
Gossypol Acetic Acid Attenuates Cardiac Ischemia/Reperfusion Injury in Rats via an Antiferroptotic Mechanism

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. H9c2 Cardiomyoblast Cell Culture
2.3. Preparation and Culture of Ventricular Myocytes
2.4. Langendorff Heart Perfusion System
2.5. Cell Viability Assay
2.6. Iron Content Analysis
2.7. Cell Death Analysis
2.8. ROS Analysis
2.9. Lipid Peroxidation Analysis
2.9.1. Thiobarbituric Acid Reactive Substances (TBARS) Assay
2.9.2. Confocal Images
2.10. Western Blot Analysis
2.11. Quantitative Real-Time Polymerase Chain Reaction
2.12. Statistics
3. Results
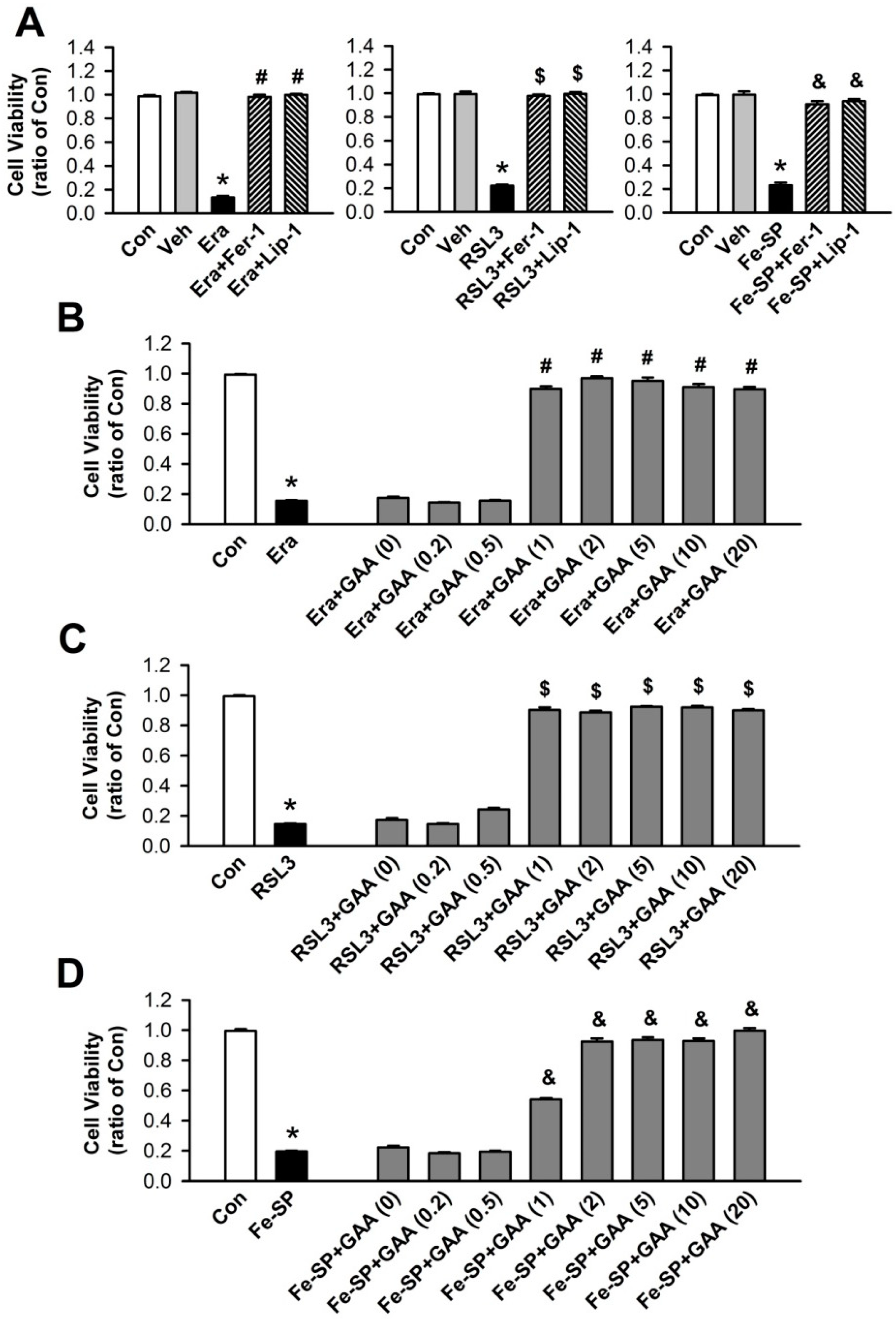
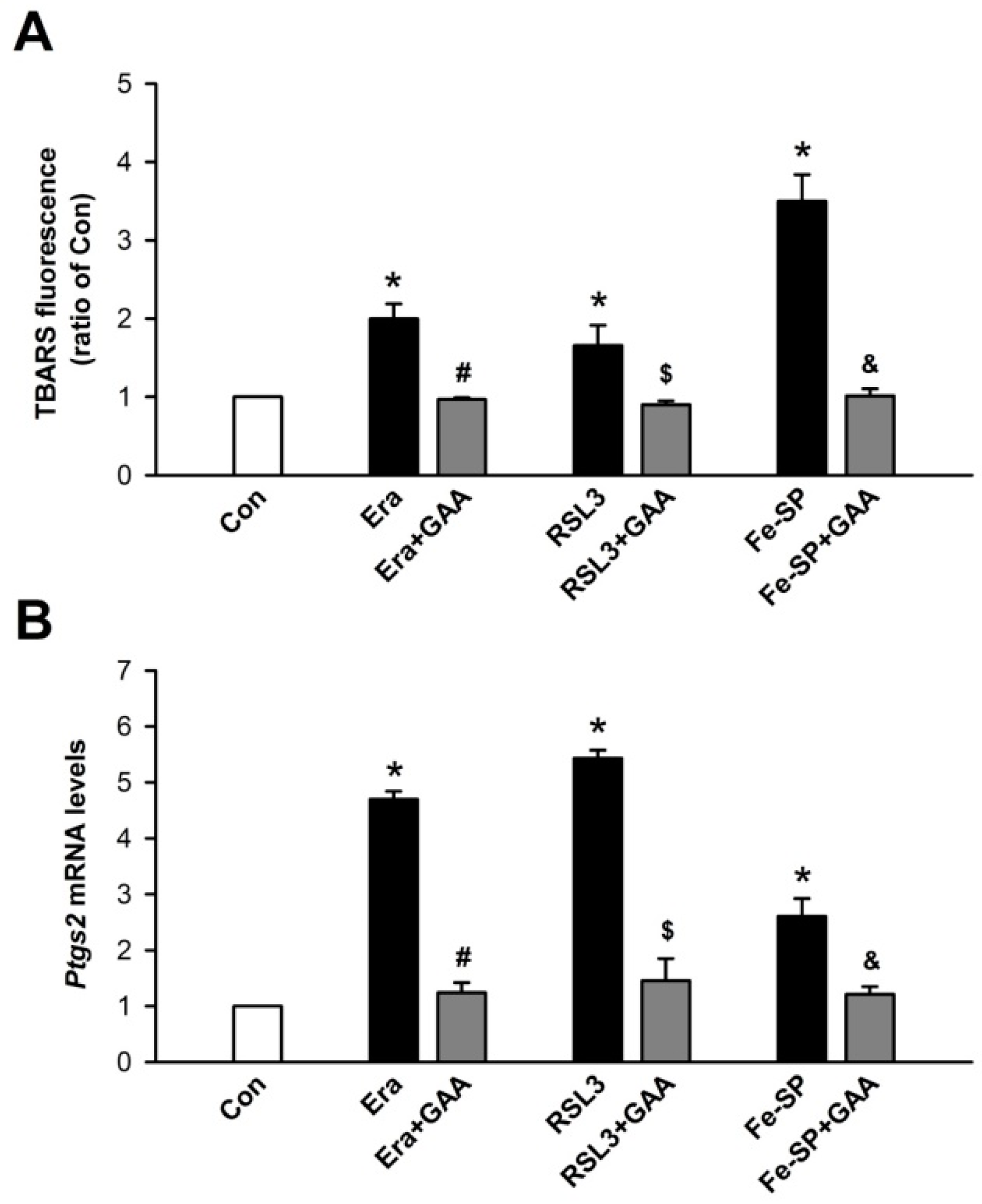
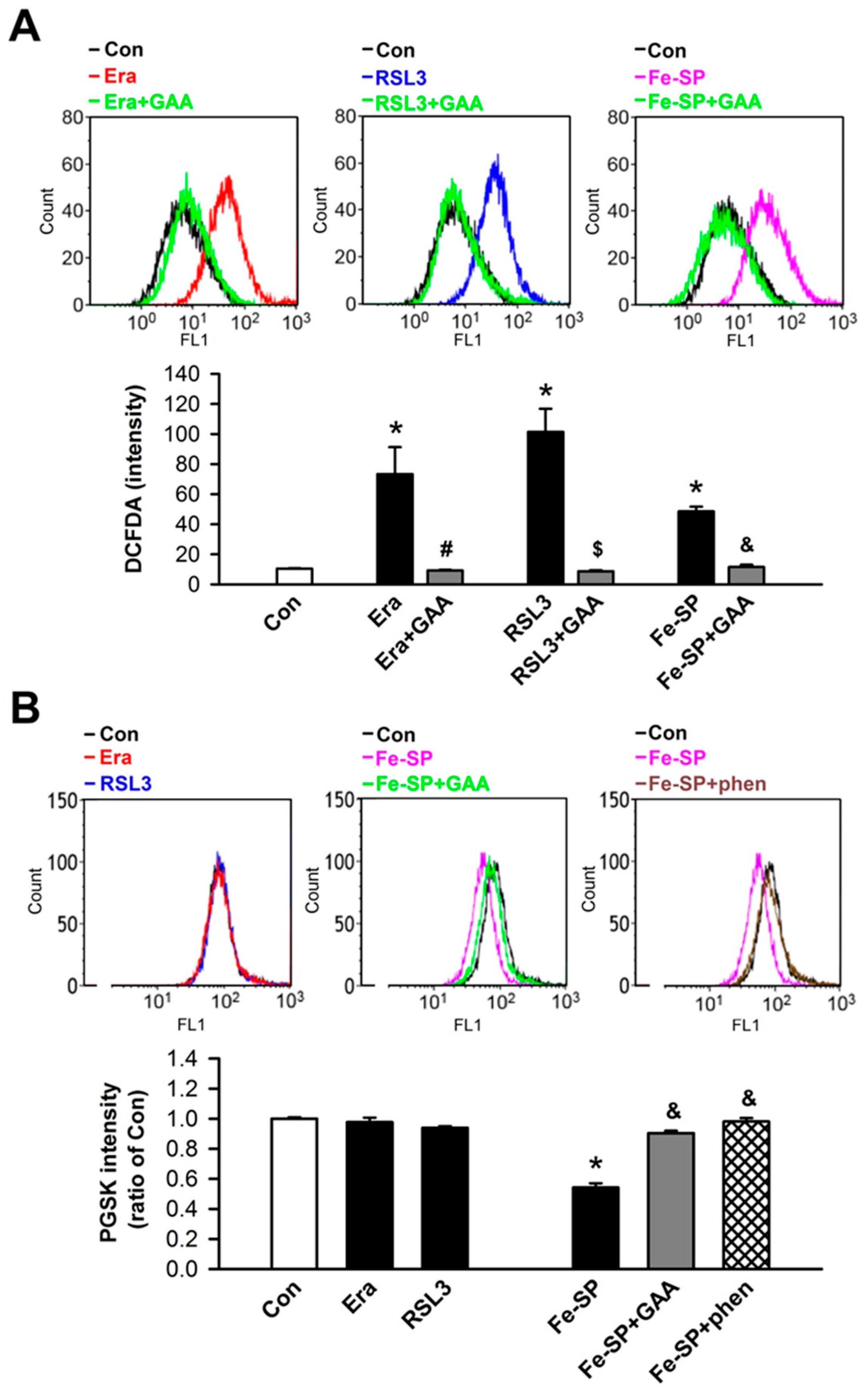
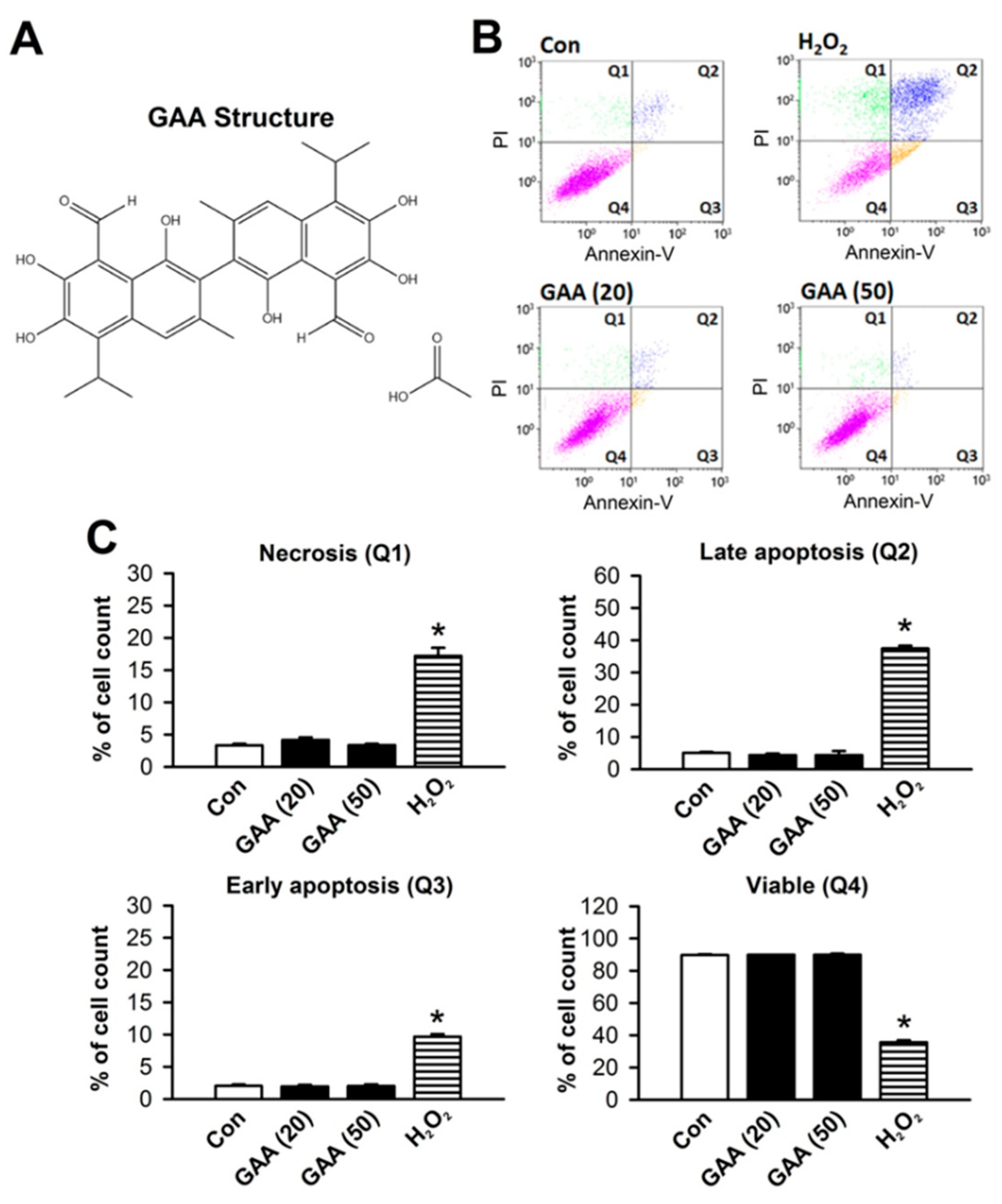
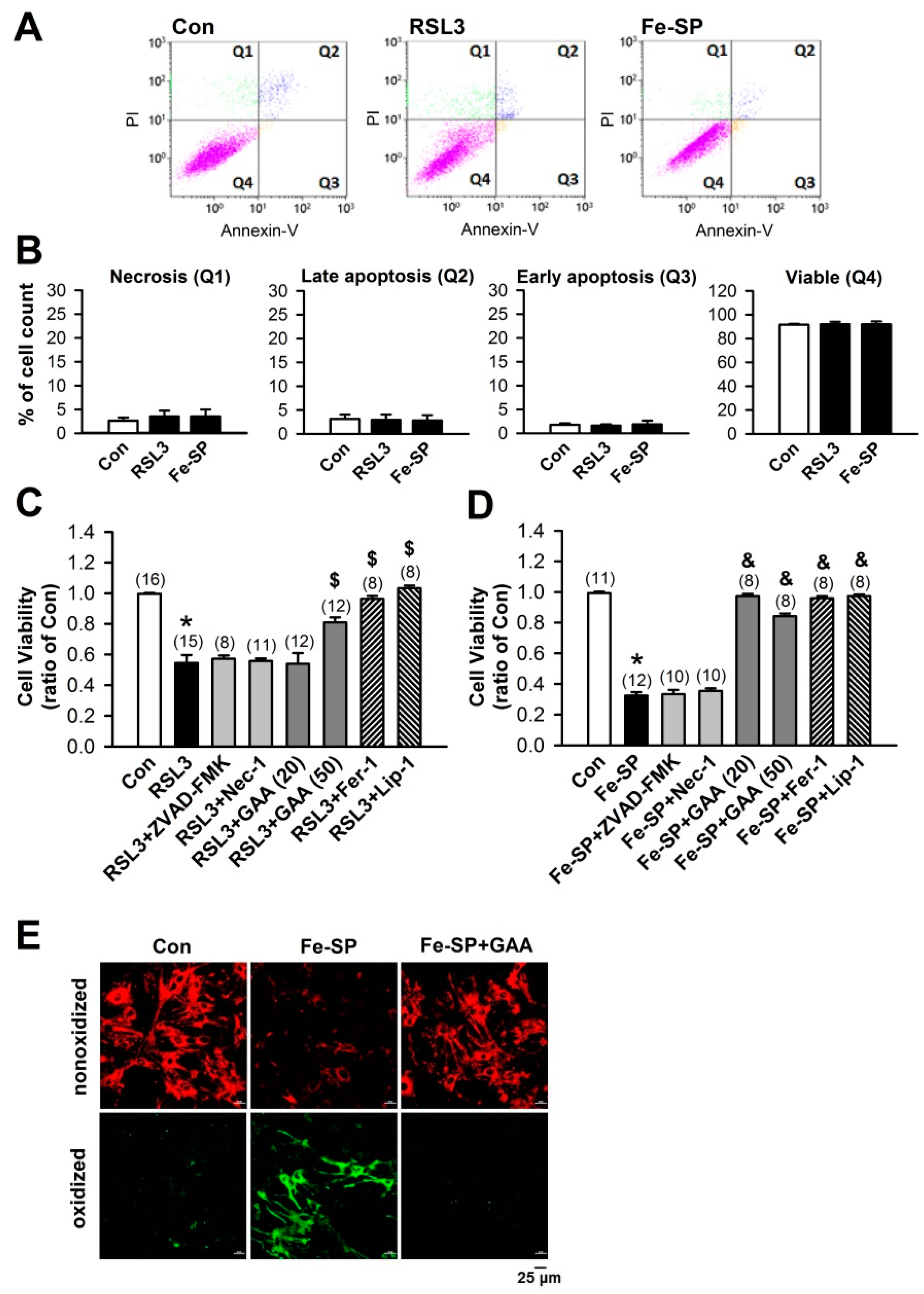
3.1. Effects of GAA on Ferroptotic Cell Death in H9c2 Cardiomyoblast Cells
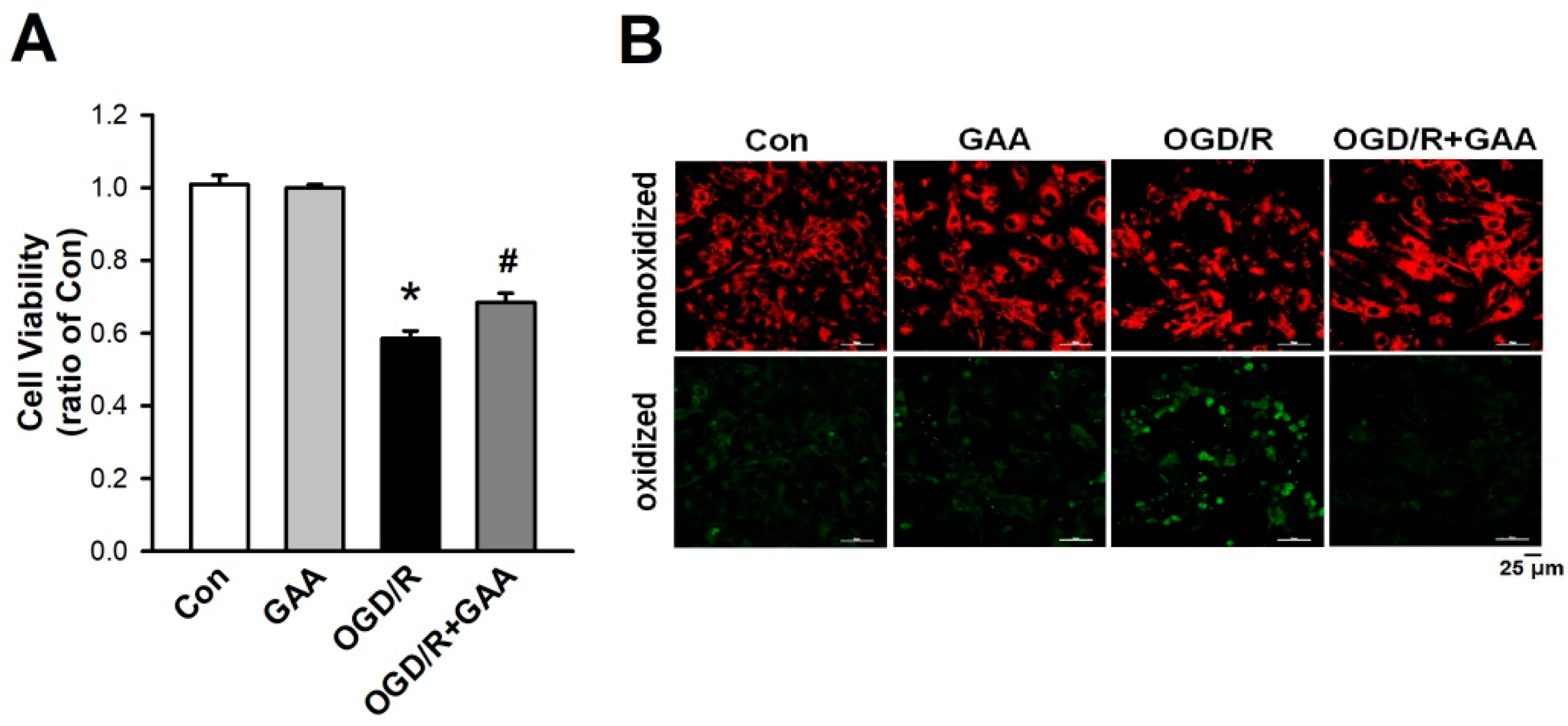
3.2. Effects of GAA on Ferroptosis-Induced Neonatal Rat Myocardial Cell Death
3.3. GAA Attenuates I/R-Induced Ferroptosis in Ex Vivo Rat Hearts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol. Res. 2021, 166, 105466. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Li, X.; Yang, X.; Yan, J.; Shi, P.; Ba, L.; Cao, Y.; Wang, P. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 2019, 235, 116795. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, W.; Leng, Y.; Xiong, Y.; Xia, Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020, 39, 210–225. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.J.; Luo, X.J.; Tu, H.; Chen, H.; Xiong, X.M.; Li, N.S.; Peng, J. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn. Schmiedebergs Arch. Pharmacol. 2021, 394, 401–410. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Tang, L.J.; Zhou, Y.J.; Xiong, X.M.; Li, N.S.; Zhang, J.J.; Luo, X.J.; Peng, J. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic. Biol. Med. 2021, 162, 339–352. [Google Scholar] [CrossRef]
- Stamenkovic, A.; O’Hara, K.A.; Nelson, D.C.; Maddaford, T.G.; Edel, A.L.; Maddaford, G.; Dibrov, E.; Aghanoori, M.; Kirshenbaum, L.A.; Fernyhough, P.; et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia/reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1170–H1184. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Zhang, H.; Chen, D.H.; Zhou, Q.; Jin, P.; Liu, Q.; Wang, S. Gossypol acetic acid prevents oxidative stress-induced retinal pigment epithelial necrosis by regulating the FoxO3/Sestrin2 pathway. Mol. Cell Biol. 2015, 35, 1952–1963. [Google Scholar] [CrossRef] [Green Version]
- El-Sharaky, A.S.; Newairy, A.A.; Elguindy, N.M.; Elwafa, A.A. Spermatotoxicity, biochemical changes and histological alteration induced by gossypol in testicular and hepatic tissues of male rats. Food Chem. Toxicol. 2010, 48, 3354–3361. [Google Scholar] [CrossRef]
- El-Sharaky, A.S.; Wahby, M.M.; Bader El-Dein, M.M.; Fawzy, R.A.; El-Shahawy, I.N. Mutual anti-oxidative effect of gossypol acetic acid and gossypol-iron complex on hepatic lipid peroxidation in male rats. Food Chem. Toxicol. 2009, 47, 2735–2741. [Google Scholar] [CrossRef]
- Reynolds, J.M.; Tone, J.N. Subchronic oral administration of gossypol-acetic acid (GAA) alters the distribution and utilization of radioiron in male rats. Drug Chem. Toxicol. 1988, 11, 135–150. [Google Scholar] [CrossRef]
- Lin, J.H.; Ting, P.C.; Lee, W.S.; Chiu, H.W.; Chien, C.A.; Liu, C.H.; Sun, L.Y.; Yang, K.T. Palmitic acid methyl ester induces G(2)/M arrest in human bone marrow-derived mesenchymal stem cells via the p53/p21 pathway. Stem. Cells Int. 2019, 2019, 7606238. [Google Scholar] [CrossRef] [Green Version]
- Gadelha, I.C.; Fonseca, N.B.; Oloris, S.C.; Melo, M.M.; Soto-Blanco, B. Gossypol toxicity from cottonseed products. Sci. World J. 2014, 2014, 231635. [Google Scholar] [CrossRef] [Green Version]
- Haasler, L.; Kondadi, A.K.; Tsigaras, T.; von Montfort, C.; Graf, P.; Stahl, W.; Brenneisen, P. The BH3 mimetic (±) gossypol induces ROS-independent apoptosis and mitochondrial dysfunction in human A375 melanoma cells in vitro. Arch. Toxicol. 2021, 95, 1349–1365. [Google Scholar] [CrossRef]
- Cao, H.; Sethumadhavan, K.; Cao, F.; Wang, T.T.Y. Gossypol decreased cell viability and down-regulated the expression of a number of genes in human colon cancer cells. Sci. Rep. 2021, 11, 5922. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wu, C.; Cui, Y.; Zhu, H.; Gao, Z.; Li, B.; Hua, J.; Zhao, B. The aldehyde group of gossypol induces mitochondrial apoptosis via ROS-SIRT1-p53-PUMA pathway in male germline stem cell. Oncotarget 2017, 8, 100128–100140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Tao, G.; Ji, L.; Tian, G. Sappanone a protects against myocardial ischemia reperfusion injury by modulation of Nrf2. Drug Des. Dev. Ther. 2020, 14, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Zhang, J.; Strom, J.; Lee, S.; Chen, Q.M. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta 2014, 1842, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Shokeir, A.A.; Hussein, A.M.; Barakat, N.; Abdelaziz, A.; Elgarba, M.; Awadalla, A. Activation of nuclear factor erythroid 2-related factor 2 (Nrf2) and Nrf-2-dependent genes by ischaemic pre-conditioning and post-conditioning: New adaptive endogenous protective responses against renal ischaemia/reperfusion injury. Acta Physiol. 2014, 210, 342–353. [Google Scholar] [CrossRef]
- Keshmiri-Neghab, H.; Goliaei, B. Therapeutic potential of gossypol: An overview. Pharm. Biol. 2014, 52, 124–128. [Google Scholar] [CrossRef]
- Flak, D.K.; Adamski, V.; Nowaczyk, G.; Szutkowski, K.; Synowitz, M.; Jurga, S.; Held-Feindt, J. AT101-loaded cubosomes as an alternative for improved glioblastoma therapy. Int. J. Nanomed. 2020, 15, 7415–7431. [Google Scholar] [CrossRef]
- Xiang, W.; Yang, C.Y.; Bai, L. MCL-1 inhibition in cancer treatment. Onco. Targets Ther. 2018, 11, 7301–7314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Cai, L.; Wang, S.; Wang, J.; Chen, B. Baicalin prevents myocardial ischemia/reperfusion injury through inhibiting ACSL4 mediated ferroptosis. Front. Pharmacol. 2021, 12, 628988. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tan, Y.; Ouyang, S.; He, J.; Liu, L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene 2022, 808, 145968. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.P.; Yu, H.Q.; Li, J.; Li, C.; Hua, X.; Sheng, X.S. Troxerutin attenuates oxygen-glucose deprivation and reoxygena-tion-induced oxidative stress and inflammation by enhancing the PI3K/AKT/HIF-1α signaling pathway in H9C2 cardiomyocytes. Mol. Med. Rep. 2020, 22, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, Y.; Hou, W.; Guo, L. MiR-153 regulates cardiomyocyte apoptosis by targeting Nrf2/HO-1 signaling. Chromo-Some Res. 2019, 27, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Mimura, J.; Ozaki, T.; Itoh, K. Emerging regulatory role of Nrf2 in iron, heme, and hemoglobin metabolism in physiology and disease. Front. Vet. Sci. 2018, 5, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masini, E.; Vannacci, A.; Marzocca, C.; Pierpaoli, S.; Giannini, L.; Fantappié, O.; Mazzanti, R.; Mannaioni, P.F. Heme oxygenase-1 and the ischemia-reperfusion injury in the rat heart. Exp. Biol. Med. 2003, 228, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.-H.; Yang, K.-T.; Ting, P.-C.; Luo, Y.-P.; Lin, D.-J.; Wang, Y.-S.; Chang, J.-C. Gossypol Acetic Acid Attenuates Cardiac Ischemia/Reperfusion Injury in Rats via an Antiferroptotic Mechanism. Biomolecules 2021, 11, 1667. https://doi.org/10.3390/biom11111667
Lin J-H, Yang K-T, Ting P-C, Luo Y-P, Lin D-J, Wang Y-S, Chang J-C. Gossypol Acetic Acid Attenuates Cardiac Ischemia/Reperfusion Injury in Rats via an Antiferroptotic Mechanism. Biomolecules. 2021; 11(11):1667. https://doi.org/10.3390/biom11111667
Chicago/Turabian StyleLin, Jian-Hong, Kun-Ta Yang, Pei-Ching Ting, Yu-Po Luo, Ding-Jyun Lin, Yi-Shun Wang, and Jui-Chih Chang. 2021. "Gossypol Acetic Acid Attenuates Cardiac Ischemia/Reperfusion Injury in Rats via an Antiferroptotic Mechanism" Biomolecules 11, no. 11: 1667. https://doi.org/10.3390/biom11111667
APA StyleLin, J.-H., Yang, K.-T., Ting, P.-C., Luo, Y.-P., Lin, D.-J., Wang, Y.-S., & Chang, J.-C. (2021). Gossypol Acetic Acid Attenuates Cardiac Ischemia/Reperfusion Injury in Rats via an Antiferroptotic Mechanism. Biomolecules, 11(11), 1667. https://doi.org/10.3390/biom11111667