
Novel Structures of Type 1 Glyceraldehyde-3-phosphate Dehydrogenase from Escherichia coli Provide New Insights into the Mechanism of Generation of 1,3-Bisphosphoglyceric Acid

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Mutagenesis
2.2. EcGAPDH1 Mutants’ Protein Expression and Purification
2.3. Enzyme Activity
2.4. Crystallization and Data Collection
2.5. Structure Solution and Refinement
2.6. Secondary Structure-Based Align
3. Results
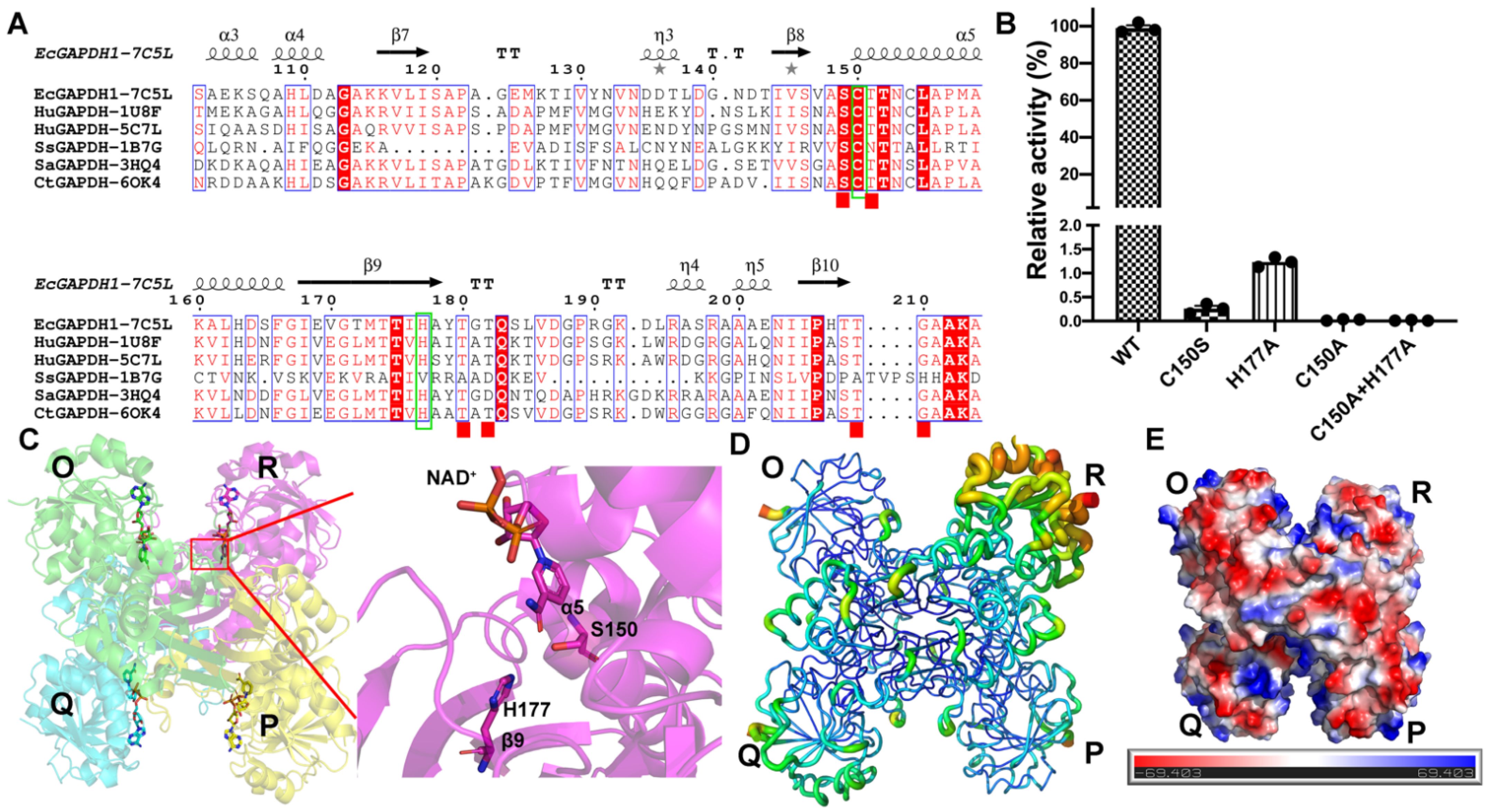
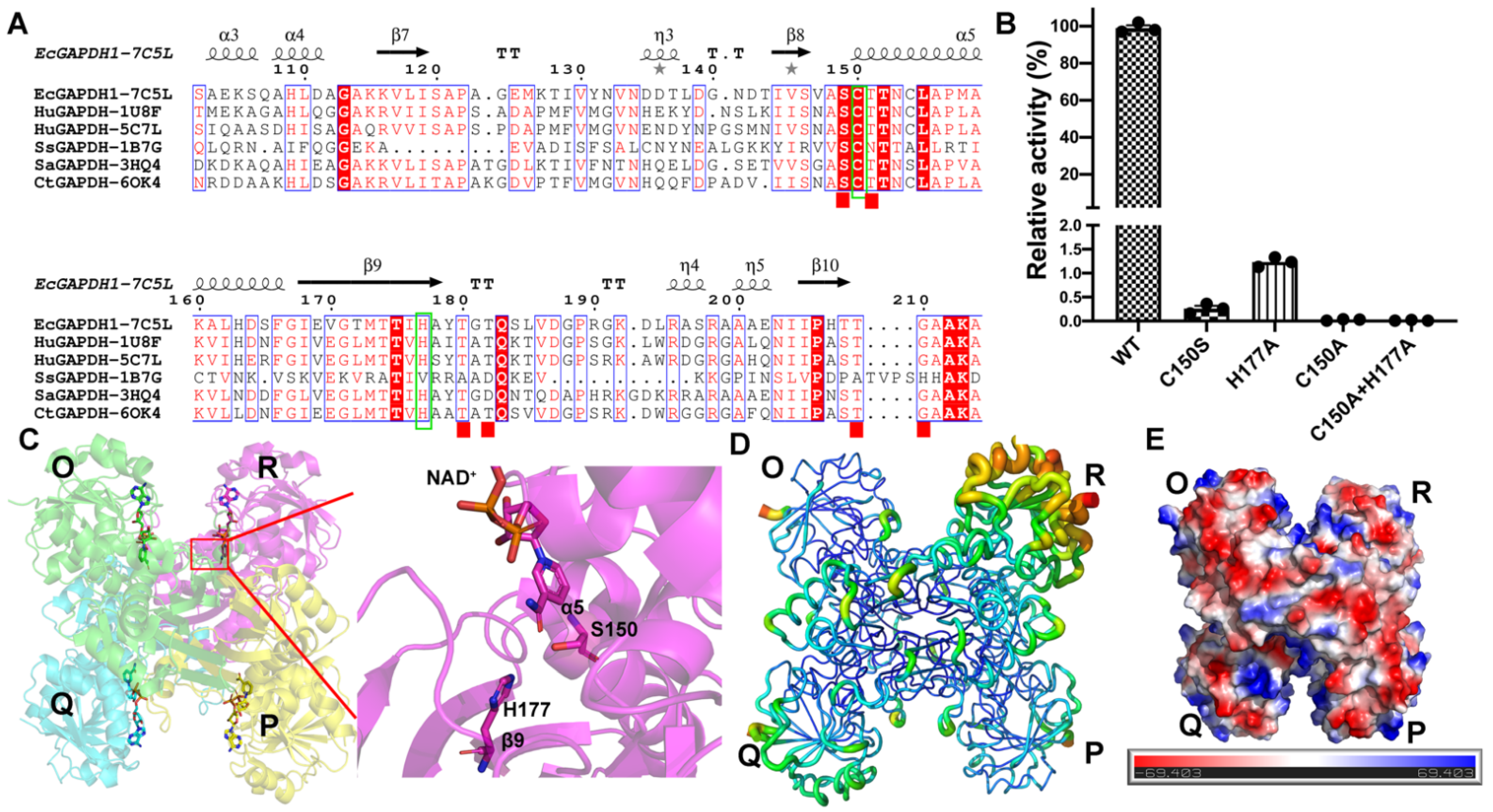
3.1. Protein Purification, Biochemical Properties, and Overall Structure of EcGAPDH1 Mutants
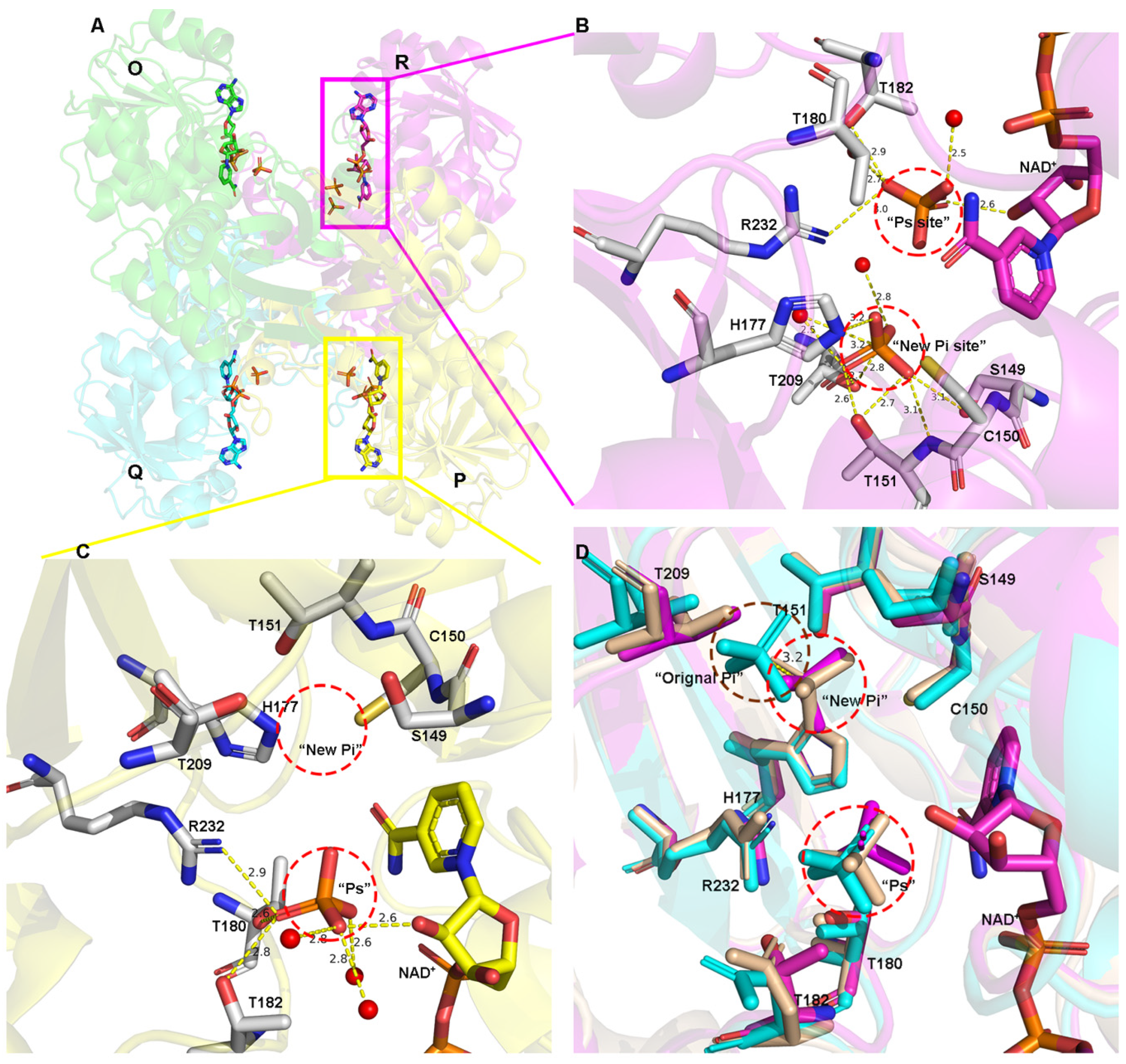
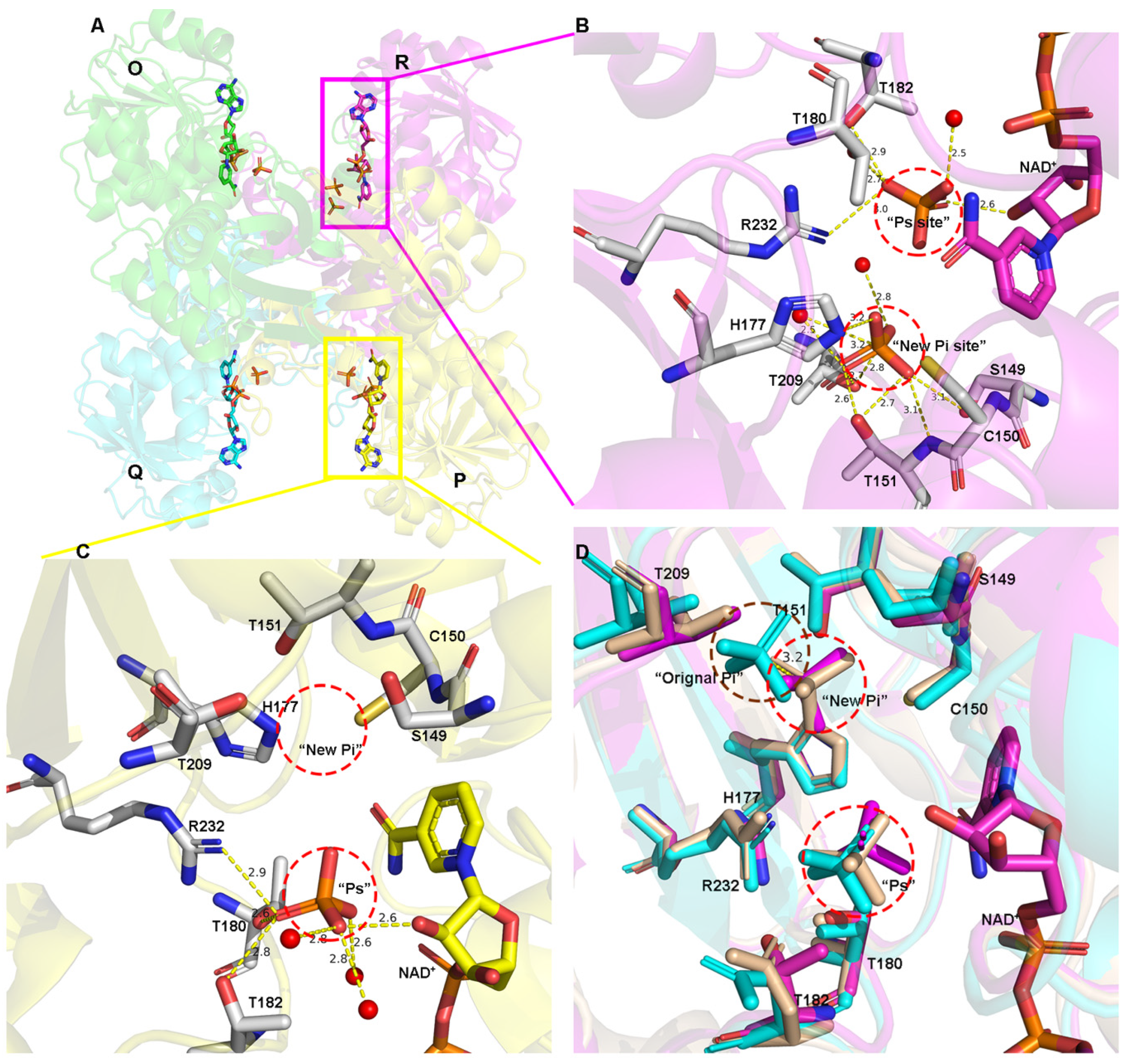
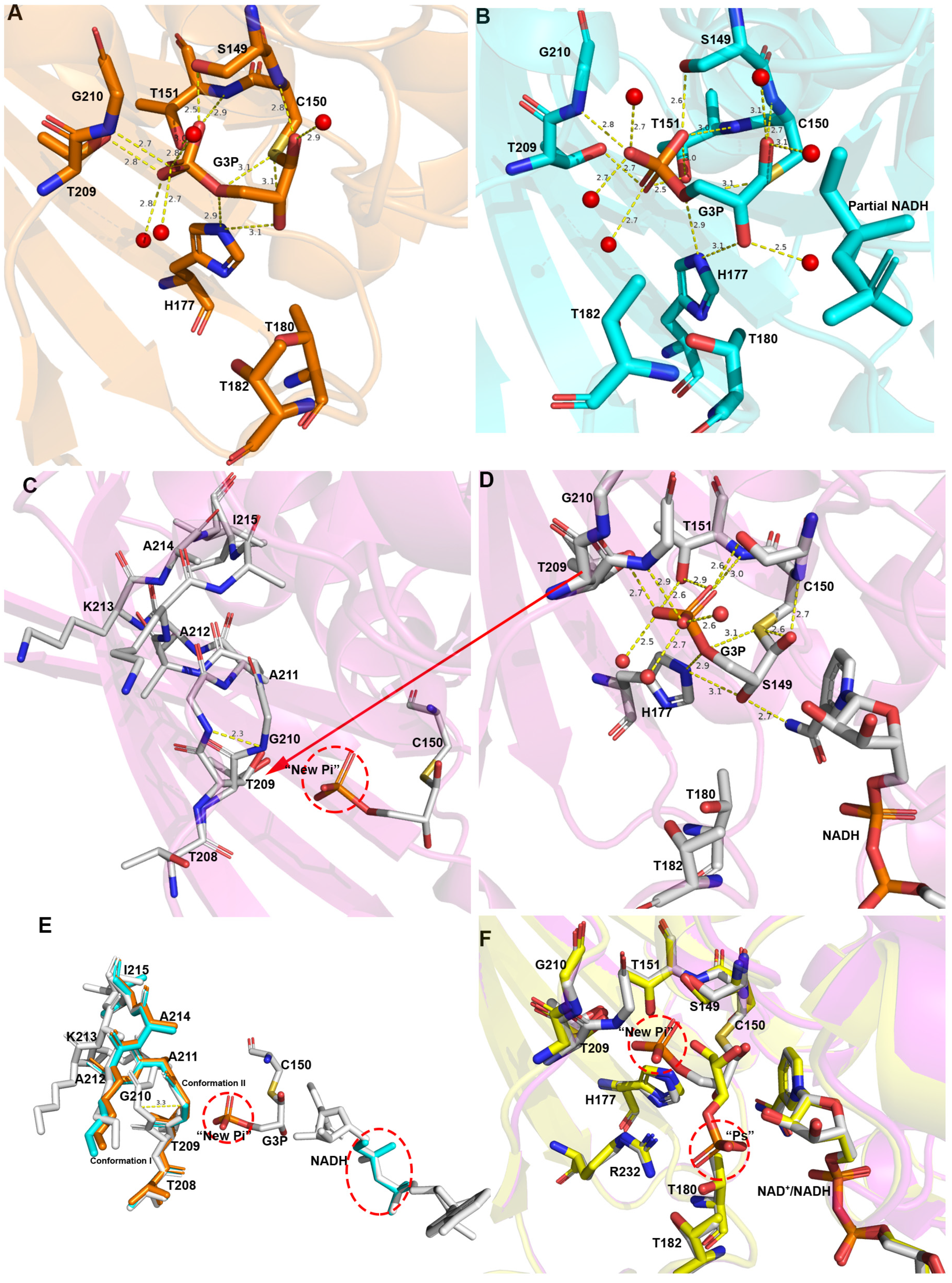
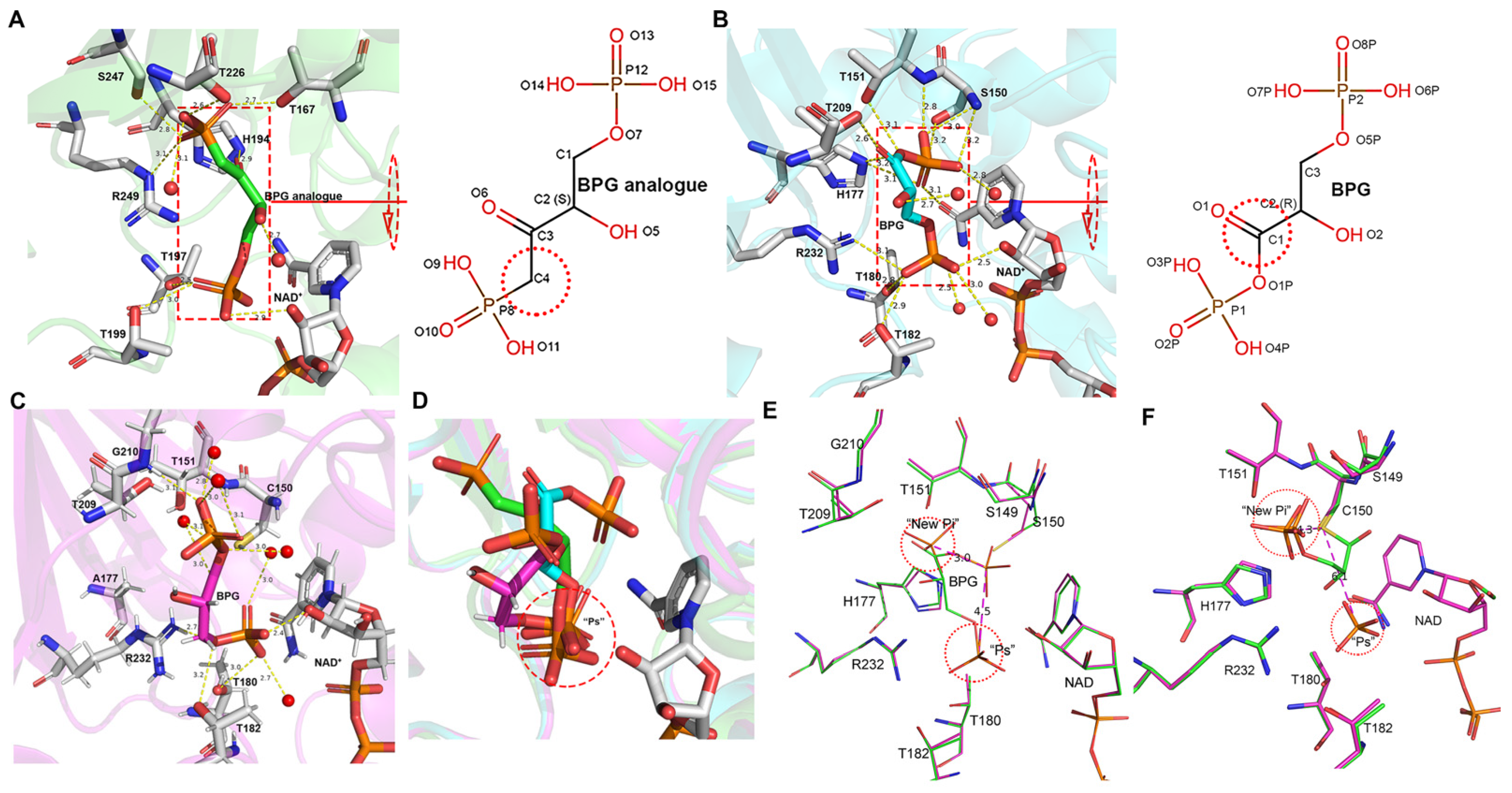
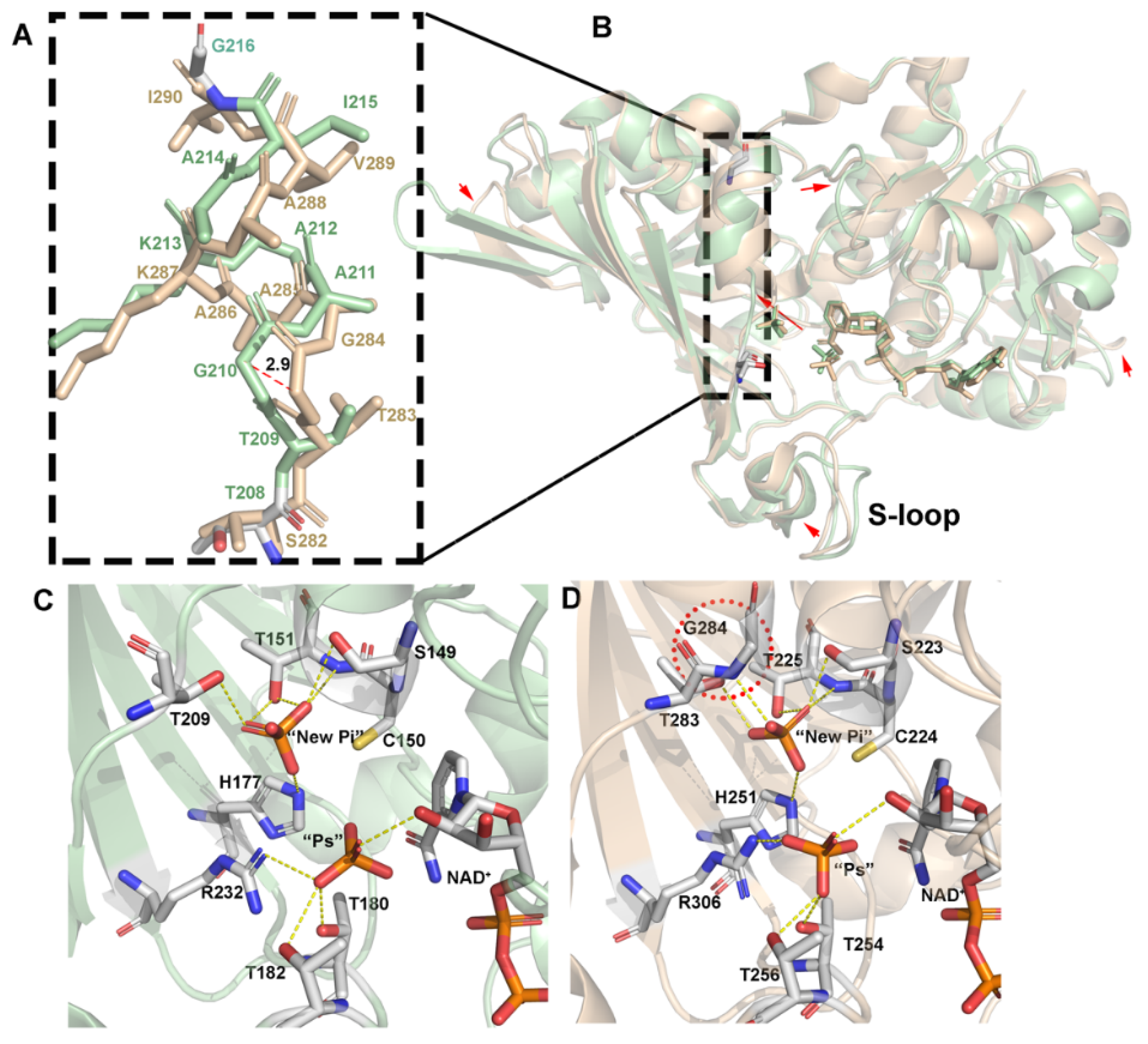
3.2. Inorganic Phosphate Binding: “New Pi” and “Ps” Site
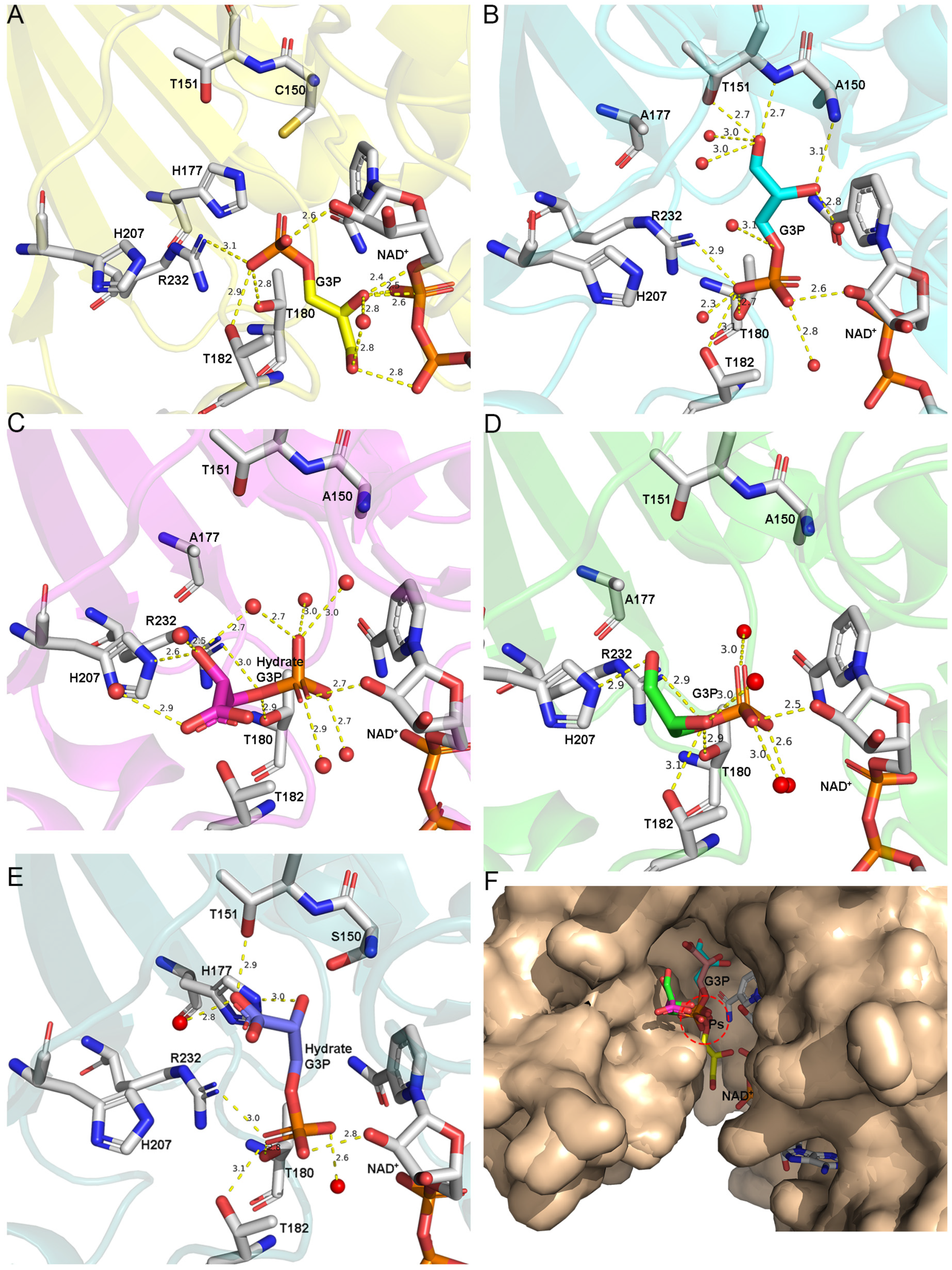
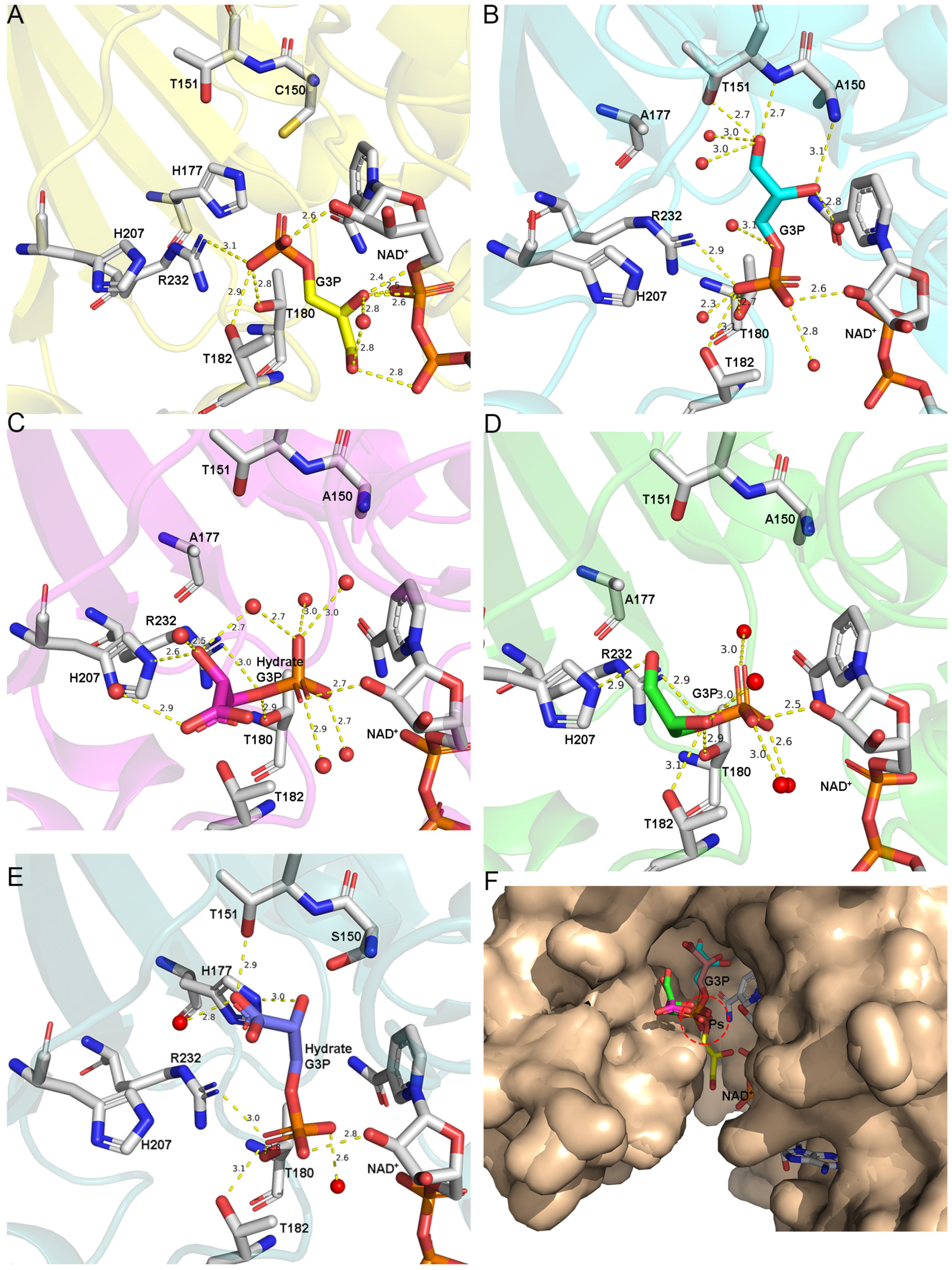
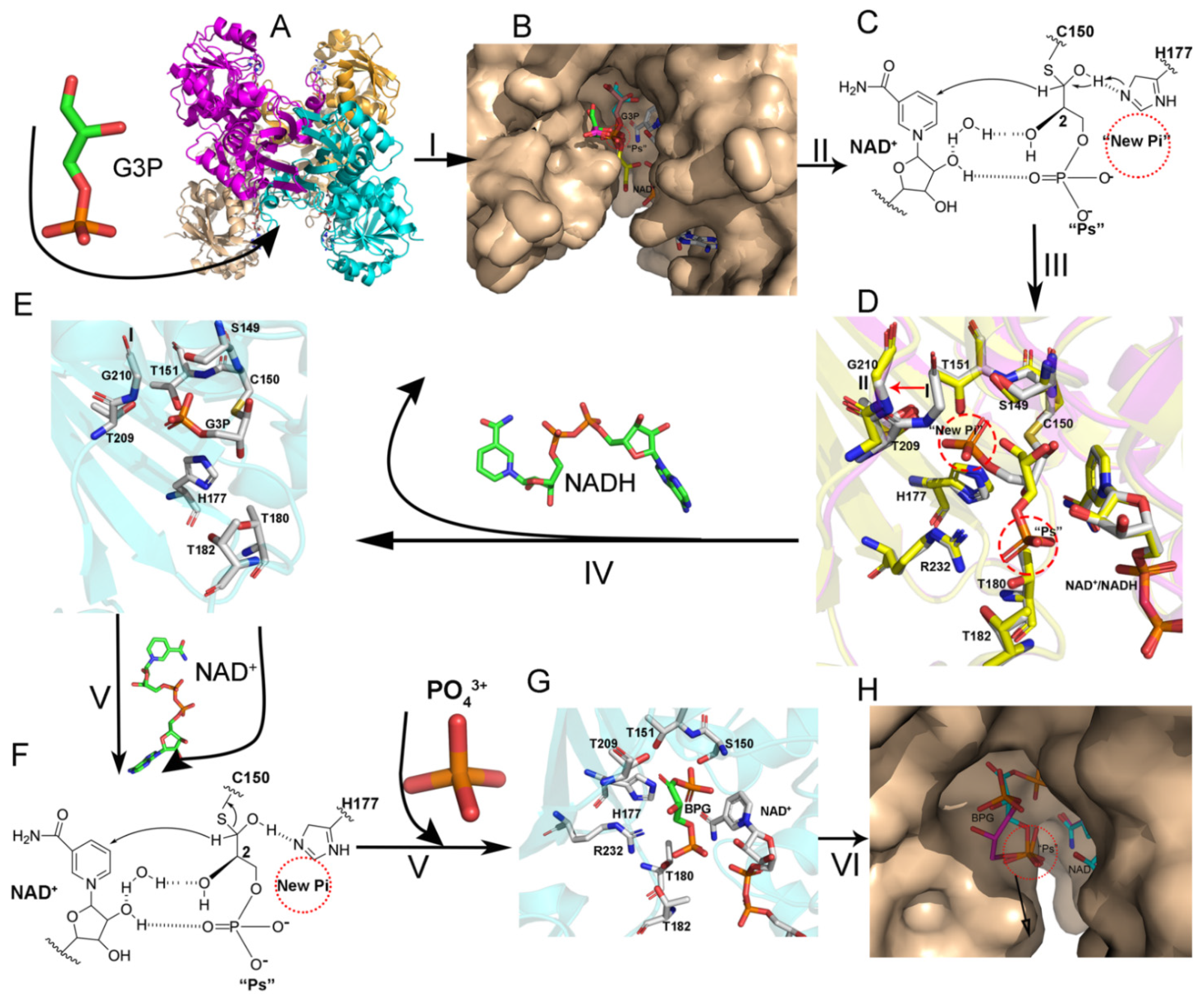
3.3. The Route Followed by G3P to Enter the Catalytic Domain
3.4. Structure of the Thioacyl Intermediate
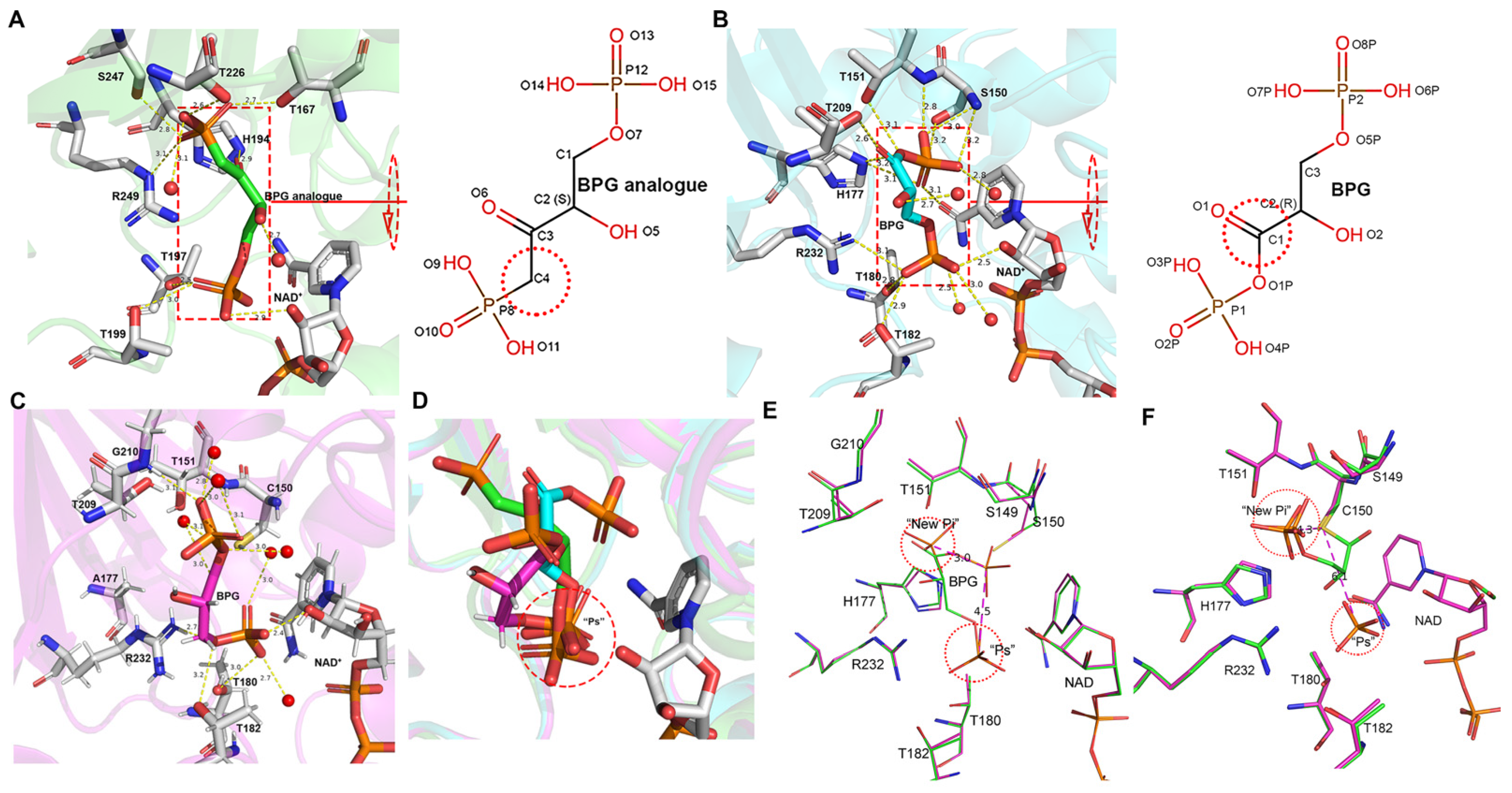
3.5. BPG Binding and Release in EcGAPDH1
4. Discussion
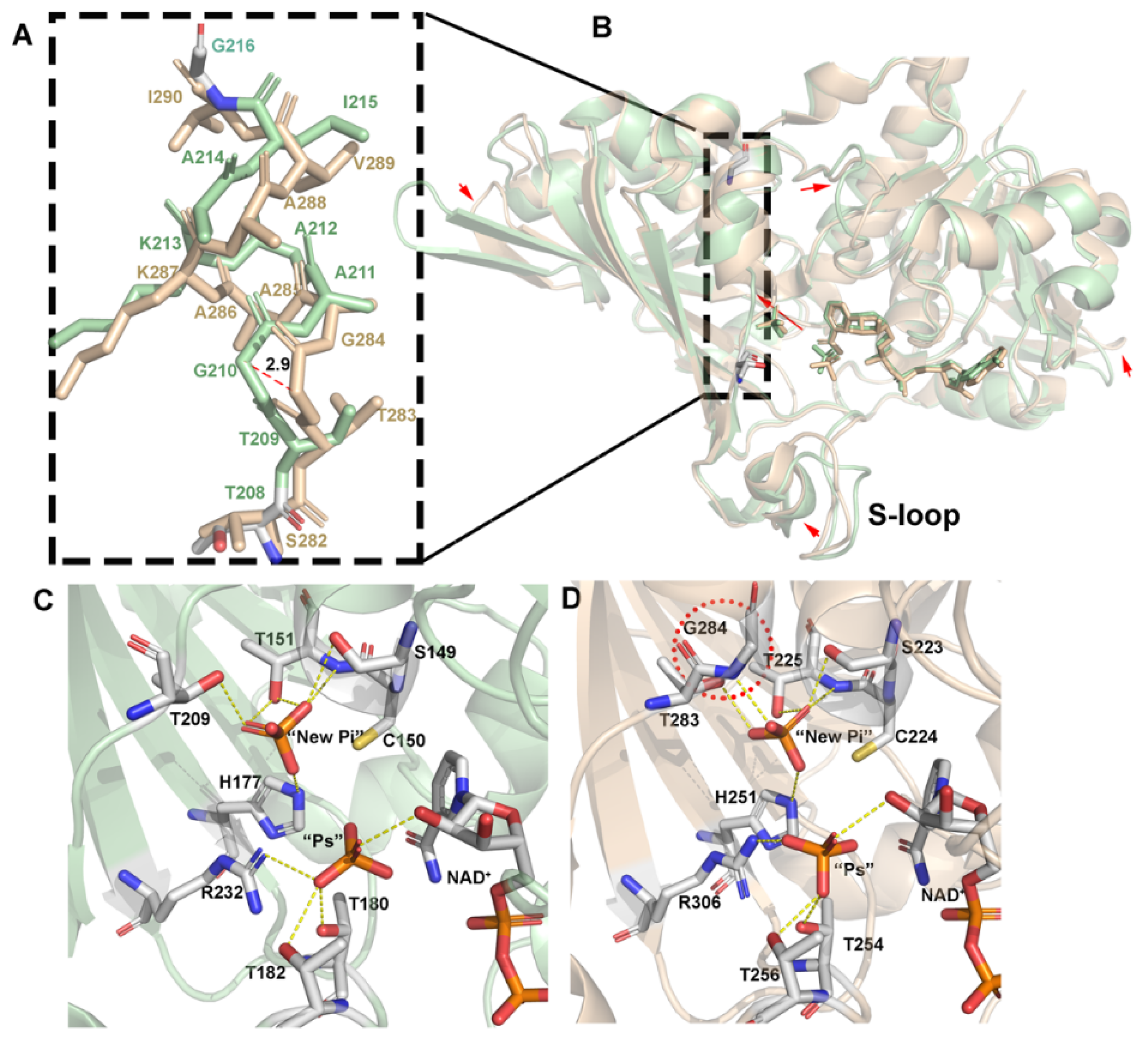
4.1. Structural Comparison of GAPDH from E. coli with Other Species
4.2. GAPDH Catalytic Process
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, M.D.; Boulton, A.A. Glyceraldehyde-3-phosphate dehydrogenase and apoptosis. J. Neurosci. Res. 2000, 60, 150–154. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Guzhova, I.V.; Margulis, B.A. Glyceraldehyde-3-phosphate Dehydrogenase Is a Multifaceted Therapeutic Target. Pharmaceutics 2020, 12, 416. [Google Scholar] [CrossRef]
- Egea, L.; Aguilera, L.; Giménez, R.; Sorolla, M.A.; Aguilar, J.; Badía, J.; Baldoma, L. Role of secreted glyceraldehyde-3-phosphate dehydrogenase in the infection mechanism of enterohemorrhagic and enteropathogenic Escherichia coli: Interaction of the extracellular enzyme with human plasminogen and fibrinogen. Int. J. Biochem. Cell Biol. 2007, 39, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Moriyón, I.; López-Goñi, I. Structure and properties of the outer membranes of Brucella abortus and Brucella melitensis. Int. Microbiol. 1998, 1, 19–26. [Google Scholar]
- Chaikuad, A.; Shafqat, N.; Al-Mokhtar, R.; Cameron, G.; Clarke, A.R.; Brady, R.L.; Oppermann, U.; Frayne, J.; Yue, W.W. Structure and kinetic characterization of human sperm-specific glyceraldehyde-3-phosphate dehydrogenase, GAPDS. Biochem. J. 2011, 435, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, W.D.; Winn, S.I.; Watson, H.C. Twinning in crystals of human skeletal muscle d-glyceraldehyde-3-phosphate dehydrogenase. J. Mol. Biol. 1976, 104, 277–283. [Google Scholar] [CrossRef]
- Park, J.B.; Park, H.; Son, J.; Ha, S.-J.; Cho, H.-S. Structural Study of Monomethyl Fumarate-Bound Human GAPDH. Mol. Cells 2019, 42, 597–603. [Google Scholar]
- Korndörfer, I.; Steipe, B.; Huber, R.; Tomschy, A.; Jaenicke, R. The crystal structure of holo-glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima at 2.5 A resolution. J. Mol. Biol. 1995, 246, 511–521. [Google Scholar] [CrossRef]
- Griffith, J.P.; Lee, B.; Murdock, A.L.; Amelunxen, R.E. Molecular symmetry of glyceraldehyde-3-phosphate dehydrogenase from Bacillus coagulans. J. Mol. Biol. 1983, 169, 963–974. [Google Scholar] [CrossRef]
- Zwickl, P.; Fabry, S.; Bogedain, C.; Haas, A.; Hensel, R. Glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic archaebacterium Pyrococcus woesei: Characterization of the enzyme, cloning and sequencing of the gene, and expression in Escherichia coli. J. Bacteriol. 1990, 172, 4329–4338. [Google Scholar] [CrossRef] [Green Version]
- Fabry, S.; Hensel, R. Purification and characterization of D-glyceraldehyde-3-phosphate dehydrogenase from the thermophilic archaebacterium Methanothermus fervidus. Eur. J. Biochem. 1987, 165, 147–155. [Google Scholar] [CrossRef]
- Ayres, C.A.; Schormann, N.; Senkovich, O.; Fry, A.; Banerjee, S.; Ulett, G.C.; Chattopadhyay, D. Structure of Streptococcus agalactiae glyceraldehyde-3-phosphate dehydrogenase holoenzyme reveals a novel surface. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 1333–1339. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J. A cryoprotectant induces conformational change in glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Park, C.-G.; Kim, J.-Y.; Park, H.-W. Structural Analysis of Glyceraldehyde 3-Phosphate Dehydrogenase from Escherichia coli: Direct Evidence of Substrate Binding and Cofactor-Induced Conformational Changes †,‡. Biochemistry 2000, 39, 10702–10710. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Dutta, D.; Saha, B.; Das, A.K. Crystal Structure of Glyceraldehyde-3-Phosphate Dehydrogenase 1 from Methicillin-Resistant Staphylococcus aureus MRSA252 Provides Novel Insights into Substrate Binding and Catalytic Mechanism. J. Mol. Biol. 2010, 401, 949–968. [Google Scholar] [CrossRef] [PubMed]
- Cook, W.J.; Senkovich, O.; Chattopadhyay, D. An unexpected phosphate binding site in glyceraldehyde 3-phosphate dehydrogenase: Crystal structures of apo, holo and ternary complex of Cryptosporidium parvum enzyme. BMC Struct. Biol. 2009, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladame, S.; Castilho, M.S.; Silva, C.H.T.P.; Denier, C.; Hannaert, V.; Périé, J.; Oliva, G.; Willson, M. Crystal structure of Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase complexed with an analogue of 1,3-bisphospho-d-glyceric acid. Eur. J. Biochem. 2003, 270, 4574–4586. [Google Scholar] [CrossRef]
- Isupov, M.N.; Fleming, T.M.; Dalby, A.R.; Crowhurst, G.S.; Bourne, P.C.; Littlechild, J.A. Crystal structure of the glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic archaeon Sulfolobus solfataricus. J. Mol. Biol. 1999, 291, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Skarzyński, T.; Moody, P.C.; Wonacott, A.J. Structure of holo-glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus at 1.8 A resolution. J. Mol. Biol. 1987, 193, 171–187. [Google Scholar] [CrossRef]
- Moniot, S.; Bruno, S.; Vonrhein, C.; Didierjean, C.; Boschi-Muller, S.; Vas, M.; Bricogne, G.; Branlant, G.; Mozzarelli, A.; Corbier, C. Trapping of the thioacylglyceraldehyde-3-phosphate dehydrogenase intermediate from Bacillus stearothermophilus. Direct evidence for a flip-flop mechanism. J. Biol. Chem. 2008, 283, 21693–21702. [Google Scholar] [CrossRef] [Green Version]
- Robien, M.A.; Bosch, J.; Buckner, F.S.; Van Voorhis, W.C.E.; Worthey, E.A.; Myler, P.; Mehlin, C.; Boni, E.E.; Kalyuzhniy, O.; Anderson, L.; et al. Crystal structure of glyceraldehyde-3-phosphate dehydrogenase from Plasmodium falciparum at 2.25 A resolution reveals intriguing extra electron density in the active site. Proteins 2006, 62, 570–577. [Google Scholar] [CrossRef]
- Wäneskog, M.; Bjerling, P. Multi-fragment site-directed mutagenic overlap extension polymerase chain reaction as a competitive alternative to the enzymatic assembly method. Anal. Biochem. 2014, 444, 32–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, M.R.; Yao, Y.C.; Bostrom, I.K.; Wang, Y.D.; Chen, A.Q.; Li, J.X.; Gu, S.H.; Ji, C.N. Characterization and structure of glyceraldehyde-3-phosphate dehydrogenase type 1 from Escherichia coli. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2020, 76, 406–413. [Google Scholar] [CrossRef]
- Ferdinand, W. The isolation and specific activity of rabbit-muscle glyceraldehyde phosphate dehydrogenase. Biochem. J. 1964, 92, 578–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Guo, Z.; Huang, J.; Liu, M.; Wang, Y.; Ji, C. Expression, purification, crystallization and preliminary X-ray crystallographic analysis of fructose-1,6-bisphosphate aldolase from Escherichia coli. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 1376–1379. [Google Scholar] [CrossRef] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol. 1997, 276, 307–326. [Google Scholar]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 1948–1954. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Chen, V.B.; Headd, J.J.; Moriarty, N.W.; Richardson, J.S.; Richardson, D.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. phenix.model_vs_data: A high-level tool for the calculation of crystallographic model and data statistics. J. Appl. Crystallogr. 2010, 43, 669–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Chistyakov, V.V.; Thornton, J.M. PDBsum more: New summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 2005, 33, D266–D268. [Google Scholar] [CrossRef] [Green Version]
- Westin, C.W.; Hanson, B.; Craig, P.A. Using PyMOL’s Align Feature to Create a Database of Ligand Binding Site Files for the Structural Analysis of Proteins. FASEB J. 2007, 21, A296. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Barrett, K.F.; Dranow, D.M.; Phan, I.Q.; Michaels, S.A.; Shaheen, S.; Navaluna, E.D.; Craig, J.K.; Tillery, L.M.; Choi, R.; Edwards, T.E.; et al. Structures of glyceraldehyde 3-phosphate dehydrogenase in Neisseria gonorrhoeae and Chlamydia trachomatis. Protein Sci. 2020, 29, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.L.; Tanner, J.J. High-resolution structure of human d-glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallography. Sect. D Biol. Crystallogr. 2006, 62, 290–301. [Google Scholar] [CrossRef]
- Danshina, P.V.; Qu, W.; Temple, B.R.; Rojas, R.J.; Miley, M.J.; Machius, M.; Betts, L.; O’Brien, D.A. Structural analyses to identify selective inhibitors of glyceraldehyde 3-phosphate dehydrogenase-S, a sperm-specific glycolytic enzyme. Mol. Hum. Reprod. 2016, 22, 410–426. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, P. Anion binding sites in protein structures. J. Mol. Biol. 1993, 234, 463–482. [Google Scholar] [CrossRef]
- Copley, R.R.; Barton, G.J. A structural analysis of phosphate and sulphate binding sites in proteins. Estimation of propensities for binding and conservation of phosphate binding sites. J. Mol. Biol. 1994, 242, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Kellogg, G.E. A Novel and Efficient Tool for Locating and Characterizing Protein Cavities and Binding Sites. Proteins 2010, 78, 825–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didierjean, C.; Corbier, C.; Fatih, M.; Favier, F.; Boschi-Muller, S.; Branlant, G.; Aubry, A. Crystal Structure of Two Ternary Complexes of Phosphorylating Glyceraldehyde-3-phosphate Dehydrogenase from Bacillus stearothermophilus with NAD and d-Glyceraldehyde 3-Phosphate. J. Biol. Chem. 2003, 278, 12968–12976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L. Using Dali for Protein Structure Comparison. Methods Mol. Biol. 2020, 2112, 29–42. [Google Scholar] [PubMed]
- Querol-García, J.; Fernández, F.J.; Marin, A.V.; Gómez, S.; Fullà, D.; Melchor-Tafur, C.; Franco-Hidalgo, V.; Albertí, S.; Juanhuix, J.; Rodríguez de Córdoba, S.; et al. Crystal Structure of Glyceraldehyde-3-Phosphate Dehydrogenase from the Gram-Positive Bacterial Pathogen A. vaginae, an Immunoevasive Factor that Interacts with the Human C5a Anaphylatoxin. Front. Microbiol. 2017, 8, 541. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ternary Complex | Thioacyl Intermediate | Ternary Complex | |||||
|---|---|---|---|---|---|---|---|
| WT.NAD.PO4 | WT.NAD.G3P | C150S.NAD.G3P | C150AH177A.NAD.G3P | C150S.NAD.BPG | H177A.NAD.BPG | ||
| Data collection | |||||||
| PDB code | 7C5H | 7C5P | 7C5K | 7C5M | 7C7K | 7C5R | 7C5Q |
| Wavelength (Å) | 0.97776 | 0.97776 | 0.97776 | 0.97853 | 0.97776 | 0.97776 | 0.97776 |
| Resolution range (Å) a | 50–2.09 (2.13–2.09) | 50–2.35 (2.39–2.35) | 50–2.68 (2.73–2.68) | 50–1.8 (1.83–1.8) | 50–1.77 (1.8–1.77) | 50–2.31 (2.35–2.31) | 50–2.13 (2.17–2.13) |
| Space group | P41212 | ||||||
| Unit cell parameters (Å°) | a = b = 89.778, c = 340.953, α = β = γ = 90 | a = b = 90.33, c = 341.547, α = β = γ = 90 | a = b = 90.378, c = 342.107, α = β = γ = 90 | a = b = 89.925, c = 342.379, α = β=γ = 90 | a = b = 90.16, c = 345.059, α = β = γ = 90 | a = b = 89.398, c = 340.821, α = β = γ = 90 | a = b = 89.674, c = 341.795, α = β = γ = 90 |
| Completeness (%) | 99.7 (99.3) | 100 (100) | 100 (100) | 100 (100) | 99.8 (98.9) | 100 (100) | 100 (100) |
| Rmerge (%) b | 15.4 (60.4) | 19 (62.4) | 13.5 (48.7) | 11.8 (75.6) | 10.9 (60.3) | 16.6 (53.5) | 15.3 (45.9) |
| Mean I/δ | 13.3 (3.14) | 14 (3.71) | 15.17 (3.6) | 22.5 (3.0) | 35.87 (4.75) | 12.5 (3.33) | 17.75 (5) |
| No. unique reflections | 83,301 | 60,156 | 41,105 | 132,236 | 139,316 | 61,720 | 79,577 |
| Redundancy | 13 | 11.3 | 12.8 | 12.6 | 26.6 | 16.6 | 19.4 |
| Refinement | |||||||
| Resolution range (Å) | 39.87–2.09 (2.17–2.09) | 48.16–2.35 (2.43–2.35) | 48.22–2.68 (2.78–2.68) | 44.96–1.8 (1.86–1.8) | 48.49–1.77 (1.83–1.77) | 47.94–2.31 (2.39–2.31) | 48.08–2.13 (2.21–2.13) |
| Rwork/Rfree (%) c | 15.81/20.23 | 16.31/20.83 | 16.8/24.1 | 15.34/19.04 | 15.37/18.42 | 15.56/20.70 | 15.35/19.71 |
| Wilson B-factor (Å2) | 19.7 | 30.3 | 36 | 22.66 | 21.38 | 26.92 | 21.84 |
| Average B factor(Å2) | 21.97 | 32.35 | 36.51 | 26.26 | 24.24 | 28.94 | 23.89 |
| B factor (Å2) | |||||||
| Protein | 21.59 | 32.48 | 36.89 | 25 | 23.17 | 28.72 | 23.3 |
| Water | 25.62 | 30.73 | 27.51 | 34.48 | 31.99 | 30.61 | 28.4 |
| NAD+, PO4, G3P, BPG | 24.21 | 31.13 | 38.8 | 28.59 | 32.69 | 31.52 | 29.63 |
| Number of NAD/PO4/G3P/BPG | 4/5/0/0 | 4/0/4/0 | 4/0/4/0 | 4/0/4/0 | 1/0/4/0 | 4/2/2/1 | 4/0/3/1 |
| Number of water molecules | 796 | 575 | 497 | 1477 | 1158 | 905 | 950 |
| RMSD bond lengths (Å) | 0.007 | 0.014 | 0.0114 | 0.019 | 0.02 | 0.015 | 0.018 |
| RMSD bond angles (deg) | 0.984 | 1.68 | 1.608 | 1.89 | 2.03 | 1.7 | 1.88 |
| Ramachandran plot (%) | |||||||
| Favored | 97 | 96 | 95 | 96 | 97 | 96 | 96 |
| Outliers | 0.15 | 0.075 | 0 | 0 | 0 | 0.075 | 0 |
| Rotamer outliers (%) | 1.1 | 0.9 | 1.1 | 0.55 | 0.5 | 0.54 | 0.82 |
| Clash core | 2.64 | 3.07 | 1.64 | 2.26 | 4.38 | 2.39 | 2.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Liu, M.; Bao, L.; Boström, K.I.; Yao, Y.; Li, J.; Gu, S.; Ji, C. Novel Structures of Type 1 Glyceraldehyde-3-phosphate Dehydrogenase from Escherichia coli Provide New Insights into the Mechanism of Generation of 1,3-Bisphosphoglyceric Acid. Biomolecules 2021, 11, 1565. https://doi.org/10.3390/biom11111565
Zhang L, Liu M, Bao L, Boström KI, Yao Y, Li J, Gu S, Ji C. Novel Structures of Type 1 Glyceraldehyde-3-phosphate Dehydrogenase from Escherichia coli Provide New Insights into the Mechanism of Generation of 1,3-Bisphosphoglyceric Acid. Biomolecules. 2021; 11(11):1565. https://doi.org/10.3390/biom11111565
Chicago/Turabian StyleZhang, Li, Meiruo Liu, Luyao Bao, Kristina I. Boström, Yucheng Yao, Jixi Li, Shaohua Gu, and Chaoneng Ji. 2021. "Novel Structures of Type 1 Glyceraldehyde-3-phosphate Dehydrogenase from Escherichia coli Provide New Insights into the Mechanism of Generation of 1,3-Bisphosphoglyceric Acid" Biomolecules 11, no. 11: 1565. https://doi.org/10.3390/biom11111565
APA StyleZhang, L., Liu, M., Bao, L., Boström, K. I., Yao, Y., Li, J., Gu, S., & Ji, C. (2021). Novel Structures of Type 1 Glyceraldehyde-3-phosphate Dehydrogenase from Escherichia coli Provide New Insights into the Mechanism of Generation of 1,3-Bisphosphoglyceric Acid. Biomolecules, 11(11), 1565. https://doi.org/10.3390/biom11111565