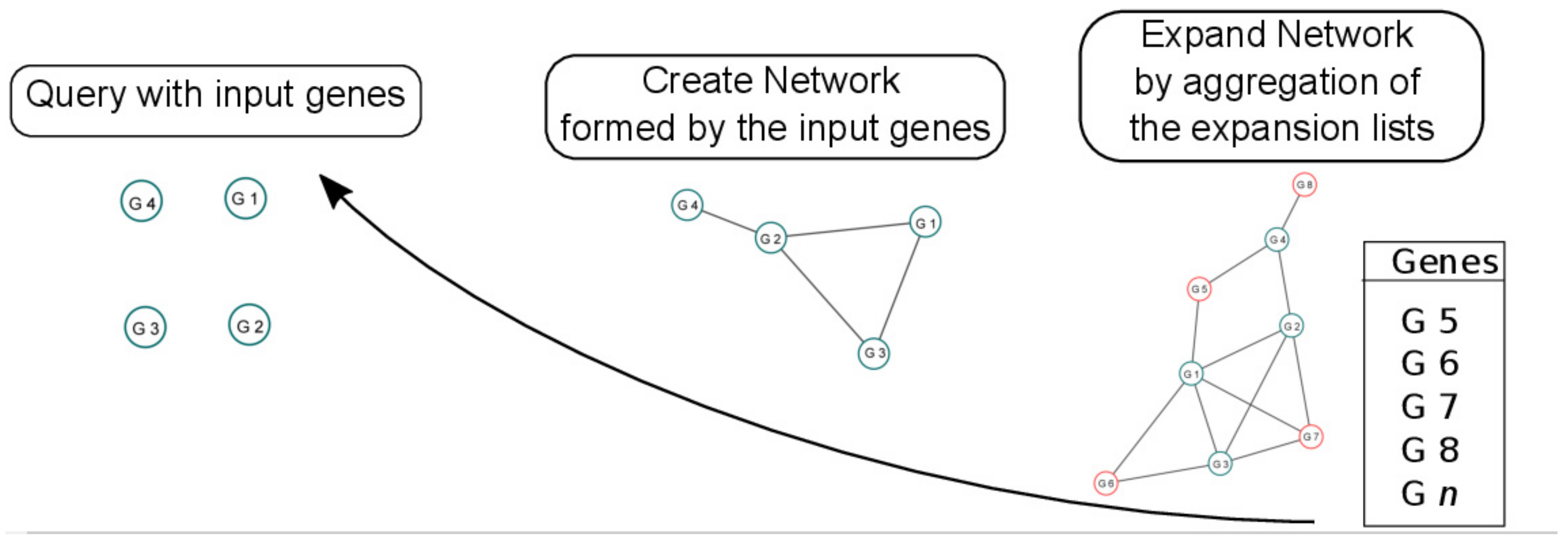
2.2.1. Create Network
2.2.2. Expand Network
2.2.3. Network Analysis
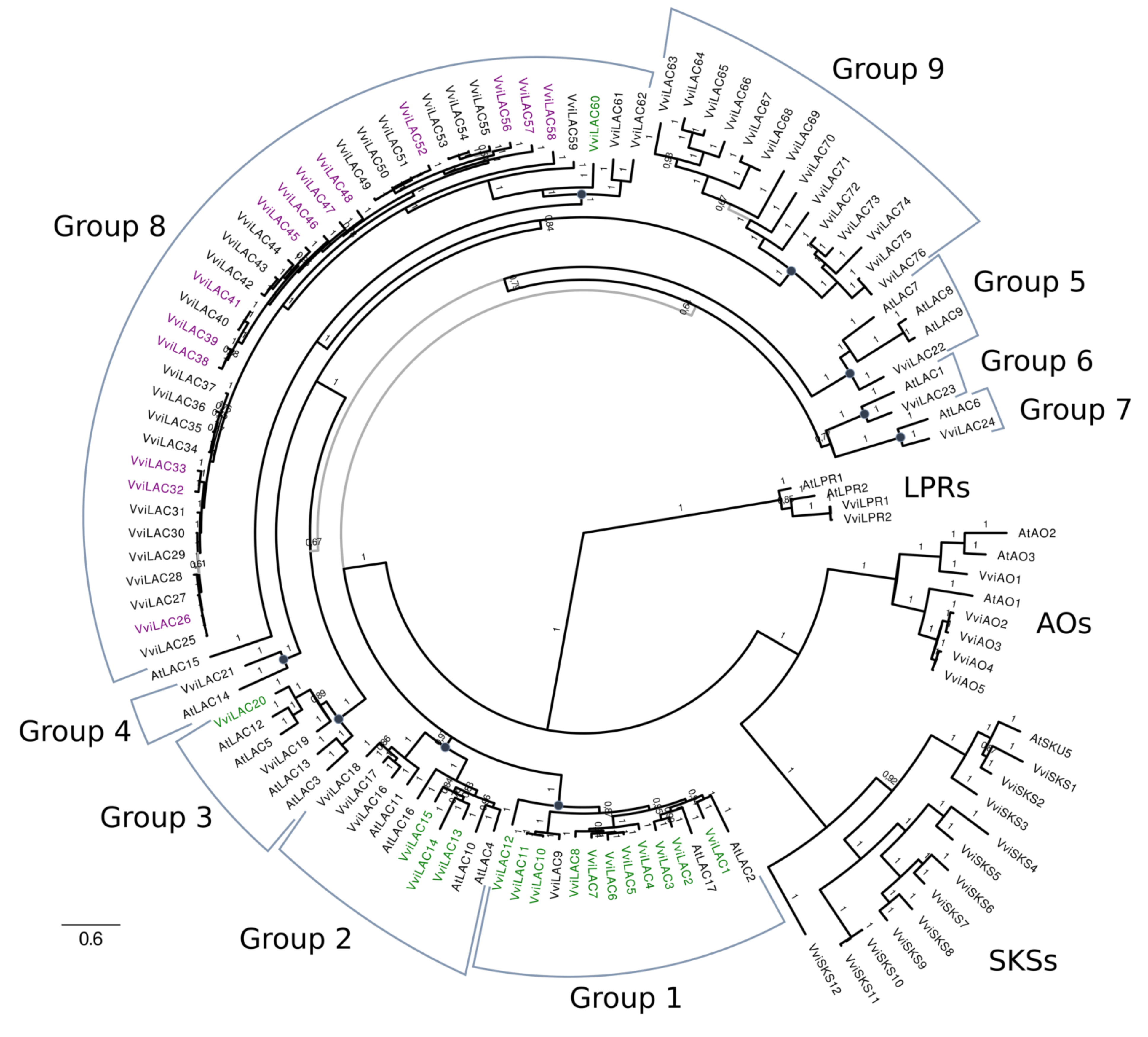
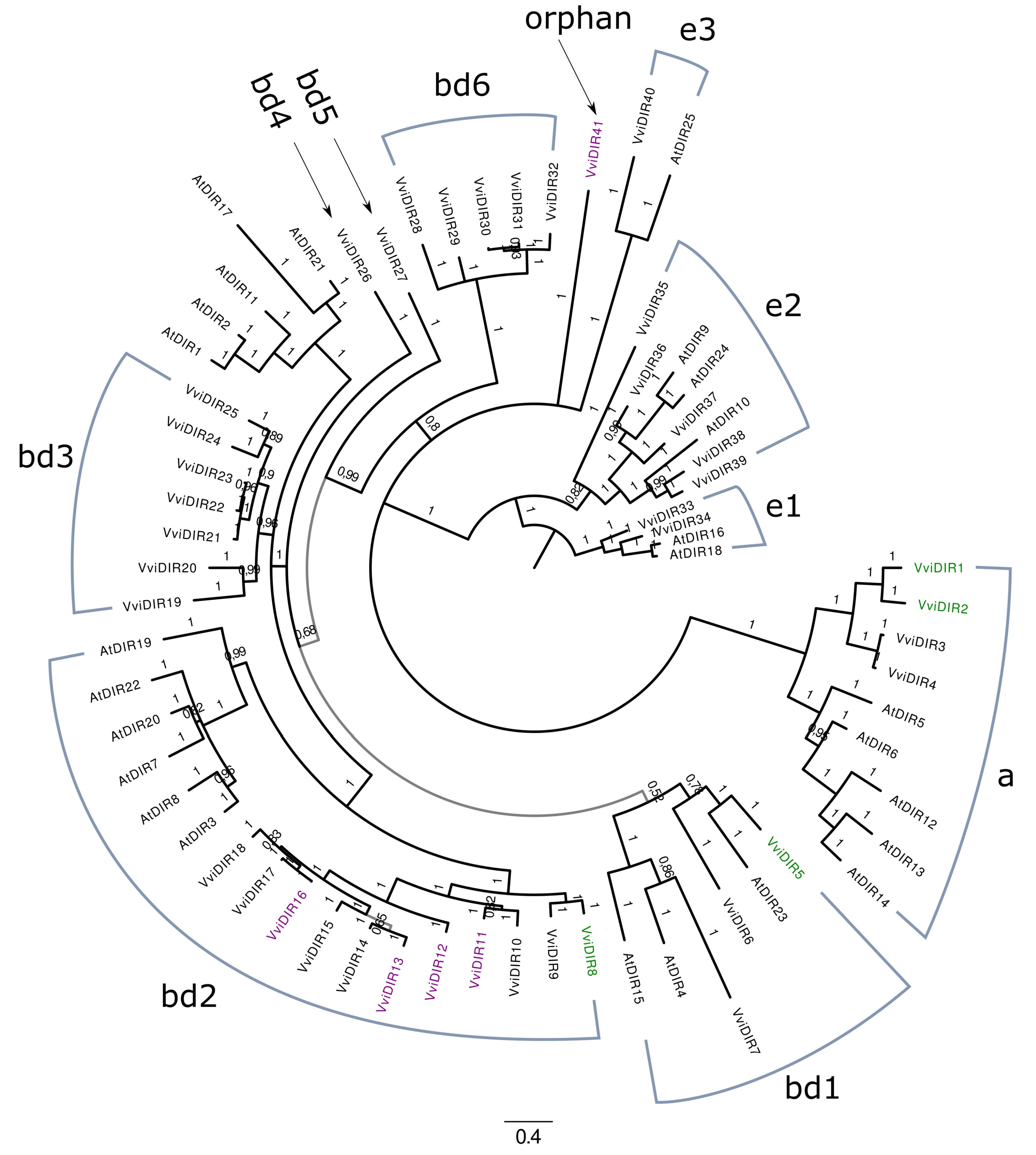
2.3. Genome-Wide Search and Gene Model Curation of LAC and DIR Gene Families
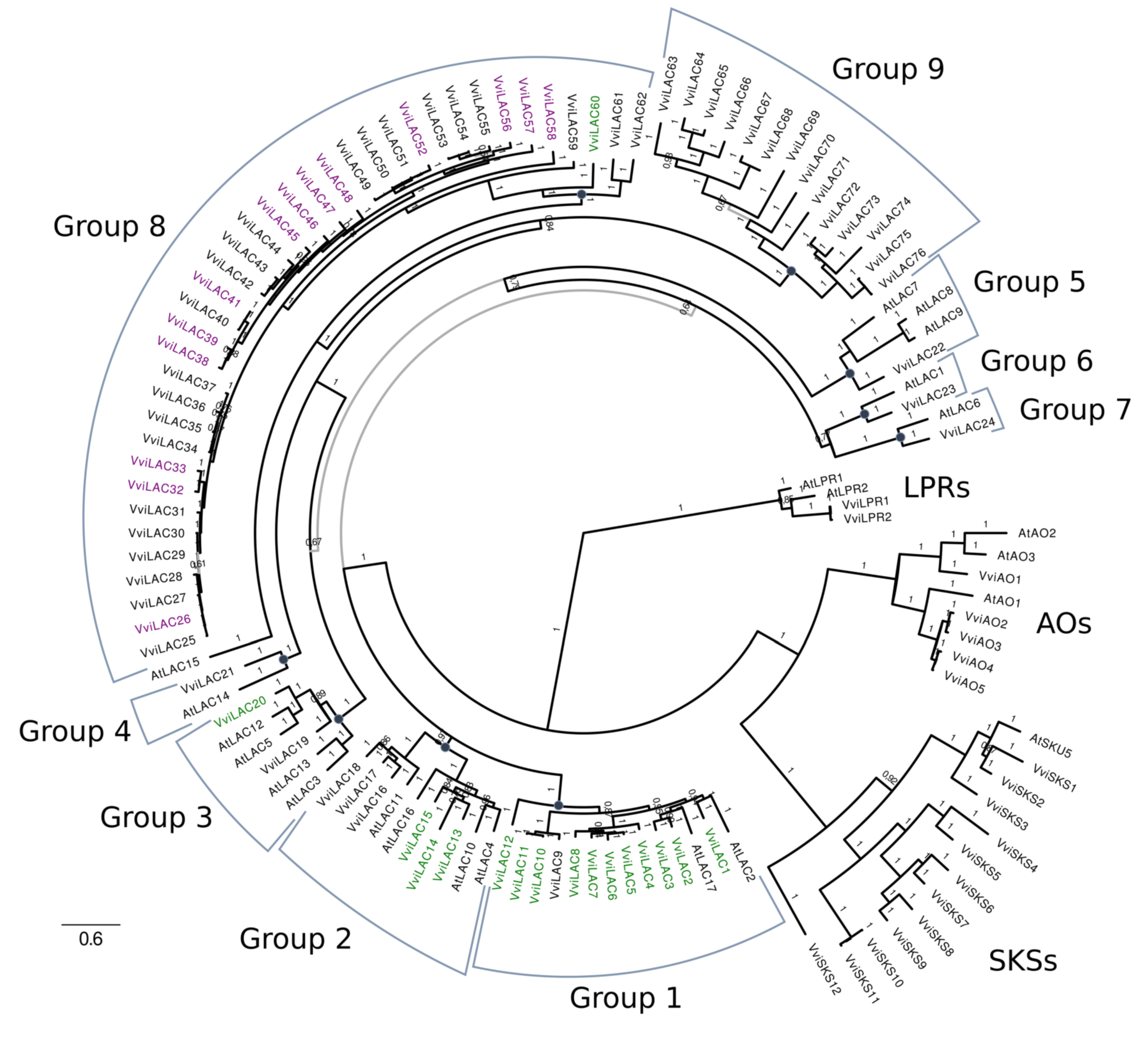
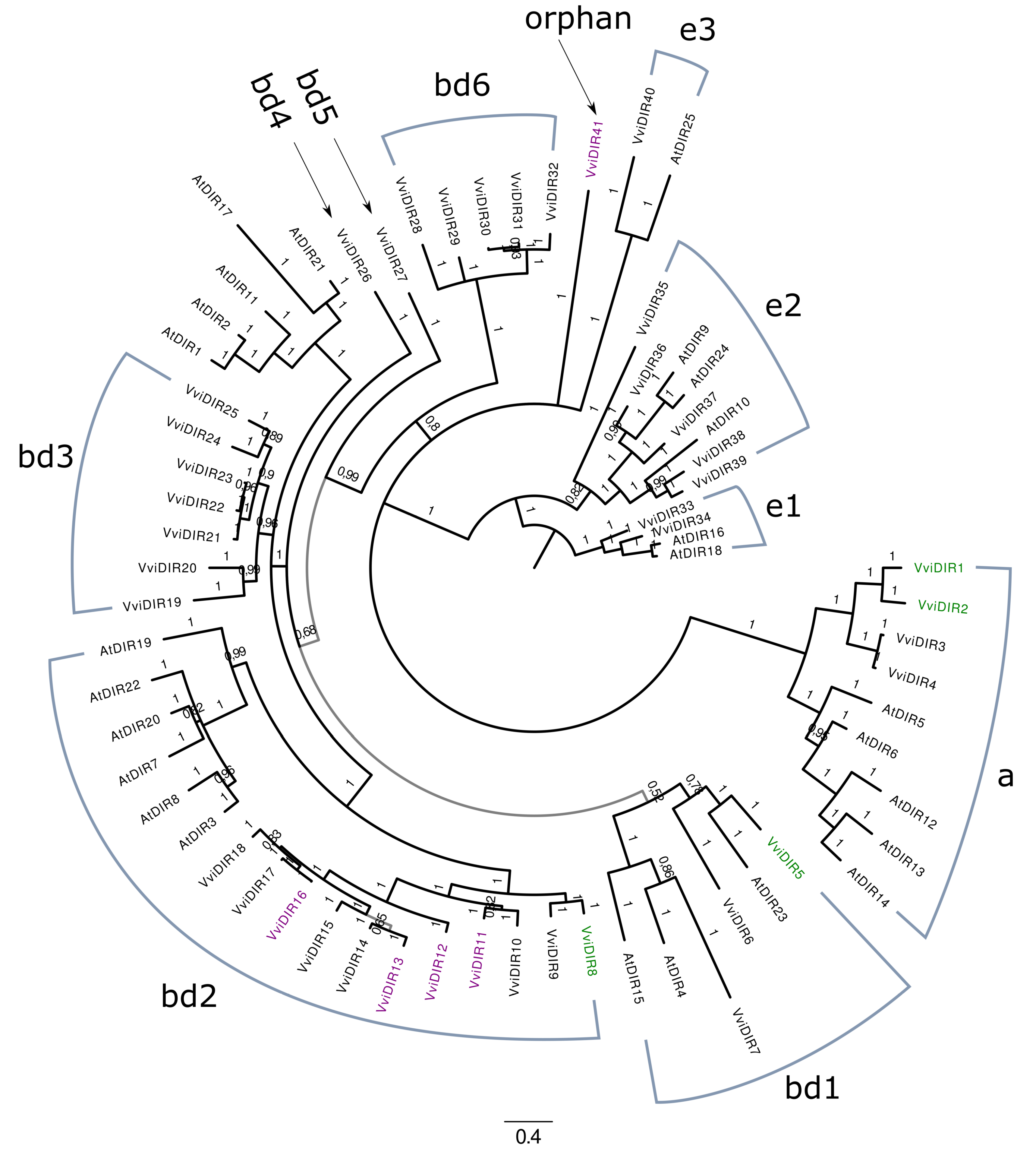
2.4. Phylogenetic Analysis of the LAC and DIR Gene Families
2.5. Data and Code Availability
3. Results
3.1. The Grapevine OneGenE Website
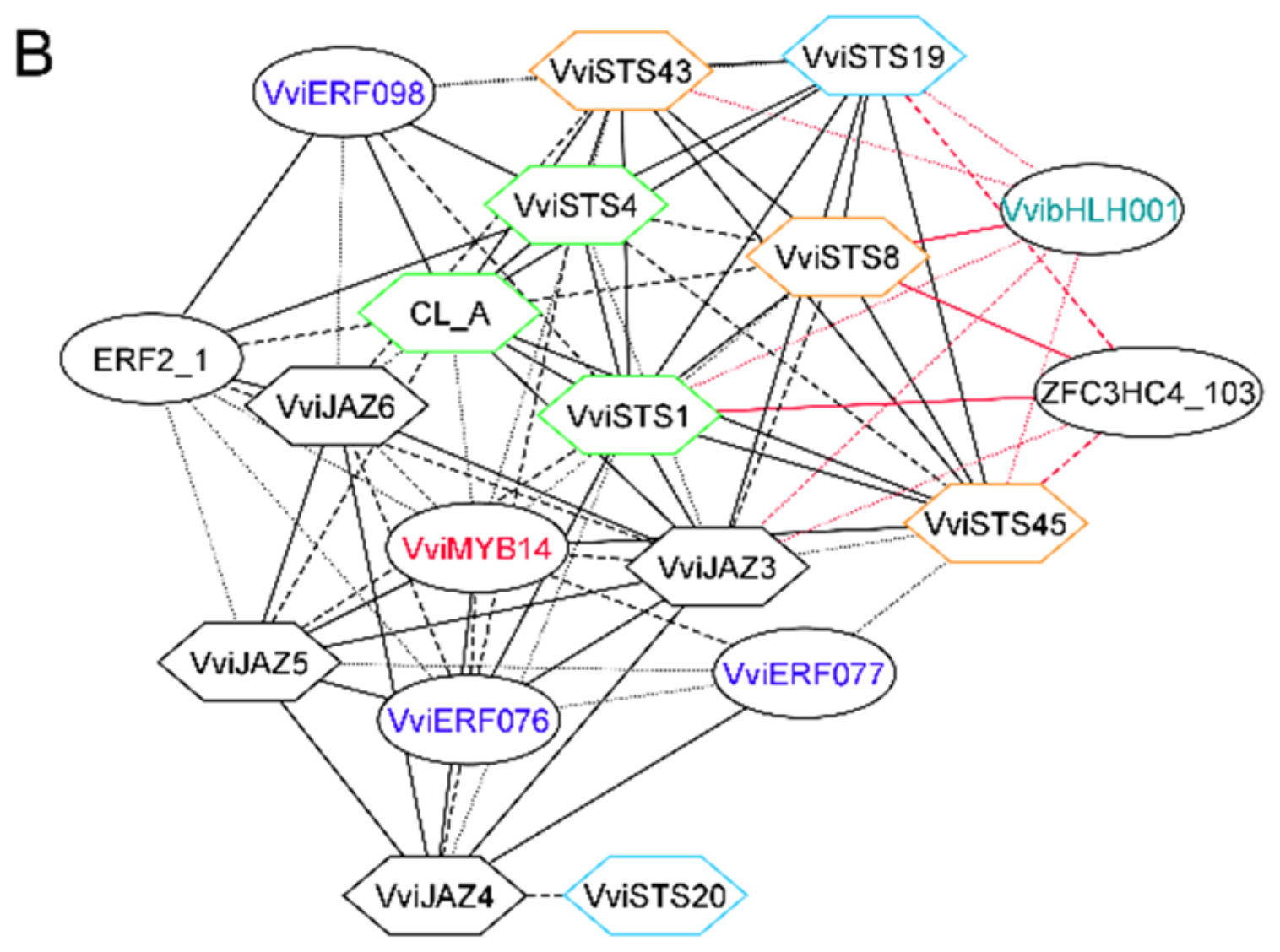
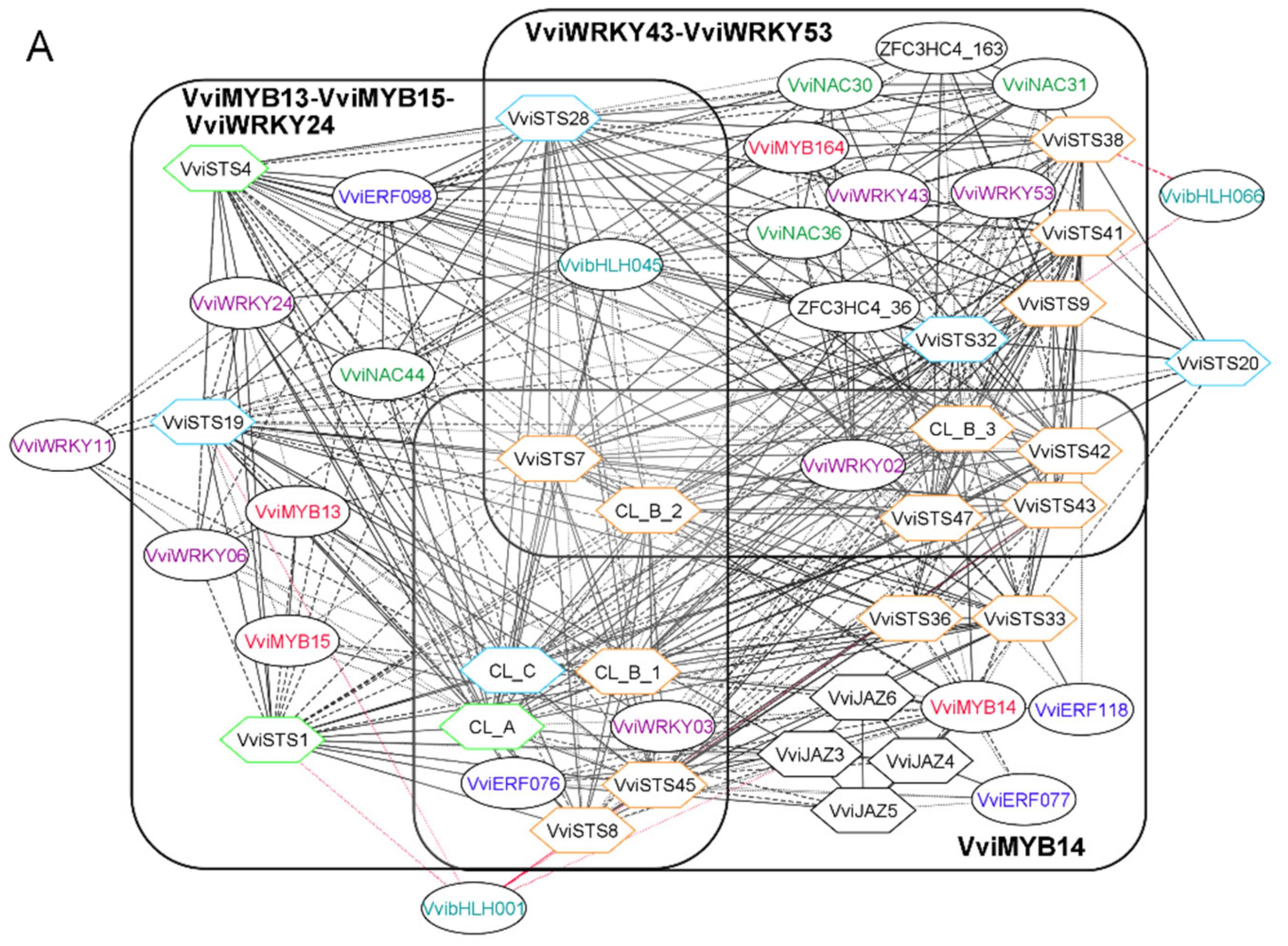
3.2. The Stilbene Synthase Gene Regulatory Network as a Proof of Concept
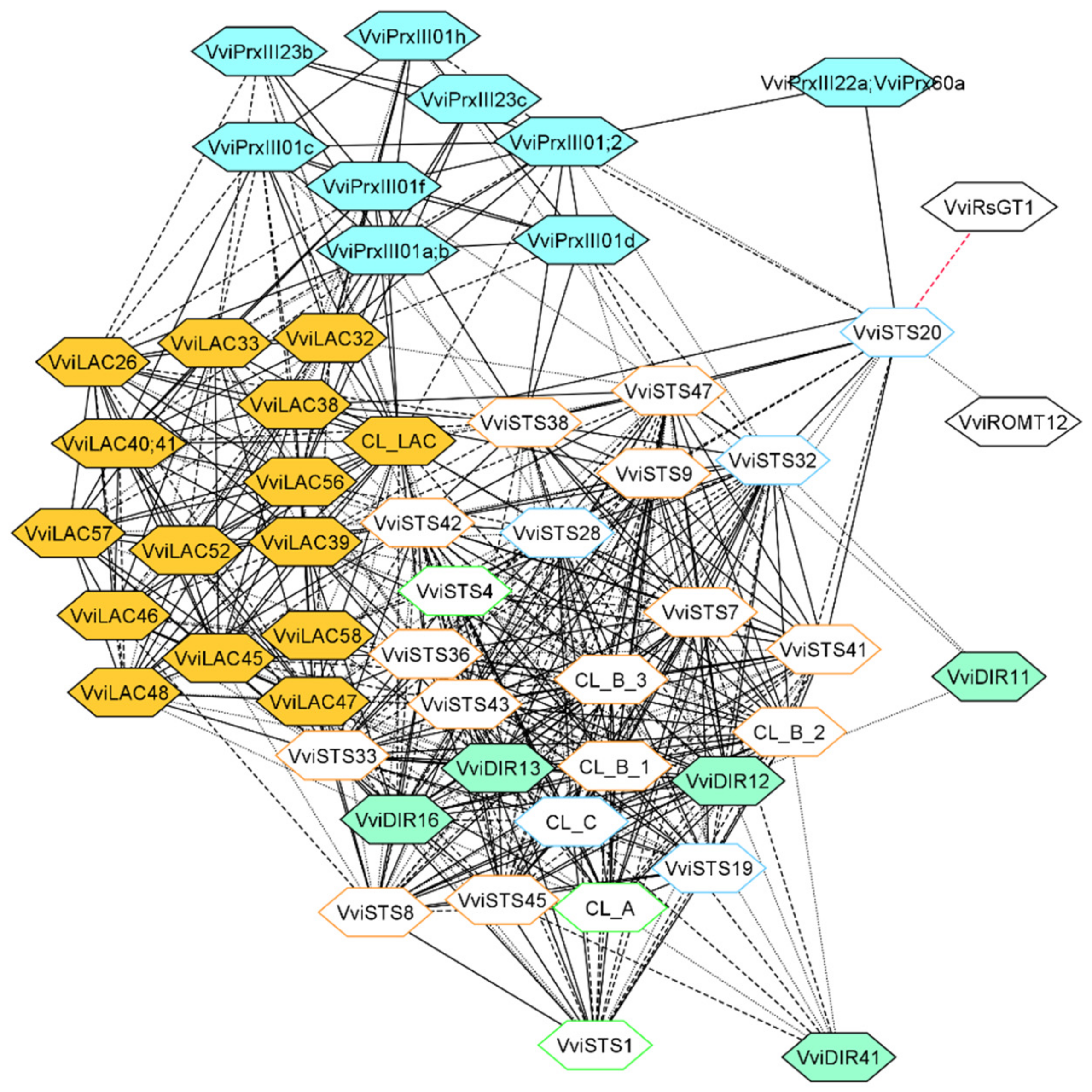
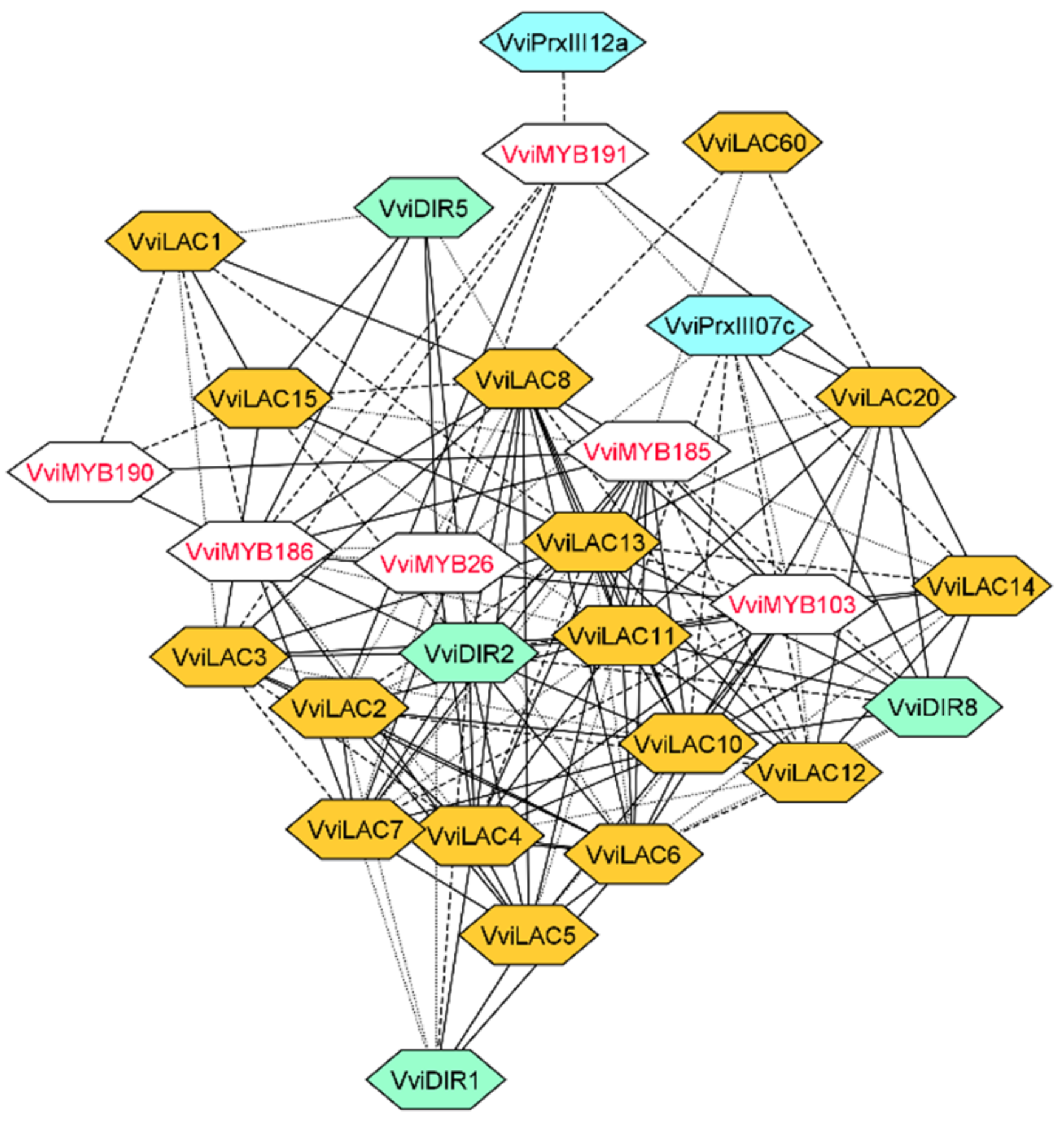
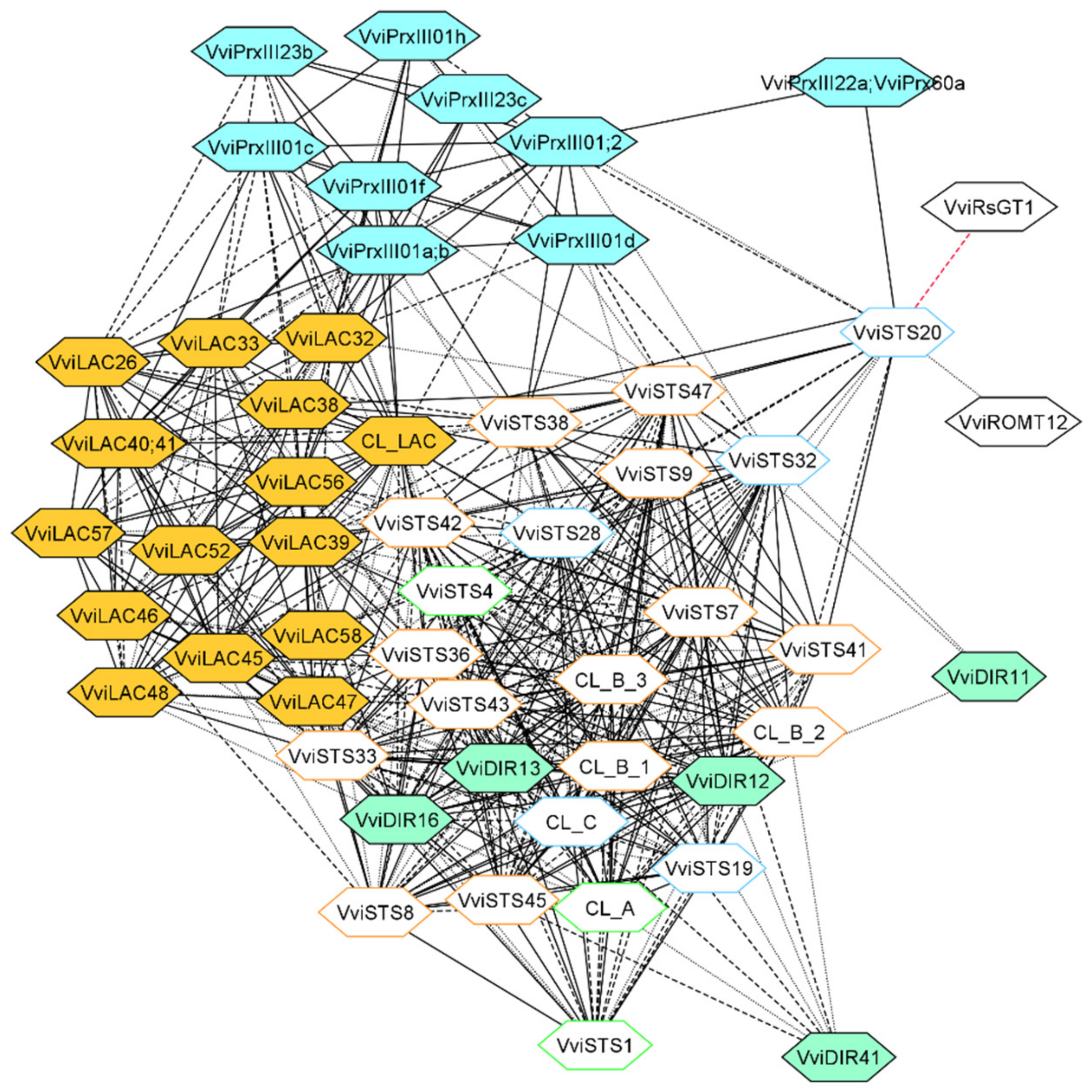
3.3. Laccase, Peroxidase and Dirigent Proteins Isoforms Involved in the Stilbenoid and Lignin Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moretto, M.; Sonego, P.; Pilati, S.; Malacarne, G.; Costantini, L.; Grzeskowiak, L.; Bagagli, G.; Grando, M.S.; Moser, C.; Engelen, K. VESPUCCI: Exploring Patterns of Gene Expression in Grapevine. Front. Plant Sci. 2016, 7, 633. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, R.; Liang, Z.; Li, S. Grape-rna: A database for the collection, evaluation, treatment, and data sharing of grape RNA-seq datasets. Genes 2020, 11, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. Comment: The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasty, J.; McMillen, D.; Isaacs, F.; Collins, J.J. Computational studies of gene regulatory networks: In numero molecular biology. Nat. Rev. Genet. 2001, 2, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pearl, S.A.; Jackson, S.A. Gene Networks in Plant Biology: Approaches in Reconstruction and Analysis. Trends Plant Sci. 2015, 20, 664–675. [Google Scholar] [CrossRef]
- Savoi, S.; Wong, D.C.J.; Degu, A.; Herrera, J.C.; Bucchetti, B.; Peterlunger, E.; Fait, A.; Mattivi, F.; Castellarin, S.D. Multi-Omics and Integrated Network Analyses Reveal New Insights into the Systems Relationships between Metabolites, Structural Genes, and Transcriptional Regulators in Developing Grape Berries (Vitis vinifera L.) Exposed to Water Deficit. Front. Plant Sci. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertazzon, N.; Bagnaresi, P.; Forte, V.; Mazzucotelli, E.; Filippin, L.; Guerra, D.; Zechini, A.; Cattivelli, L.; Angelini, E. Grapevine comparative early transcriptomic profiling suggests that Flavescence dorée phytoplasma represses plant responses induced by vector feeding in susceptible varieties. BMC Genomics 2019, 20, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, T.V.M.; Quecini, V. Comparative transcriptome analyses between cultivated and wild grapes reveal conservation of expressed genes but extensive rewiring of co-expression networks. Plant Mol. Biol. 2021, 1, 3. [Google Scholar] [CrossRef]
- Toups, H.S.; Cochetel, N.; Gray, D.; Cramer, G.R. VviERF6Ls: An expanded clade in Vitis responds transcriptionally to abiotic and biotic stresses and berry development. BMC Genomics 2020, 21, 1–27. [Google Scholar] [CrossRef]
- Ghan, R.; Petereit, J.; Tillett, R.L.; Schlauch, K.A.; Toubiana, D.; Fait, A.; Cramer, G.R. The common transcriptional subnetworks of the grape berry skin in the late stages of ripening. BMC Plant Biol. 2017, 17, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Hopper, D.W.; Ghan, R.; Schlauch, K.A.; Cramer, G.R. Transcriptomic network analyses of leaf dehydration responses identify highly connected ABA and ethylene signaling hubs in three grapevine species differing in drought tolerance. BMC Plant Biol. 2016, 16, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, G.R.; Cochetel, N.; Ghan, R.; Destrac-Irvine, A.; Delrot, S. A sense of place: Transcriptomics identifies environmental signatures in Cabernet Sauvignon berry skins in the late stages of ripening. BMC Plant Biol. 2020, 20, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.C.J.; Matus, J.T. Constructing Integrated Networks for Identifying New Secondary Metabolic Pathway Regulators in Grapevine: Recent Applications and Future Opportunities. Front. Plant Sci. 2017, 8, 505. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.C.J. Network aggregation improves gene function prediction of grapevine gene co-expression networks. Plant Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Spirtes, P.; Glymour, C. An Algorithm for Fast Recovery of Sparse Causal Graphs. Soc. Sci. Comput. Rev. 1991, 9, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Malacarne, G.; Pilati, S.; Valentini, S.; Asnicar, F.; Moretto, M.; Sonego, P.; Masera, L.; Cavecchia, V.; Blanzieri, E.; Moser, C. Discovering Causal Relationships in Grapevine Expression Data to Expand Gene Networks. A Case Study: Four Networks Related to Climate Change. Front. Plant Sci. 2018, 9, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asnicar, F.; Sella, N.; Masera, L.; Morettin, P.; Tolio, T.; Semeniuta, S.; Moser, C.; Blanzieri, E.; Cavecchia, V. TN-Grid and gene@home project: Volunteer Computing for Bioinformatics. In Proceedings of the International Conference BOINC:FAST 2015, Petrozavodsk, Russia, 14–18 September 2015; Ivashko, E., Ed.; Aachen University: Aachen, Germany, 2015. [Google Scholar]
- Blanzieri, E.; Tebaldi, T.; Cavecchia, V.; Asnicar, F.; Masera, L.; Tome, G.; Nigro, E.; Colasurdo, E.; Ciciani, M.; Mazzoni, C.; et al. A Computing System for Discovering Causal Relationships among Human Genes to Improve Drug Repositioning. IEEE Trans. Emerg. Top. Comput. 2020. [Google Scholar] [CrossRef]
- Jeandet, P.; Douillet-Breuil, A.C.; Bessis, R.; Debord, S.; Sbaghi, M.; Adrian, M. Phytoalexins from the vitaceae: Biosynthesis, phytoalexin gene expression in transgenic plants, antifungal activity, and metabolism. J. Agric. Food Chem. 2002, 50, 2731–2741. [Google Scholar] [CrossRef]
- Pezet, R.; Gindro, K.; Viret, O.; Spring, J.L. Glycosylation and oxidative dimerization of resveratrol are respectively associated to sensitivity and resistance of grapevine cultivars to downy mildew. Physiol. Mol. Plant Pathol. 2004, 65, 297–303. [Google Scholar] [CrossRef]
- Mattivi, F.; Vrhovsek, U.; Malacarne, G.; Masuero, D.; Zulini, L.; Stefanini, M.; Mose, C.; Velasco, R.; Guella, G. Profiling of resveratrol oligomers, important stress metabolites, accumulating in the leaves of hybrid Vitis vinifera (Merzling × Teroldego) genotypes infected with Plasmopara viticola. J. Agric. Food Chem. 2011, 59, 5364–5375. [Google Scholar] [CrossRef]
- Vannozzi, A.; Dry, I.B.; Fasoli, M.; Zenoni, S.; Lucchin, M. Genome-wide analysis of the grapevine stilbene synthase multigenic family: Genomic organization and expression profiles upon biotic and abiotic stresses. BMC Plant Biol. 2012, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Gatto, P.; Vrhovsek, U.; Muth, J.; Segala, C.; Romualdi, C.; Fontana, P.; Pruefer, D.; Stefanini, M.; Moser, C.; Mattivi, F.; et al. Ripening and Genotype Control Stilbene Accumulation in Healthy Grapes. J. Agric. Food Chem. 2008, 56, 11773–11785. [Google Scholar] [CrossRef] [PubMed]
- Malacarne, G.; Vrhovsek, U.; Zulini, L.; Masuero, D.; Cestaro, A.; Stefanini, M.; Delledonne, M.; Velasco, R.; Guella, G.; Mattivi, F.; et al. Resistance to Plasmopara viticola is associated with a complex pattern of stilbenoids and with specific host transcriptional responses. BMC Plant Biol. 2011, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höll, J.; Vannozzi, A.; Czemmel, S.; D’Onofrio, C.; Walker, A.R.; Rausch, T.; Lucchin, M.; Boss, P.K.; Dry, I.B.; Bogs, J. The R2R3-MYB transcription factors MYB14 and MYB15 regulate stilbene biosynthesis in Vitis vinifera. Plant Cell 2013, 25, 4135–4149. [Google Scholar] [CrossRef] [Green Version]
- Vezzulli, S.; Malacarne, G.; Masuero, D.; Vecchione, A.; Dolzani, C.; Goremykin, V.; Mehari, Z.H.; Banchi, E.; Velasco, R.; Stefanini, M.; et al. The Rpv3-3 haplotype and stilbenoid induction mediate downy mildew resistance in a grapevine interspecific population. Front. Plant Sci. 2019, 10, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keylor, M.H.; Matsuura, B.S.; Stephenson, C.R.J. Chemistry and Biology of Resveratrol-Derived Natural Products. Chem. Rev. 2015, 115, 8976–9027. [Google Scholar] [CrossRef] [PubMed]
- Herrero, J.; Esteban-Carrasco, A.; Zapata, J.M. Looking for Arabidopsis thaliana peroxidases involved in lignin biosynthesis. Plant Physiol. Biochem. 2013, 67, 77–86. [Google Scholar] [CrossRef]
- Davin, L.B.; Lewis, N.G. Dirigent proteins and dirigent sites explain the mystery of specificity of radical precursor coupling in lignan and lignin biosynthesis. Plant Physiol. 2000, 123, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Paniagua, C.; Bilkova, A.; Jackson, P.; Dabravolski, S.; Riber, W.; Didi, V.; Houser, J.; Gigli-Bisceglia, N.; Wimmerova, M.; Budínská, E.; et al. Dirigent proteins in plants: Modulating cell wall metabolism during abiotic and biotic stress exposure. J. Exp. Bot. 2017, 68, 3287–3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, N.; Kaneta, T.; Sato, S.; Sato, Y. Analysis of expression profiles of three peroxidase genes associated with lignification in Arabidopsis thaliana. Physiol. Plant. 2009, 136, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, M.; Demura, T. The quest for transcriptional hubs of lignin biosynthesis: Beyond the NAC-MYB-gene regulatory network model. Curr. Opin. Biotechnol. 2019, 56, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.C.J.; Schlechter, R.; Vannozzi, A.; Höll, J.; Hmmam, I.; Bogs, J.; Tornielli, G.B.; Castellarin, S.D.; Matus, J.T. A systems-oriented analysis of the grapevine R2R3-MYB transcription factor family uncovers new insights into the regulation of stilbene accumulation. DNA Res. 2016, 23, 451. [Google Scholar] [CrossRef] [PubMed]
- Ohman, D.; Demedts, B.; Kumar, M.; Gerber, L.; Gorzsas, A.; Goeminne, G.; Hedenström, M.; Ellis, B.; Boerjan, W.; Sundberg, B. MYB103 is required for FERULATE-5-HYDROXYLASE expression and syringyl lignin biosynthesis in Arabidopsis stems. Plant J. 2013, 73, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Song, J.; Ferguson, A.C.; Klisch, D.; Simpson, K.; Mo, R.; Taylor, B.; Mitsuda, N.; Wilson, Z.A. Transcription factor MYB26 is key to spatial specificity in anther secondary thickening formation. Plant Physiol. 2017, 175, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Zhang, S.; Liu, J.; Zhao, C.; Wu, J.; Cao, Y.; Fu, C.; Han, X.; He, H.; Zhao, Q. MYB20, MYB42, MYB43, and MYB85 regulate phenylalanine and lignin biosynthesis during secondary cell wall formation1[OPEN]. Plant Physiol. 2020, 182, 1272–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, R.; Richardson, E.A.; Ye, Z.H. The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis. Plant Cell 2007, 19, 2776–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asnicar, F.; Masera, L.; Pistore, D.; Valentini, S.; Cavecchia, V.; Blanzieri, E. OneGenE: Regulatory Gene Network Expansion via Distributed Volunteer Computing on BOINC. In Proceedings of the 27th Euromicro International Conference on Parallel, Distributed and Network-Based Processing (PDP), Pavia, Italy, 13–15 February 2019. [Google Scholar]
- Kalisch, M.; Bühlmann, P. Estimating high-dimensional directed acyclic graphs with the PC-algorithm. J. Mach. Learn. Res. 2007, 8, 613–636. [Google Scholar]
- Anderson, D.P. BOINC: A System for Public-Resource Computing and Storage. In Proceedings of the 5th IEEE/ACM International Workshop on Grid Computing, Washington, DC, USA, 8 November 2004; IEEE Computer Society: Washington, DC, USA, 2004; pp. 4–10. [Google Scholar]
- Grimplet, J.; Van Hemert, J.; Carbonell-Bejerano, P.; Díaz-Riquelme, J.; Dickerson, J.; Fennell, A.; Pezzotti, M.; Martínez-Zapater, J.M. Comparative analysis of grapevine whole-genome gene predictions, functional annotation, categorization and integration of the predicted gene sequences. BMC Res. Notes 2012, 5, 213. [Google Scholar] [CrossRef] [Green Version]
- Canaguier, A.; Grimplet, J.; di Gaspero, G.; Scalabrin, S.; Duchêne, E.; Choisne, N.; Mohellibi, N.; Guichard, C.; Rombauts, S.; le Clainche, I.; et al. A new version of the grapevine reference genome assembly (12X.v2) and of its annotation (VCost.v3). Genomics Data 2017, 14, 56–62. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Lopes, C.T.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape.js: A graph theory library for visualisation and analysis. Bioinformatics 2016, 32, 309–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexa, A.; Rahnenfuehrer, J. Gene Set Enrichment Analysis with topGO. 2015, pp. 1–27. Available online: https://bioconductor.org/packages/release/bioc/vignettes/topGO/inst/doc/topGO.pdf (accessed on 10 March 2020).
- Bailey, T.L. STREME: Accurate and versatile sequence motif discovery. Bioinformatics 2021, 37, 2834–2840. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Helt, G.A.; Reese, J.T.; Munoz-Torres, M.C.; Childers, C.P.; Buels, R.M.; Stein, L.; Holmes, I.H.; Elsik, C.G.; Lewis, S.E. Web Apollo: A web-based genomic annotation editing platform. Genome Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Vannozzi, A.; Wong, D.C.J.; Höll, J.; Hmmam, I.; Matus, J.T.; Bogs, J.; Ziegler, T.; Dry, I.; Barcaccia, G.; Lucchin, M. Combinatorial Regulation of Stilbene Synthase Genes by WRKY and MYB Transcription Factors in Grapevine (Vitis vinifera L.). Plant Cell Physiol. 2018, 59, 1043–1059. [Google Scholar] [CrossRef]
- Eisenmann, B.; Czemmel, S.; Ziegler, T.; Buchholz, G.; Kortekamp, A.; Trapp, O.; Rausch, T.; Dry, I.; Bogs, J. Rpv3-1 mediated resistance to grapevine downy mildew is associated with specific host transcriptional responses and the accumulation of stilbenes. BMC Plant Biol. 2019, 19, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Jiang, C.; Liu, W.; Wang, Y.; Hancock, R. The WRKY53 transcription factor enhances stilbene synthesis and disease resistance by interacting with MYB14 and MYB15 in Chinese wild grape. J. Exp. Bot. 2020, 71, 3211–3226. [Google Scholar] [CrossRef] [PubMed]
- Orduña, L.; Li, M.; Navarro-Payá, D.; Zhang, C.; Santiago, A.; Romero, P.; Ramšak, Ž.; Gruden, K.; Höll, J.; Merz, P.; et al. Major orchestration of shikimate, early phenylpropanoid and stilbenoid pathways by Subgroup 2 R2R3-MYBs in grapevine. bioRxiv 2021, 1–16. [Google Scholar] [CrossRef]
- Corso, M.; Vannozzi, A.; Maza, E.; Vitulo, N.; Meggio, F.; Pitacco, A.; Telatin, A.; D’Angelo, M.; Feltrin, E.; Negri, A.S.; et al. Comprehensive transcript profiling of two grapevine rootstock genotypes contrasting in drought susceptibility links the phenylpropanoid pathway to enhanced tolerance. J. Exp. Bot. 2015, 66, 5739–5752. [Google Scholar] [CrossRef]
- Turlapati, P.V.; Kim, K.W.; Davin, L.B.; Lewis, N.G. The laccase multigene family in Arabidopsis thaliana: Towards addressing the mystery of their gene function(s). Planta 2011, 233, 439–470. [Google Scholar] [CrossRef]
- Xu, X.; Zhou, Y.; Wang, B.; Ding, L.; Wang, Y.; Luo, L.; Zhang, Y.; Kong, W. Genome-wide identification and characterization of laccase gene family in Citrus sinensis. Gene 2019, 689, 114–123. [Google Scholar] [CrossRef]
- Grimplet, J.; Adam-Blondon, A.F.; Bert, P.F.; Bitz, O.; Cantu, D.; Davies, C.; Delrot, S.; Pezzotti, M.; Rombauts, S.; Cramer, G.R. The grapevine gene nomenclature system. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralph, S.; Park, J.Y.; Bohlmann, J.; Mansfield, S.D. Dirigent proteins in conifer defense: Gene discovery, phylogeny, and differential wound- and insect-induced expression of a family of DIR and DIR-like genes in spruce (Picea spp.). Plant Mol. Biol. 2006, 60, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.G.; Jancsik, S.; Bohlmann, J. Dirigent proteins in conifer defense II: Extended gene discovery, phylogeny, and constitutive and stress-induced gene expression in spruce (Picea spp.). Phytochemistry 2007, 68, 1975–1991. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, A.; Figueiredo, J.; Sousa Silva, M.; Figueiredo, A. Linking jasmonic acid to grapevine resistance against the biotrophic oomycete Plasmopara viticola. Front. Plant Sci. 2016, 7, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassoni, A.; Fornalè, S.; Franceschetti, M.; Musiani, F.; Michael, A.J.; Perry, B.; Bagni, N. Jasmonates and Na-orthovanadate promote resveratrol production in Vitis vinifera cv. Barbera cell cultures. New Phytol. 2005, 166, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Pezet, R.; Gindro, K.; Viret, O.; Richter, H. Effects of resveratrol, viniferins and pterostilbene on Plasmopara viticola zoospore mobility and disease development. Vitis -J. Grapevine Res. 2004, 43, 145–148. [Google Scholar]
- Cichewicz, R.H.; Kouzi, S.A. Resveratrol oligomers: Structure, chemistry, and biological activity. Stud. Nat. Prod. Chem. 2002, 26, 507–579. [Google Scholar] [CrossRef]
- Pezet, R.; Pont, V.; Hoang-Van, K. Evidence for oxidative detoxication of pterostilbene and resveratrol by a laccase-like stilbene oxidase produced by Botrytis cinerea. Physiol. Mol. Plant Pathol. 1991, 39, 441–450. [Google Scholar] [CrossRef]
- Breuil, A.C.; Jeandet, P.; Adrian, M.; Chopin, F.; Pirio, N.; Meunier, P.; Bessis, R. Characterization of a pterostilbene dehydrodimer produced by laccase of Botrytis cinerea. Phytopathology 1999, 89, 298–302. [Google Scholar] [CrossRef]
- Di Gaspero, G.; Copetti, D.; Coleman, C.; Castellarin, S.D.; Eibach, R.; Kozma, P.; Lacombe, T.; Gambetta, G.; Zvyagin, A.; Cindrić, P.; et al. Selective sweep at the Rpv3 locus during grapevine breeding for downy mildew resistance. Theor. Appl. Genet. 2012, 124, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Chitarrini, G.; Soini, E.; Riccadonna, S.; Franceschi, P.; Zulini, L.; Masuero, D.; Vecchione, A.; Stefanini, M.; Di Gaspero, G.; Mattivi, F.; et al. Identification of Biomarkers for Defense Response to Plasmopara viticola in a Resistant Grape Variety. Front. Plant Sci. 2017, 8, 1524. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N. of Connected VviSTS | 12X.v1 | Gene Name | Reference | N. of Connected VviSTS | 12X.v1 | Gene Name | Reference |
|---|---|---|---|---|---|---|---|
| 15 | Vitvi11g00165 | VvibHLH045 | [49,50] | 2 | Vitvi17g00025 | VviERF118 | [49] |
| 14 | Vitvi07g00598 | VviMYB14 | [13,49,51] | 2 | Vitvi15g00948 | VvibHLH066, MYC7E | - |
| 13 | Vitvi07g02070 | VviERF098 | [49,50] | 2 | Vitvi15g00938 | VviMYBPA1 | - |
| 9 | Vitvi14g01523 | VviWRKY43 | [13,49,52] | 2 | Vitvi08g01412 | VviRAV5 | - |
| 8 | Vitvi08g01931 | VviHsfB3a | [49,50] | 2 | Vitvi08g01224 | ZFDHHC_8 | [52] |
| 7 | Vitvi05g01732 | VviMYB13 | [49] | 1 | Vitvi14g01617 | C2H2FAM_25 | - |
| 7 | Vitvi17g00309 | VviMYB164 | - | 1 | Vitvi11g00715 | C2H2FAM_28 | - |
| 7 | Vitvi06g00007 | VviNAC44 | [49,52] | 1 | Vitvi11g00436 | CCCHFAM_12 | - |
| 7 | Vitvi08g00793 | VviWRKY24 | [49,50,53] | 1 | Vitvi15g00636 | GATA2 | - |
| 7 | Vitvi13g00620 | ZFC3HC4_163 | - | 1 | Vitvi13g01879 | GCN5_18 | - |
| 6 | Vitvi15g01203 | ERF2_1 | [50] | 1 | IAA22D_2 | - | |
| 6 | Vitvi04g02175 | VviJAZ3 | - | 1 | Vitvi06g00167 | JMJD5 | - |
| 6 | Vitvi19g02272 | VviNAC31 | [49] | 1 | Vitvi15g00736 | VviLBDIa2 LBD19 | - |
| 6 | Vitvi12g00076 | VviNAC36 | [49,50] | 1 | Vitvi13g00085 | VviLBDIc3 LBD21 | [50,52] |
| 5 | Vitvi05g00523 | GCN5_7 | [50] | 1 | Vitvi00g01060 | VviLBDIc5 | - |
| 5 | Vitvi08g01525 | HMGIY_1 | [52] | 1 | Vitvi01g01921 | MYB3R1_5 | - |
| 5 | Vitvi01g01287 | VvibHLH001, BIM3 | - | 1 | Vitvi14g00217 | NFYB5_3 | - |
| 5 | Vitvi05g01733 | VviMYB15 | [13,49,50,51] | 1 | Vitvi18g01322 | RGL2_1 | - |
| 5 | Vitvi04g00133 | VviWRKY06 | [50] | 1 | Vitvi01g00185 | SAP7_3 | - |
| 5 | Vitvi17g00556 | VviWRKY53 | [49,50,51] | 1 | Vitvi13g00311 | SCL14_1 | [49,52] |
| 4 | Vitvi10g00053 | JMJD1B_5 | [50] | 1 | Vitvi07g01762 | TFB3_4 | - |
| 4 | Vitvi19g02273 | VviNAC30 | [52] | 1 | Vitvi07g01529 | VvibHLH038 | - |
| 4 | Vitvi17g00801 | ZFC3HC4_103 | - | 1 | Vitvi15g01027 | VvibZIP40 | - |
| 3 | Vitvi07g02168 | MTERF_10 | - | 1 | Vitvi05g01722 | VviERF109 | - |
| 3 | Vitvi01g00940 | VviWRKY02 | [13,49,50] | 1 | Vitvi15g01206 | VviERF073 | [49] |
| 3 | Vitvi01g01680 | VviWRKY03 | [49,50,52] | 1 | Vitvi19g01669 | VviMYB139 | [49,52] |
| 3 | Vitvi04g00760 | VviWRKY11 | [49] | 1 | Vitvi07g03055 | VviMYB148 | [49] |
| 3 | Vitvi19g00155 | ZFC3HC4_158 | [52] | 1 | Vitvi19g01564 | VviNAC29 | [52] |
| 3 | Vitvi12g00220 | ZFC3HC4_36 | [50] | 1 | Vitvi04g00756 | VviWRKY10 | - |
| 2 | Vitvi10g00572 | - | - | 1 | Vitvi06g01574 | VviWRKY15 | [50] |
| 2 | Vitvi13g01958 | ERUNK_7 | - | 1 | Vitvi10g00063 | VviWRKY29 | [49,53] |
| 2 | Vitvi09g00064 | VviJAZ4 | - | 1 | Vitvi13g00189 | VviWRKY40 | [52] |
| 2 | Vitvi10g00826 | VviJAZ5 | - | 1 | Vitvi16g01213 | VviWRKY51 | [50] |
| 2 | Vitvi10g01879 | VviJAZ6 | - | 1 | Vitvi05g00368 | ZFC3HC4_164 | - |
| 2 | Vitvi16g00349 | VviERF077 | - | 1 | Vitvi08g00923 | ZFC3HC4_43 | - |
| 2 | Vitvi02g01780 | VviERF076 | [50] | 1 | Vitvi16g02032 | ZFC3HC4_45 | - |
| Source | Initial Genes | Corrected CDS | Split Genes | Merged Genes | Curated Gene Models | Identified as Pseudogenes or Non-Laccases | Final Gene Models |
|---|---|---|---|---|---|---|---|
| Present in 12X.v1 | 77 | 16 | 2 | 3 | 76 | 14 | 62 |
| HMMER search in 12X.v2 | 44 | 4 | 0 | 0 | 44 | 11 | 33 |
| Total | 121 | 20 | 2 | 3 | 120 | 25 | 95 |
| Species | LAC (Group 1 to 7) | LAC (Group 8 and 9) | Total LACs | Low Phosphate Root | Ascorbate Oxidases | SKU5 Similar |
|---|---|---|---|---|---|---|
| Vitis vinifera | 24 | 52 | 76 | 2 | 5 | 12 |
| Arabidopsis thaliana | 17 | 0 | 17 | 2 | 3 | 19 |
| Source | Initial Genes | Corrected CDS | Split Genes | Merged Genes | Curated Gene Models |
|---|---|---|---|---|---|
| Present in 12X.v1 | 32 | 6 | 0 | 0 | 32 |
| HMMER search in 12X.v2 | 9 | 2 | 0 | 0 | 9 |
| Total | 41 | 8 | 0 | 0 | 41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilati, S.; Malacarne, G.; Navarro-Payá, D.; Tomè, G.; Riscica, L.; Cavecchia, V.; Matus, J.T.; Moser, C.; Blanzieri, E. Vitis OneGenE: A Causality-Based Approach to Generate Gene Networks in Vitis vinifera Sheds Light on the Laccase and Dirigent Gene Families. Biomolecules 2021, 11, 1744. https://doi.org/10.3390/biom11121744
Pilati S, Malacarne G, Navarro-Payá D, Tomè G, Riscica L, Cavecchia V, Matus JT, Moser C, Blanzieri E. Vitis OneGenE: A Causality-Based Approach to Generate Gene Networks in Vitis vinifera Sheds Light on the Laccase and Dirigent Gene Families. Biomolecules. 2021; 11(12):1744. https://doi.org/10.3390/biom11121744
Chicago/Turabian StylePilati, Stefania, Giulia Malacarne, David Navarro-Payá, Gabriele Tomè, Laura Riscica, Valter Cavecchia, José Tomás Matus, Claudio Moser, and Enrico Blanzieri. 2021. "Vitis OneGenE: A Causality-Based Approach to Generate Gene Networks in Vitis vinifera Sheds Light on the Laccase and Dirigent Gene Families" Biomolecules 11, no. 12: 1744. https://doi.org/10.3390/biom11121744
APA StylePilati, S., Malacarne, G., Navarro-Payá, D., Tomè, G., Riscica, L., Cavecchia, V., Matus, J. T., Moser, C., & Blanzieri, E. (2021). Vitis OneGenE: A Causality-Based Approach to Generate Gene Networks in Vitis vinifera Sheds Light on the Laccase and Dirigent Gene Families. Biomolecules, 11(12), 1744. https://doi.org/10.3390/biom11121744