
Targeting Hypoxia: Revival of Old Remedies



{kind=link}
Abstract
:1. Introduction
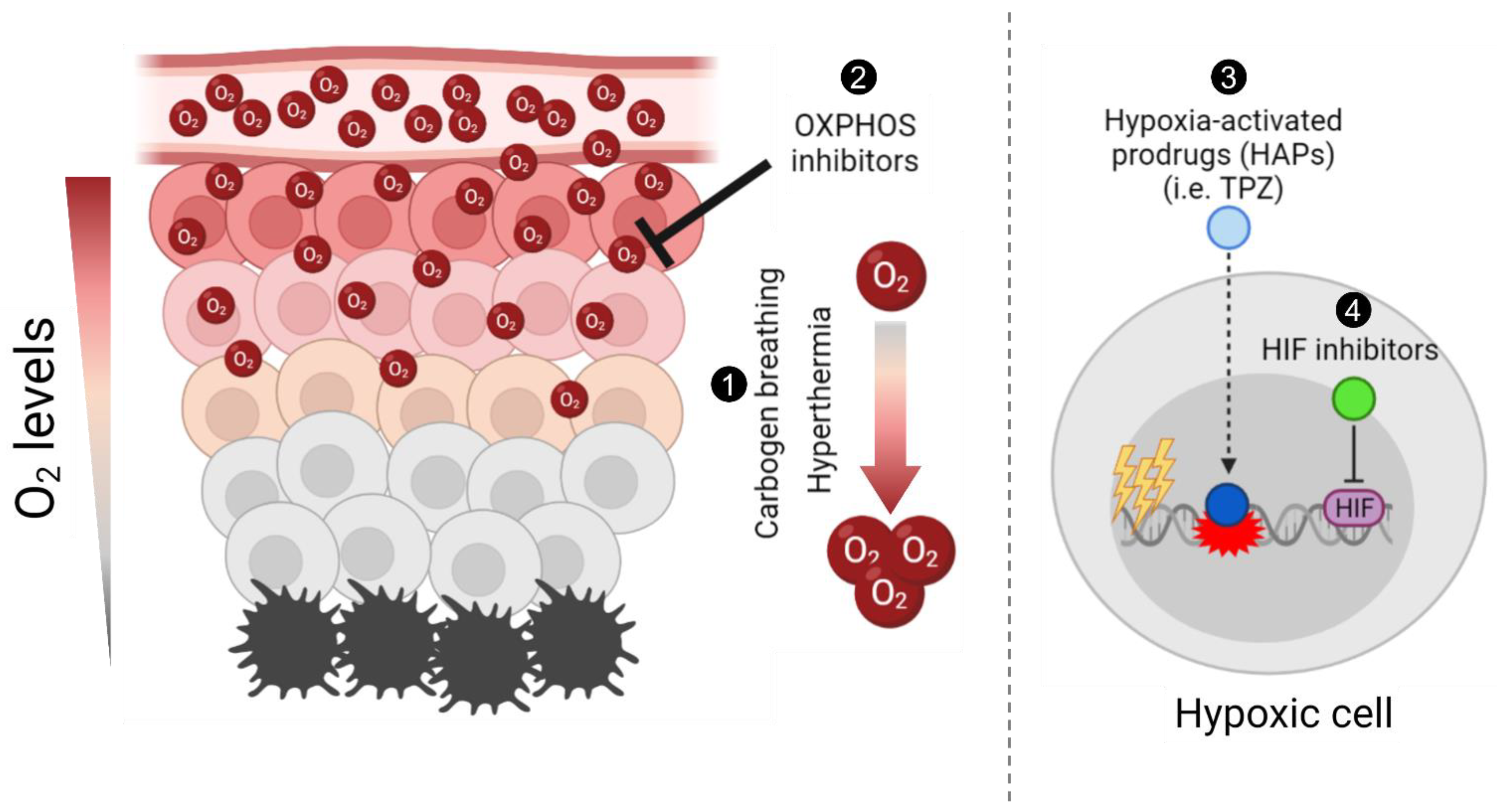
2. Carbogen Breathing
3. Hyperthermia
4. Hypoxia-Activated Prodrugs
5. Tirapazamine
6. Hypoxia-Inducible Factor (HIF) Inhibitors
7. Targeting Metabolism
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res. 2016, 57 (Suppl. 1), i90–i98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsman, M.R.; Sorensen, B.S.; Busk, M.; Siemann, D.W. Therapeutic Modification of Hypoxia. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.H.; Feldmeier, J.; Smee, R.; Milross, C. Hyperbaric oxygenation for tumour sensitisation to radiotherapy. Cochrane Database Syst. Rev. 2018, 4, CD005007. [Google Scholar] [CrossRef]
- Chaplin, D.J.; Horsman, M.R.; Siemann, D.W. Further evaluation of nicotinamide and carbogen as a strategy to reoxygenate hypoxic cells in vivo: Importance of nicotinamide dose and pre-irradiation breathing time. Br. J. Cancer 1993, 68, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Siemann, D.W.; Hill, R.P.; Bush, R.S. The importance of the pre-irradiation breathing times of oxygen and carbogen (5% CO2: 95% O2) on the in vivo radiation response of a murine sarcoma. Int. J. Radiat. Oncol. Biol. Phys. 1977, 2, 903–911. [Google Scholar] [CrossRef]
- Aquino-Parsons, C.; Hukin, J.; Green, A. Concurrent carbogen and radiation therapy in children with high-risk brainstem gliomas. Pediatr. Blood Cancer 2008, 50, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Surjana, D.; Halliday, G.M.; Damian, D.L. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J. Nucleic Acids 2010, 2010, 157591. [Google Scholar] [CrossRef] [Green Version]
- van Laarhoven, H.W.; Bussink, J.; Lok, J.; Punt, C.J.; Heerschap, A.; van Der Kogel, A.J. Effects of nicotinamide and carbogen in different murine colon carcinomas: Immunohistochemical analysis of vascular architecture and microenvironmental parameters. Int. J. Radiat. Oncol. Biol. Phys. 2004, 60, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Kaanders, J.H.; Bussink, J.; van der Kogel, A.J. ARCON: A novel biology-based approach in radiotherapy. Lancet Oncol. 2002, 3, 728–737. [Google Scholar] [CrossRef]
- Janssens, G.O.; Rademakers, S.E.; Terhaard, C.H.; Doornaert, P.A.; Bijl, H.P.; van den Ende, P.; Chin, A.; Marres, H.A.; de Bree, R.; van der Kogel, A.J.; et al. Accelerated radiotherapy with carbogen and nicotinamide for laryngeal cancer: Results of a phase III randomized trial. J. Clin. Oncol. 2012, 30, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, P.; Rojas, A.; Saunders, M. Accelerated radiotherapy, carbogen, and nicotinamide (ARCON) in the treatment of advanced bladder cancer: Mature results of a Phase II nonrandomized study. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Janssens, G.O.; Rademakers, S.E.; Terhaard, C.H.; Doornaert, P.A.; Bijl, H.P.; van den Ende, P.; Chin, A.; Takes, R.P.; de Bree, R.; Hoogsteen, I.J.; et al. Improved recurrence-free survival with ARCON for anemic patients with laryngeal cancer. Clin. Cancer Res. 2014, 20, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Kaanders, J.H.; Pop, L.A.; Marres, H.A.; Bruaset, I.; van den Hoogen, F.J.; Merkx, M.A.; van der Kogel, A.J. ARCON: Experience in 215 patients with advanced head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 769–778. [Google Scholar] [CrossRef]
- Brizel, D.M.; Hage, W.D.; Dodge, R.K.; Munley, M.T.; Piantadosi, C.A.; Dewhirst, M.W. Hyperbaric oxygen improves tumor radiation response significantly more than carbogen/nicotinamide. Radiat. Res. 1997, 147, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Brizel, D.M.; Lin, S.; Johnson, J.L.; Brooks, J.; Dewhirst, M.W.; Piantadosi, C.A. The mechanisms by which hyperbaric oxygen and carbogen improve tumour oxygenation. Br. J. Cancer 1995, 72, 1120–1124. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, M.W.; Vujaskovic, Z.; Jones, E.; Thrall, D. Re-setting the biologic rationale for thermal therapy. Int. J. Hyperth. 2005, 21, 779–790. [Google Scholar] [CrossRef]
- Elming, P.B.; Sorensen, B.S.; Oei, A.L.; Franken, N.A.P.; Crezee, J.; Overgaard, J.; Horsman, M.R. Hyperthermia: The Optimal Treatment to Overcome Radiation Resistant Hypoxia. Cancers 2019, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhirst, M.W.; Viglianti, B.L.; Lora-Michiels, M.; Hanson, M.; Hoopes, P.J. Basic principles of thermal dosimetry and thermal thresholds for tissue damage from hyperthermia. Int. J. Hyperth. 2003, 19, 267–294. [Google Scholar] [CrossRef]
- Streffer, C. Metabolic changes during and after hyperthermia. Int. J. Hyperth. 1985, 1, 305–319. [Google Scholar] [CrossRef]
- Oei, A.L.; Vriend, L.E.; Crezee, J.; Franken, N.A.; Krawczyk, P.M. Effects of hyperthermia on DNA repair pathways: One treatment to inhibit them all. Radiat. Oncol. 2015, 10, 165. [Google Scholar] [CrossRef] [Green Version]
- Lepock, J.R. Role of nuclear protein denaturation and aggregation in thermal radiosensitization. Int. J. Hyperth. 2004, 20, 115–130. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Dikomey, E. Hyperthermic radiosensitization: Mode of action and clinical relevance. Int. J. Radiat. Biol. 2001, 77, 399–408. [Google Scholar] [CrossRef]
- Oleson, J.R. Eugene Robertson Special Lecture. Hyperthermia from the clinic to the laboratory: A hypothesis. Int. J. Hyperth. 1995, 11, 315–322. [Google Scholar] [CrossRef]
- Lee, S.Y.; Fiorentini, G.; Szasz, A.M.; Szigeti, G.; Szasz, A.; Minnaar, C.A. Quo Vadis Oncological Hyperthermia (2020)? Front. Oncol. 2020, 10, 1690. [Google Scholar] [CrossRef]
- Roussakow, S. The History of Hyperthermia Rise and Decline. Conf. Pap. Med. 2013, 213, 1–40. [Google Scholar] [CrossRef]
- Emami, B.; Myerson, R.J.; Cardenes, H.; Paris, K.G.; Perez, C.A.; Straube, W.; Leybovich, L.; Mildenberger, M.; Kuske, R.R.; Devineni, V.R.; et al. Combined hyperthermia and irradiation in the treatment of superficial tumors: Results of a prospective randomized trial of hyperthermia fractionation (1/wk vs. 2/wk). Int. J. Radiat. Oncol. Biol. Phys. 1992, 24, 145–152. [Google Scholar] [CrossRef]
- Jones, E.L.; Oleson, J.R.; Prosnitz, L.R.; Samulski, T.V.; Vujaskovic, Z.; Yu, D.; Sanders, L.L.; Dewhirst, M.W. Randomized trial of hyperthermia and radiation for superficial tumors. J. Clin. Oncol. 2005, 23, 3079–3085. [Google Scholar] [CrossRef] [Green Version]
- Kapp, D.S.; Petersen, I.A.; Cox, R.S.; Hahn, G.M.; Fessenden, P.; Prionas, S.D.; Lee, E.R.; Meyer, J.L.; Samulski, T.V.; Bagshaw, M.A. Two or six hyperthermia treatments as an adjunct to radiation therapy yield similar tumor responses: Results of a randomized trial. Int. J. Radiat. Oncol. Biol. Phys. 1990, 19, 1481–1495. [Google Scholar] [CrossRef]
- Myerson, R.J.; Scott, C.B.; Emami, B.; Sapozink, M.D.; Samulski, T.V. A phase I/II study to evaluate radiation therapy and hyperthermia for deep-seated tumours: A report of RTOG 89-08. Int. J. Hyperth. 1996, 12, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J.; Gonzalez Gonzalez, D.; Hulshof, M.C.; Arcangeli, G.; Dahl, O.; Mella, O.; Bentzen, S.M. Hyperthermia as an adjuvant to radiation therapy of recurrent or metastatic malignant melanoma. A multicentre randomized trial by the European Society for Hyperthermic Oncology. Int. J. Hyperth. 1996, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; Pajak, T.; Emami, B.; Hornback, N.B.; Tupchong, L.; Rubin, P. Randomized phase III study comparing irradiation and hyperthermia with irradiation alone in superficial measurable tumors. Final report by the Radiation Therapy Oncology Group. Am. J. Clin. Oncol. 1991, 14, 133–141. [Google Scholar] [CrossRef]
- Valdagni, R.; Amichetti, M. Report of long-term follow-up in a randomized trial comparing radiation therapy and radiation therapy plus hyperthermia to metastatic lymph nodes in stage IV head and neck patients. Int. J. Radiat. Oncol. Biol. Phys. 1994, 28, 163–169. [Google Scholar] [CrossRef]
- Valdagni, R.; Italia, C.; Montanaro, P.; Lanceni, A.; Lattuada, P.; Magnani, T.; Fiorino, C.; Nahum, A. Is the alpha-beta ratio of prostate cancer really low? A prospective, non-randomized trial comparing standard and hyperfractionated conformal radiation therapy. Radiother. Oncol. 2005, 75, 74–82. [Google Scholar] [CrossRef]
- van der Zee, J.; Gonzalez Gonzalez, D.; van Rhoon, G.C.; van Dijk, J.D.; van Putten, W.L.; Hart, A.A. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: A prospective, randomised, multicentre trial. Dutch Deep Hyperthermia Group. Lancet 2000, 355, 1119–1125. [Google Scholar] [CrossRef]
- Franckena, M.; Stalpers, L.J.; Koper, P.C.; Wiggenraad, R.G.; Hoogenraad, W.J.; van Dijk, J.D.; Warlam-Rodenhuis, C.C.; Jobsen, J.J.; van Rhoon, G.C.; van der Zee, J. Long-term improvement in treatment outcome after radiotherapy and hyperthermia in locoregionally advanced cervix cancer: An update of the Dutch Deep Hyperthermia Trial. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 1176–1182. [Google Scholar] [CrossRef]
- Vernon, C.C.; Hand, J.W.; Field, S.B.; Machin, D.; Whaley, J.B.; van der Zee, J.; van Putten, W.L.; van Rhoon, G.C.; van Dijk, J.D.; Gonzalez Gonzalez, D.; et al. Radiotherapy with or without hyperthermia in the treatment of superficial localized breast cancer: Results from five randomized controlled trials. International Collaborative Hyperthermia Group. Int. J. Radiat. Oncol. Biol. Phys. 1996, 35, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Seifert, G.; Budach, V.; Keilholz, U.; Wust, P.; Eggert, A.; Ghadjar, P. Regional hyperthermia combined with chemotherapy in paediatric, adolescent and young adult patients: Current and future perspectives. Radiat. Oncol. 2016, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Tydings, C.; Sharma, K.V.; Kim, A.; Yarmolenko, P.S. Emerging hyperthermia applications for pediatric oncology. Adv. Drug Deliv. Rev. 2020, 163–164, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Li, G.C.; Mivechi, N.F.; Weitzel, G. Heat shock proteins, thermotolerance, and their relevance to clinical hyperthermia. Int. J. Hyperth. 1995, 11, 459–488. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J.; Nielsen, O.S. The importance of thermotolerance for the clinical treatment with hyperthermia. Radiother. Oncol. 1983, 1, 167–178. [Google Scholar] [CrossRef]
- Ozhinsky, E.; Salgaonkar, V.A.; Diederich, C.J.; Rieke, V. MR thermometry-guided ultrasound hyperthermia of user-defined regions using the ExAblate prostate ablation array. J. Ther. Ultrasound 2018, 6, 7. [Google Scholar] [CrossRef]
- Stakhursky, V.L.; Arabe, O.; Cheng, K.S.; Macfall, J.; Maccarini, P.; Craciunescu, O.; Dewhirst, M.; Stauffer, P.; Das, S.K. Real-time MRI-guided hyperthermia treatment using a fast adaptive algorithm. Phys. Med. Biol. 2009, 54, 2131–2145. [Google Scholar] [CrossRef]
- Datta, N.R.; Puric, E.; Klingbiel, D.; Gomez, S.; Bodis, S. Hyperthermia and Radiation Therapy in Locoregional Recurrent Breast Cancers: A Systematic Review and Meta-analysis. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 1073–1087. [Google Scholar] [CrossRef]
- Datta, N.R.; Rogers, S.; Klingbiel, D.; Gomez, S.; Puric, E.; Bodis, S. Hyperthermia and radiotherapy with or without chemotherapy in locally advanced cervical cancer: A systematic review with conventional and network meta-analyses. Int. J. Hyperth. 2016, 32, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Datta, N.R.; Rogers, S.; Ordonez, S.G.; Puric, E.; Bodis, S. Hyperthermia and radiotherapy in the management of head and neck cancers: A systematic review and meta-analysis. Int. J. Hyperth. 2016, 32, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Park, H.; Griffin, R.J. Improvement of tumor oxygenation by mild hyperthermia. Radiat. Res. 2001, 155, 515–528. [Google Scholar] [CrossRef]
- Brizel, D.M.; Scully, S.P.; Harrelson, J.M.; Layfield, L.J.; Dodge, R.K.; Charles, H.C.; Samulski, T.V.; Prosnitz, L.R.; Dewhirst, M.W. Radiation therapy and hyperthermia improve the oxygenation of human soft tissue sarcomas. Cancer Res. 1996, 56, 5347–5350. [Google Scholar] [PubMed]
- Vujaskovic, Z.; Rosen, E.L.; Blackwell, K.L.; Jones, E.L.; Brizel, D.M.; Prosnitz, L.R.; Samulski, T.V.; Dewhirst, M.W. Ultrasound guided pO2 measurement of breast cancer reoxygenation after neoadjuvant chemotherapy and hyperthermia treatment. Int. J. Hyperth. 2003, 19, 498–506. [Google Scholar] [CrossRef]
- Jones, E.L.; Prosnitz, L.R.; Dewhirst, M.W.; Marcom, P.K.; Hardenbergh, P.H.; Marks, L.B.; Brizel, D.M.; Vujaskovic, Z. Thermochemoradiotherapy improves oxygenation in locally advanced breast cancer. Clin. Cancer Res. 2004, 10, 4287–4293. [Google Scholar] [CrossRef] [Green Version]
- Vujaskovic, Z.; Song, C.W. Physiological mechanisms underlying heat-induced radiosensitization. Int. J. Hyperth. 2004, 20, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Shakil, A.; Osborn, J.L.; Song, C.W. Changes in oxygenation status and blood flow in a rat tumor model by mild temperature hyperthermia. Int. J. Radiat. Oncol. Biol. Phys. 1999, 43, 859–865. [Google Scholar] [CrossRef]
- Vujaskovic, Z.; Poulson, J.M.; Gaskin, A.A.; Thrall, D.E.; Page, R.L.; Charles, H.C.; MacFall, J.R.; Brizel, D.M.; Meyer, R.E.; Prescott, D.M.; et al. Temperature-dependent changes in physiologic parameters of spontaneous canine soft tissue sarcomas after combined radiotherapy and hyperthermia treatment. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 179–185. [Google Scholar] [CrossRef]
- Vaupel, P.W.; Kelleher, D.K. Pathophysiological and vascular characteristics of tumours and their importance for hyperthermia: Heterogeneity is the key issue. Int. J. Hyperth. 2010, 26, 211–223. [Google Scholar] [CrossRef]
- Lepock, J.R.; Cheng, K.H.; Al-Qysi, H.; Sim, I.; Koch, C.J.; Kruuv, J. Hyperthermia-induced inhibition of respiration and mitochondrial protein denaturation in CHL cells. Int. J. Hyperth. 1987, 3, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Willis, W.T.; Jackman, M.R.; Bizeau, M.E.; Pagliassotti, M.J.; Hazel, J.R. Hyperthermia impairs liver mitochondrial function in vitro. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R1240–R1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dressler, C.; Beuthan, J.; Mueller, G.; Zabarylo, U.; Minet, O. Fluorescence imaging of heat-stress induced mitochondrial long-term depolarization in breast cancer cells. J. Fluoresc. 2006, 16, 689–695. [Google Scholar] [CrossRef]
- Tamulevicius, P.; Streffer, C. Bioluminescence imaging of metabolites in a human tumour xenograft after treatment with hyperthermia and/or the radiosensitizer pimonidazole. Int. J. Hyperth. 1997, 13, 235–245. [Google Scholar] [CrossRef]
- Kelleher, D.K.; Engel, T.; Vaupel, P.W. Changes in microregional perfusion, oxygenation, ATP and lactate distribution in subcutaneous rat tumours upon water-filtered IR-A hyperthermia. Int. J. Hyperth. 1995, 11, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.J.; Sonveaux, P.; Porporato, P.E.; Danhier, P.; Gallez, B.; Batinic-Haberle, I.; Nien, Y.C.; Schroeder, T.; Dewhirst, M.W. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc. Natl. Acad. Sci. USA 2010, 107, 20477–20482. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, K.E.; Jeong, S.W.; Hwang, S.W.; Jo, H.; Lee, J.; Cho, D.; Park, H.J. Effects of the Ultra-High-Frequency Electrical Field Radiofrequency Device on Mouse Skin: A Histologic and Molecular Study. Plast. Reconstr. Surg. 2016, 138, 248e–255e. [Google Scholar] [CrossRef]
- van Leeuwen, C.M.; Oei, A.L.; Chin, K.; Crezee, J.; Bel, A.; Westermann, A.M.; Buist, M.R.; Franken, N.A.P.; Stalpers, L.J.A.; Kok, H.P. A short time interval between radiotherapy and hyperthermia reduces in-field recurrence and mortality in women with advanced cervical cancer. Radiat. Oncol. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; van Leeuwen, C.M.; ten Cate, R.; Rodermond, H.M.; Buist, M.R.; Stalpers, L.J.; Crezee, J.; Kok, H.P.; Medema, J.P.; Franken, N.A. Hyperthermia Selectively Targets Human Papillomavirus in Cervical Tumors via p53-Dependent Apoptosis. Cancer Res. 2015, 75, 5120–5129. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Stavridi, E.; Leeper, D.B.; Iliakis, G. Effects of hyperthermia on p53 protein expression and activity. J. Cell. Physiol. 2002, 190, 365–374. [Google Scholar] [CrossRef]
- Hunt, C.R.; Pandita, R.K.; Laszlo, A.; Higashikubo, R.; Agarwal, M.; Kitamura, T.; Gupta, A.; Rief, N.; Horikoshi, N.; Baskaran, R.; et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res. 2007, 67, 3010–3017. [Google Scholar] [CrossRef] [Green Version]
- Burgman, P.; Ouyang, H.; Peterson, S.; Chen, D.J.; Li, G.C. Heat inactivation of Ku autoantigen: Possible role in hyperthermic radiosensitization. Cancer Res. 1997, 57, 2847–2850. [Google Scholar]
- Ito, A.; Shinkai, M.; Honda, H.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Augmentation of MHC class I antigen presentation via heat shock protein expression by hyperthermia. Cancer Immunol. Immunother. 2001, 50, 515–522. [Google Scholar] [CrossRef]
- Burd, R.; Dziedzic, T.S.; Xu, Y.; Caligiuri, M.A.; Subjeck, J.R.; Repasky, E.A. Tumor cell apoptosis, lymphocyte recruitment and tumor vascular changes are induced by low temperature, long duration (fever-like) whole body hyperthermia. J. Cell. Physiol. 1998, 177, 137–147. [Google Scholar] [CrossRef]
- Mace, T.A.; Zhong, L.; Kokolus, K.M.; Repasky, E.A. Effector CD8+ T cell IFN-gamma production and cytotoxicity are enhanced by mild hyperthermia. Int. J. Hyperth. 2012, 28, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan Mohd Zawawi, W.F.A.; Hibma, M.H.; Salim, M.I.; Jemon, K. Hyperthermia by near infrared radiation induced immune cells activation and infiltration in breast tumor. Sci. Rep. 2021, 11, 10278. [Google Scholar] [CrossRef]
- Corry, P.M.; Dewhirst, M.W. Thermal medicine, heat shock proteins and cancer. Int. J. Hyperth. 2005, 21, 675–677. [Google Scholar] [CrossRef]
- Park, C.H.; Lee, M.J.; Ahn, J.; Kim, S.; Kim, H.H.; Kim, K.H.; Eun, H.C.; Chung, J.H. Heat shock-induced matrix metalloproteinase (MMP)-1 and MMP-3 are mediated through ERK and JNK activation and via an autocrine interleukin-6 loop. J. Investig. Dermatol. 2004, 123, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.L.; Paulson, D.F.; Mickey, G.H.; Webb, K.S. Spontaneous metastasis of cells of the human prostate carcinoma cell line PC-3 in athymic nude mice. J. Urol. 1982, 128, 1064–1067. [Google Scholar] [CrossRef]
- Lee, T.H.; Bu, J.; Kim, B.H.; Poellmann, M.J.; Hong, S.; Hyun, S.H. Sub-lethal hyperthermia promotes epithelial-to-mesenchymal-like transition of breast cancer cells: Implication of the synergy between hyperthermia and chemotherapy. RSC Adv. 2019, 9, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Koong, A. Therapeutic advantage of hypoxic cells in tumors: A theoretical study. J. Natl. Cancer Inst. 1991, 83, 178–185. [Google Scholar] [CrossRef]
- Kennedy, K.A. Hypoxic cells as specific drug targets for chemotherapy. Anticancer Drug Des. 1987, 2, 181–194. [Google Scholar] [PubMed]
- Guise, C.P.; Mowday, A.M.; Ashoorzadeh, A.; Yuan, R.; Lin, W.H.; Wu, D.H.; Smaill, J.B.; Patterson, A.V.; Ding, K. Bioreductive prodrugs as cancer therapeutics: Targeting tumor hypoxia. Chin. J. Cancer 2014, 33, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.E.; Cooke, M.S. Electron-affinic sensitization. I. A structural basis for chemical radiosensitizers in bacteria. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1969, 15, 457–471. [Google Scholar] [CrossRef]
- Oronsky, B.T.; Knox, S.J.; Scicinski, J. Six degrees of separation: The oxygen effect in the development of radiosensitizers. Transl. Oncol. 2011, 4, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Kappen, L.S.; Lee, T.R.; Yang, C.C.; Goldberg, I.H.J.B. Oxygen transfer from the nitro group of a nitroaromatic radiosensitizer to a DNA sugar damage product. Biochemistry 1989, 28, 4540–4542. [Google Scholar] [CrossRef]
- Asquith, J.C.; Foster, J.L.; Willson, R.L.; Ings, R.; McFadzean, J.A. Metronidazole (“Flagyl”). A radiosensitizer of hypoxic cells. Br. J. Radiol. 1974, 47, 474–481. [Google Scholar] [CrossRef]
- Dische, S.; Saunders, M.; Lee, M.E.; Adams, G.; Flockhart, I.J.B.j.o.c. Clinical testing of the radiosensitizer Ro 07-0582: Experience with multiple doses. Br. J. Cancer 1977, 35, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urtasun, R.; Feldstein, M.; Partington, J.; Tanasichuk, H.; Miller, J.; Russell, D.; Agboola, O.; Mielke, B.J.B.j.o.c. Radiation and nitroimidazoles in supratentorial high grade gliomas: A second clinical trial. Br. J. Cancer 1982, 46, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Dische, S.J.R. Chemical sensitizers for hypoxic cells: A decade of experience in clinical radiotherapy. Radiother. Oncol. 1985, 3, 97–115. [Google Scholar] [CrossRef]
- Eschwège, F.; Sancho-Garnier, H.; Chassagne, D.; Brisgand, D.; Guerra, M.; Philippe Malaise, E.; Bey, P.; Busutti, L.; Cionini, L.; N’Guyen, T.; et al. Results of a european randomized trial of Etanidazole combined with radiotherapy in head and neck carcinomas. Int. J. Radiat. Oncol. 1997, 39, 275–281. [Google Scholar] [CrossRef]
- Urtasun, R.C.; Palmer, M.; Kinney, B.; Belch, A.; Hewitt, J.; Hanson, J. Intervention with the hypoxic tumor cell sensitizer etanidazole in the combined modality treatment of limited stage small-cell lung cancer. A one-institution study. Int. J. Radiat. Oncol. Biol. Phys. 1998, 40, 337–342. [Google Scholar] [CrossRef]
- Dische, S.; Chassagne, D.; Hope-Stone, H.F.; Dawes, P.J.D.K.; Roberts, J.T.; Yosef, H.; Bey, P.; Horiot, J.C.; Jacobson, A.; Frankendal, B.; et al. A trial of Ro 03-8799 (pimonidazole) in carcinoma of the uterine cervix: An interim report from the Medical Research Council Working Party on advanced carcinoma of the cervix. Radiother. Oncol. 1993, 26, 93–103. [Google Scholar] [CrossRef]
- Krohn, K.A.; Link, J.M.; Mason, R.P. Molecular imaging of hypoxia. J. Nucl. Med. 2008, 49 (Suppl. 2), 129s–148s. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, J.; Sand Hansen, H.; Overgaard, M.; Bastholt, L.; Berthelsen, A.; Specht, L.; Lindeløv, B.; Jørgensen, K. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother. Oncol. 1998, 46, 135–146. [Google Scholar] [CrossRef]
- Saksø, M.; Jensen, K.; Andersen, M.; Hansen, C.R.; Eriksen, J.G.; Overgaard, J. DAHANCA 28: A phase I/II feasibility study of hyperfractionated, accelerated radiotherapy with concomitant cisplatin and nimorazole (HART-CN) for patients with locally advanced, HPV/p16-negative squamous cell carcinoma of the oropharynx, hypopharynx, larynx and oral cavity. Radiother. Oncol. 2020, 148, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypoxia-Activated Prodrugs in Combination With Radiation Therapy. Int. J. Radiat. Oncol. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.K.; Liew, L.P.; Hay, M.P. Overcoming Radioresistance: Small Molecule Radiosensitisers and Hypoxia-activated Prodrugs. Clin. Oncol. 2019, 31, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Biedermann, K.A.; Wolf, C.R.; Brown, J.M. Metabolism of the bioreductive cytotoxin SR 4233 by tumour cells: Enzymatic studies. Br. J. Cancer 1993, 67, 321–325. [Google Scholar] [CrossRef] [Green Version]
- Daniels, J.S.; Gates, K.S.; Tronche, C.; Greenberg, M.M. Direct evidence for bimodal DNA damage induced by tirapazamine. Chem. Res. Toxicol. 1998, 11, 1254–1257. [Google Scholar] [CrossRef]
- Baker, M.A.; Zeman, E.M.; Hirst, V.K.; Brown, J.M. Metabolism of SR 4233 by Chinese hamster ovary cells: Basis of selective hypoxic cytotoxicity. Cancer Res. 1988, 48, 5947–5952. [Google Scholar]
- Moriwaki, T.; Okamoto, S.; Sasanuma, H.; Nagasawa, H.; Takeda, S.; Masunaga, S.I.; Tano, K. Cytotoxicity of Tirapazamine (3-Amino-1,2,4-benzotriazine-1,4-dioxide)-Induced DNA Damage in Chicken DT40 Cells. Chem. Res. Toxicol. 2017, 30, 699–704. [Google Scholar] [CrossRef]
- Lartigau, E.; Guichard, M. Does tirapazamine (SR-4233) have any cytotoxic or sensitizing effect on three human tumour cell lines at clinically relevant partial oxygen pressure? Int. J. Radiat. Biol. 1995, 67, 211–216. [Google Scholar] [CrossRef]
- Zeman, E.M.; Brown, J.M.; Lemmon, M.J.; Hirst, V.K.; Lee, W.W. SR-4233: A new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1239–1242. [Google Scholar] [CrossRef]
- Marcu, L.; Olver, I. Tirapazamine: From bench to clinical trials. Curr. Clin. Pharmacol. 2006, 1, 71–79. [Google Scholar] [CrossRef]
- Masunaga, S.; Ono, K.; Hori, H.; Shibata, T.; Suzuki, M.; Kinashi, Y.; Takagaki, M.; Akaboshi, M. Effects of bioreductive agents, tirapazamine and mitomycin C, on quiescent cell populations in solid tumors, evaluated by micronucleus assay. Jpn. J. Cancer Res. 1997, 88, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Masunaga, S.; Ono, K.; Hori, H.; Suzuki, M.; Kinashi, Y.; Takagaki, M.; Kasai, S.; Nagasawa, H.; Uto, Y. Change in oxygenation status in intratumour total and quiescent cells following gamma-ray irradiation, tirapazamine administration, cisplatin injection and bleomycin treatment. Br. J. Radiol. 2000, 73, 978–986. [Google Scholar] [CrossRef]
- Lambin, P.; Guichard, M.; Chavaudra, N.; Malaise, E.P. The effect of the hypoxic cell drug SR-4233 alone or combined with the ionizing radiations on two human tumor cell lines having different radiosensitivity. Radiother. Oncol. 1992, 24, 201–204. [Google Scholar] [CrossRef]
- Dorie, M.J.; Brown, J.M. Tumor-specific, schedule-dependent interaction between tirapazamine (SR 4233) and cisplatin. Cancer Res. 1993, 53, 4633–4636. [Google Scholar]
- Rischin, D.; Peters, L.; Hicks, R.; Hughes, P.; Fisher, R.; Hart, R.; Sexton, M.; D’Costa, I.; von Roemeling, R. Phase I trial of concurrent tirapazamine, cisplatin, and radiotherapy in patients with advanced head and neck cancer. J. Clin. Oncol. 2001, 19, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Brown, C.; O’Flaherty, C.; Fleischauer, A.; Curtin, J.; Roemeling, R.; Spriggs, D.R. Phase I study of tirapazamine and cisplatin in patients with recurrent cervical cancer. Gynecol. Oncol. 1997, 67, 127–130. [Google Scholar] [CrossRef]
- Craighead, P.S.; Pearcey, R.; Stuart, G. A phase I/II evaluation of tirapazamine administered intravenously concurrent with cisplatin and radiotherapy in women with locally advanced cervical cancer. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 791–795. [Google Scholar] [CrossRef]
- Le, Q.T.; McCoy, J.; Williamson, S.; Ryu, J.; Gaspar, L.E.; Edelman, M.J.; Dakhil, S.R.; Sides, S.D.; Crowley, J.J.; Gandara, D.R.; et al. Phase I study of tirapazamine plus cisplatin/etoposide and concurrent thoracic radiotherapy in limited-stage small cell lung cancer (S0004): A Southwest Oncology Group study. Clin. Cancer Res. 2004, 10, 5418–5424. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.A.; Kilpatrick, D.; von Roemeling, R.; Langer, C.; Graham, M.A.; Greenslade, D.; Kennedy, G.; Keenan, E.; O’Dwyer, P.J. Phase I trial of tirapazamine in combination with cisplatin in a single dose every 3 weeks in patients with solid tumors. J. Clin. Oncol. 1997, 15, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Senan, S.; Rampling, R.; Graham, M.A.; Wilson, P.; Robin, H., Jr.; Eckardt, N.; Lawson, N.; McDonald, A.; von Roemeling, R.; Workman, P.; et al. Phase I and pharmacokinetic study of tirapazamine (SR 4233) administered every three weeks. Clin. Cancer Res. 1997, 3, 31–38. [Google Scholar]
- Shulman, L.N.; Buswell, L.; Riese, N.; Doherty, N.; Loeffler, J.S.; von Roemeling, R.W.; Coleman, C.N. Phase I trial of the hypoxic cell cytotoxin tirapazamine with concurrent radiation therapy in the treatment of refractory solid tumors. Int. J. Radiat. Oncol. Biol. Phys. 1999, 44, 349–353. [Google Scholar] [CrossRef]
- Bedikian, A.Y.; Legha, S.S.; Eton, O.; Buzaid, A.C.; Papadopoulos, N.; Coates, S.; Simmons, T.; Neefe, J.; von Roemeling, R. Phase II trial of tirapazamine combined with cisplatin in chemotherapy of advanced malignant melanoma. Ann. Oncol. 1997, 8, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Rischin, D.; Peters, L.; Fisher, R.; Macann, A.; Denham, J.; Poulsen, M.; Jackson, M.; Kenny, L.; Penniment, M.; Corry, J.; et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin, and Radiation in patients with locally advanced head and neck cancer: A randomized phase II trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02). J. Clin. Oncol. 2005, 23, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.J.; Trotti, A.; Spencer, S.; Rostock, R.; Fisher, C.; von Roemeling, R.; Harvey, E.; Groves, E. Concurrent tirapazamine and radiotherapy for advanced head and neck carcinomas: A Phase II study. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 811–815. [Google Scholar] [CrossRef]
- Miller, V.A.; Ng, K.K.; Grant, S.C.; Kindler, H.; Pizzo, B.; Heelan, R.T.; von Roemeling, R.; Kris, M.G. Phase II study of the combination of the novel bioreductive agent, tirapazamine, with cisplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 1997, 8, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Treat, J.; Johnson, E.; Langer, C.; Belani, C.; Haynes, B.; Greenberg, R.; Rodriquez, R.; Drobins, P.; Miller, W., Jr.; Meehan, L.; et al. Tirapazamine with cisplatin in patients with advanced non-small-cell lung cancer: A phase II study. J. Clin. Oncol. 1998, 16, 3524–3527. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; von Pawel, J.; Nimmermann, C.; Groth, G.; Gatzemeier, U. [Phase II-trial of tirapazamine in combination with cisplatin and gemcitabine in patients with advanced non-small-cell-lung-cancer (NSCLC)]. Pneumologie 2004, 58, 845–849. [Google Scholar] [CrossRef] [Green Version]
- Maluf, F.C.; Leiser, A.L.; Aghajanian, C.; Sabbatini, P.; Pezzulli, S.; Chi, D.S.; Wolf, J.K.; Levenback, C.; Loh, E.; Spriggs, D.R. Phase II study of tirapazamine plus cisplatin in patients with advanced or recurrent cervical cancer. Int. J. Gynecol. Cancer 2006, 16, 1165–1171. [Google Scholar] [CrossRef]
- Le, Q.T.; Moon, J.; Redman, M.; Williamson, S.K.; Lara, P.N., Jr.; Goldberg, Z.; Gaspar, L.E.; Crowley, J.J.; Moore, D.F., Jr.; Gandara, D.R. Phase II study of tirapazamine, cisplatin, and etoposide and concurrent thoracic radiotherapy for limited-stage small-cell lung cancer: SWOG 0222. J. Clin. Oncol. 2009, 27, 3014–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Rowe, J.; Scott, C.; Werner-Wasik, M.; Bahary, J.P.; Curran, W.J.; Urtasun, R.C.; Fisher, B. Single-arm, open-label phase II study of intravenously administered tirapazamine and radiation therapy for glioblastoma multiforme. J. Clin. Oncol. 2000, 18, 1254–1259. [Google Scholar] [CrossRef]
- DiSilvestro, P.A.; Ali, S.; Craighead, P.S.; Lucci, J.A.; Lee, Y.C.; Cohn, D.E.; Spirtos, N.M.; Tewari, K.S.; Muller, C.; Gajewski, W.H.; et al. Phase III randomized trial of weekly cisplatin and irradiation versus cisplatin and tirapazamine and irradiation in stages IB2, IIA, IIB, IIIB, and IVA cervical carcinoma limited to the pelvis: A Gynecologic Oncology Group study. J. Clin. Oncol. 2014, 32, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Rischin, D.; Peters, L.J.; O’Sullivan, B.; Giralt, J.; Fisher, R.; Yuen, K.; Trotti, A.; Bernier, J.; Bourhis, J.; Ringash, J.; et al. Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): A phase III trial of the Trans-Tasman Radiation Oncology Group. J. Clin. Oncol. 2010, 28, 2989–2995. [Google Scholar] [CrossRef] [PubMed]
- von Pawel, J.; von Roemeling, R.; Gatzemeier, U.; Boyer, M.; Elisson, L.O.; Clark, P.; Talbot, D.; Rey, A.; Butler, T.W.; Hirsh, V.; et al. Tirapazamine plus cisplatin versus cisplatin in advanced non-small-cell lung cancer: A report of the international CATAPULT I study group. Cisplatin and Tirapazamine in Subjects with Advanced Previously Untreated Non-Small-Cell Lung Tumors. J. Clin. Oncol. 2000, 18, 1351–1359. [Google Scholar] [CrossRef]
- Shepherd, F.; Koschel, G.; Von Pawel, J.; Gatzmeier, U.; Van Zandwiyk, N.; Woll, P.; Van Klavren, R.; Krasko, P.; DeSimone, P.; Nicolson, M.; et al. Comparison of Tirazone (Tirapazamine) and cisplatin vs. etoposide and cisplatin in advanced non-small cell lung cancer (NSCLC): Final results of the international Phase III CATAPULT II Trial. Lung Cancer 2000, 29, 28. [Google Scholar] [CrossRef]
- Aquino, V.M.; Weitman, S.D.; Winick, N.J.; Blaney, S.; Furman, W.L.; Kepner, J.L.; Bonate, P.; Krailo, M.; Qu, W.; Bernstein, M. Phase I trial of tirapazamine and cyclophosphamide in children with refractory solid tumors: A pediatric oncology group study. J. Clin. Oncol. 2004, 22, 1413–1419. [Google Scholar] [CrossRef]
- Mascarenhas, L.; Lyden, E.R.; Breitfeld, P.P.; Walterhouse, D.O.; Donaldson, S.S.; Rodeberg, D.A.; Parham, D.M.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Risk-based treatment for patients with first relapse or progression of rhabdomyosarcoma: A report from the Children’s Oncology Group. Cancer 2019, 125, 2602–2609. [Google Scholar] [CrossRef]
- Korga, A.; Iwan, M.; Matosiuk, D.; Rzadkowska, M.; Szacon, E.; Humeniuk, E.; Sysa, M.; Ostrowska, M.; Dudka, J. New tirapazamine derivatives protect cardiomyocytes from doxorubicin toxicity. Curr. Issues Pharm. Med. Sci. 2020, 33, 1–5. [Google Scholar] [CrossRef]
- Mehibel, M.; Xu, Y.; Li, C.G.; Moon, E.J.; Thakkar, K.N.; Diep, A.N.; Kim, R.K.; Bloomstein, J.D.; Xiao, Y.; Bacal, J.; et al. Eliminating hypoxic tumor cells improves response to PARP inhibitors in homologous recombination-deficient cancer models. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Wang, J.; Biedermann, K.A.; Brown, J.M. Repair of DNA and chromosome breaks in cells exposed to SR 4233 under hypoxia or to ionizing radiation. Cancer Res. 1992, 52, 4473–4477. [Google Scholar] [PubMed]
- Parveen, I.; Naughton, D.P.; Whish, W.J.; Threadgill, M.D. 2-nitroimidazol-5-ylmethyl as a potential bioreductively activated prodrug system: Reductively triggered release of the PARP inhibitor 5-bromoisoquinolinone. Bioorg. Med. Chem. Lett. 1999, 9, 2031–2036. [Google Scholar] [CrossRef]
- Pruijn, F.B.; Patel, K.; Hay, M.P.; Wilson, W.R.; Hicks, K.O. Prediction of Tumour Tissue Diffusion Coefficients of Hypoxia-Activated Prodrugs from Physicochemical Parameters. Aust. J. Chem. 2008, 61, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Gong, M.; Deng, Z.; Liu, H.; Chang, Y.; Yang, Z.; Cai, L. Tirapazamine suppress osteosarcoma cells in part through SLC7A11 mediated ferroptosis. Biochem. Biophys. Res. Commun. 2021, 567, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.H.; Yeh, S.H.; Yeh, K.H.; Chen, K.W.; Cheng, Y.W.; Su, T.H.; Jao, P.; Ni, L.C.; Chen, P.J.; Chen, D.S. Hypoxia-activated cytotoxic agent tirapazamine enhances hepatic artery ligation-induced killing of liver tumor in HBx transgenic mice. Proc. Natl. Acad. Sci. USA 2016, 113, 11937–11942. [Google Scholar] [CrossRef] [Green Version]
- Abi-Jaoudeh, N.; Dayyani, F.; Chen, P.J.; Fernando, D.; Fidelman, N.; Javan, H.; Liang, P.C.; Hwang, J.I.; Imagawa, D.K. Phase I Trial on Arterial Embolization with Hypoxia Activated Tirapazamine for Unresectable Hepatocellular Carcinoma. J. Hepatocell Carcinoma 2021, 8, 421–434. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.J.; Brizel, D.M.; Chi, J.T.; Dewhirst, M.W. The potential role of intrinsic hypoxia markers as prognostic variables in cancer. Antioxid. Redox Signal. 2007, 9, 1237–1294. [Google Scholar] [CrossRef]
- Semenza, G.L. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Harada, H.; Kizaka-Kondoh, S.; Li, G.; Itasaka, S.; Shibuya, K.; Inoue, M.; Hiraoka, M. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene 2007, 26, 7508–7516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.F.; Poon, R.T.; To, J.; Ho, D.W.; Fan, S.T. The potential role of hypoxia inducible factor 1alpha in tumor progression after hypoxia and chemotherapy in hepatocellular carcinoma. Cancer Res. 2004, 64, 5496–5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levina, V.; Su, Y.; Nolen, B.; Liu, X.; Gordin, Y.; Lee, M.; Lokshin, A.; Gorelik, E. Chemotherapeutic drugs and human tumor cells cytokine network. Int. J. Cancer 2008, 123, 2031–2040. [Google Scholar] [CrossRef] [Green Version]
- Viola, R.J.; Provenzale, J.M.; Li, F.; Li, C.Y.; Yuan, H.; Tashjian, J.; Dewhirst, M.W. In vivo bioluminescence imaging monitoring of hypoxia-inducible factor 1alpha, a promoter that protects cells, in response to chemotherapy. AJR Am. J. Roentgenol. 2008, 191, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Befani, C.D.; Vlachostergios, P.J.; Hatzidaki, E.; Patrikidou, A.; Bonanou, S.; Simos, G.; Papandreou, C.N.; Liakos, P. Bortezomib represses HIF-1alpha protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. 2012, 90, 45–54. [Google Scholar] [CrossRef]
- Bertozzi, D.; Marinello, J.; Manzo, S.G.; Fornari, F.; Gramantieri, L.; Capranico, G. The natural inhibitor of DNA topoisomerase I, camptothecin, modulates HIF-1alpha activity by changing miR expression patterns in human cancer cells. Mol. Cancer Ther. 2014, 13, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Shirai, Y.; Chow, C.C.T.; Kambe, G.; Suwa, T.; Kobayashi, M.; Takahashi, I.; Harada, H.; Nam, J.M. An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor. Cancers 2021, 13, 2813. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.M.; Rizzi, J.P.; Han, G.; Wehn, P.M.; Cao, Z.; Du, X.; Cheng, T.; Czerwinski, R.M.; Dixon, D.D.; Goggin, B.S.; et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016, 76, 5491–5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renfrow, J.J.; Soike, M.H.; West, J.L.; Ramkissoon, S.H.; Metheny-Barlow, L.; Mott, R.T.; Kittel, C.A.; D’Agostino, R.B., Jr.; Tatter, S.B.; Laxton, A.W.; et al. Attenuating hypoxia driven malignant behavior in glioblastoma with a novel hypoxia-inducible factor 2 alpha inhibitor. Sci. Rep. 2020, 10, 15195. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients with Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Ma, Y.; Diaz de Leon, A.; Christie, A.; Xie, Z.; Woolford, L.; Singla, N.; Joyce, A.; Hill, H.; Madhuranthakam, A.J.; et al. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Albiges, L.; Fan, L.; Perini, R.F.; Zojwalla, N.J.; Powles, T.; Rini, B.I. Phase III study of the hypoxia-inducible factor 2α (HIF-2α) inhibitor MK-6482 versus everolimus in previously treated patients with advanced clear cell renal cell carcinoma (ccRCC). J. Clin. Oncol. 2020, 38, TPS5094. [Google Scholar] [CrossRef]
- Xu, R.; Wang, K.; Rizzi, J.P.; Huang, H.; Grina, J.A.; Schlachter, S.T.; Wang, B.; Wehn, P.M.; Yang, H.; Dixon, D.D.; et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzo nitrile (PT2977), a Hypoxia-Inducible Factor 2alpha (HIF-2alpha) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. J. Med. Chem. 2019, 62, 6876–6893. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, B.J.; Richardson, R.A.; Dewhirst, M.W. Hypoxia and radiotherapy: Opportunities for improved outcomes in cancer treatment. Cancer Metastasis Rev. 2007, 26, 241–248. [Google Scholar] [CrossRef]
- Secomb, T.W.; Hsu, R.; Ong, E.T.; Gross, J.F.; Dewhirst, M.W. Analysis of the Effects of Oxygen Supply and Demand on Hypoxic Fraction in Tumors. Acta Oncol. 1995, 34, 313–316. [Google Scholar] [CrossRef]
- Koritzinsky, M. Metformin: A Novel Biological Modifier of Tumor Response to Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased Cancer-Related Mortality for Patients with Type 2 Diabetes Who Use Sulfonylureas or Insulin. J. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Hussien, K.; Fokas, E.; Kannan, P.; Shipley, R.J.; Ashton, T.M.; Stratford, M.; Pearson, N.; Muschel, R.J. Regulation of O2 consumption by the PI3K and mTOR pathways contributes to tumor hypoxia. Radiother. Oncol. 2014, 111, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Yung, E.; Pintilie, M.; Muaddi, H.; Chaib, S.; Yeung, M.; Fusciello, M.; Sykes, J.; Pitcher, B.; Hagenkort, A.; et al. MATE2 Expression Is Associated with Cancer Cell Response to Metformin. PLoS ONE 2016, 11, e0165214. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming Metabolism with Metformin Improves Tumor Oxygenation and Radiotherapy Response. Clin. Cancer Res. 2013, 19, 6741–6750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.L.; Kolozsvary, A.; Isrow, D.M.; Al Feghali, K.; Lapanowski, K.; Jenrow, K.A.; Kim, J.H. A Novel Mechanism of High Dose Radiation Sensitization by Metformin. Front. Oncol. 2019, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- de Mey, S.; Jiang, H.; Corbet, C.; Wang, H.; Dufait, I.; Law, K.; Bastien, E.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Antidiabetic Biguanides Radiosensitize Hypoxic Colorectal Cancer Cells Through a Decrease in Oxygen Consumption. Front. Pharmacol. 2018, 9, 1073. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18226–18231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Appleyard, M.V.; Murray, K.E.; Coates, P.J.; Wullschleger, S.; Bray, S.E.; Kernohan, N.M.; Fleming, S.; Alessi, D.R.; Thompson, A.M. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br. J. Cancer 2012, 106, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Abdel Hadi, N.; Gicquel, T.; El Kaoutari, A.; Diémé, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef]
- Ellinghaus, P.; Heisler, I.; Unterschemmann, K.; Haerter, M.; Beck, H.; Greschat, S.; Ehrmann, A.; Summer, H.; Flamme, I.; Oehme, F.; et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013, 2, 611–624. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Ye, Z.; Townsend, D.M.; Tew, K.D. Pharmacology of ME-344, a novel cytotoxic isoflavone. Adv. Cancer Res. 2019, 142, 187–207. [Google Scholar] [CrossRef]
- Lissanu Deribe, Y.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.A.; Khor, T.; Feng, N.; et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, J.R.; Goff, B.; Forster, M.D.; Bendell, J.C.; Britten, C.D.; Gordon, M.S.; Gabra, H.; Waterhouse, D.M.; Poole, M.; Ross Camidge, D.; et al. Phase Ib study of the mitochondrial inhibitor ME-344 plus topotecan in patients with previously treated, locally advanced or metastatic small cell lung, ovarian and cervical cancers. Investig. New Drugs 2017, 35, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Wu, J.; Ding, C.C.; Lin, C.C.; Pan, S.; Bossa, N.; Xu, Y.; Yang, W.H.; Mathey-Prevot, B.; Chi, J.T. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020, 27, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Diepart, C.; Karroum, O.; Magat, J.; Feron, O.; Verrax, J.; Calderon, P.B.; Grégoire, V.; Leveque, P.; Stockis, J.; Dauguet, N.; et al. Arsenic Trioxide Treatment Decreases the Oxygen Consumption Rate of Tumor Cells and Radiosensitizes Solid Tumors. Cancer Res. 2012, 72, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilaplana-Lopera, N.; Besh, M.; Moon, E.J. Targeting Hypoxia: Revival of Old Remedies. Biomolecules 2021, 11, 1604. https://doi.org/10.3390/biom11111604
Vilaplana-Lopera N, Besh M, Moon EJ. Targeting Hypoxia: Revival of Old Remedies. Biomolecules. 2021; 11(11):1604. https://doi.org/10.3390/biom11111604
Chicago/Turabian StyleVilaplana-Lopera, Nuria, Maxym Besh, and Eui Jung Moon. 2021. "Targeting Hypoxia: Revival of Old Remedies" Biomolecules 11, no. 11: 1604. https://doi.org/10.3390/biom11111604
APA StyleVilaplana-Lopera, N., Besh, M., & Moon, E. J. (2021). Targeting Hypoxia: Revival of Old Remedies. Biomolecules, 11(11), 1604. https://doi.org/10.3390/biom11111604