
RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma



Abstract
:1. Introduction
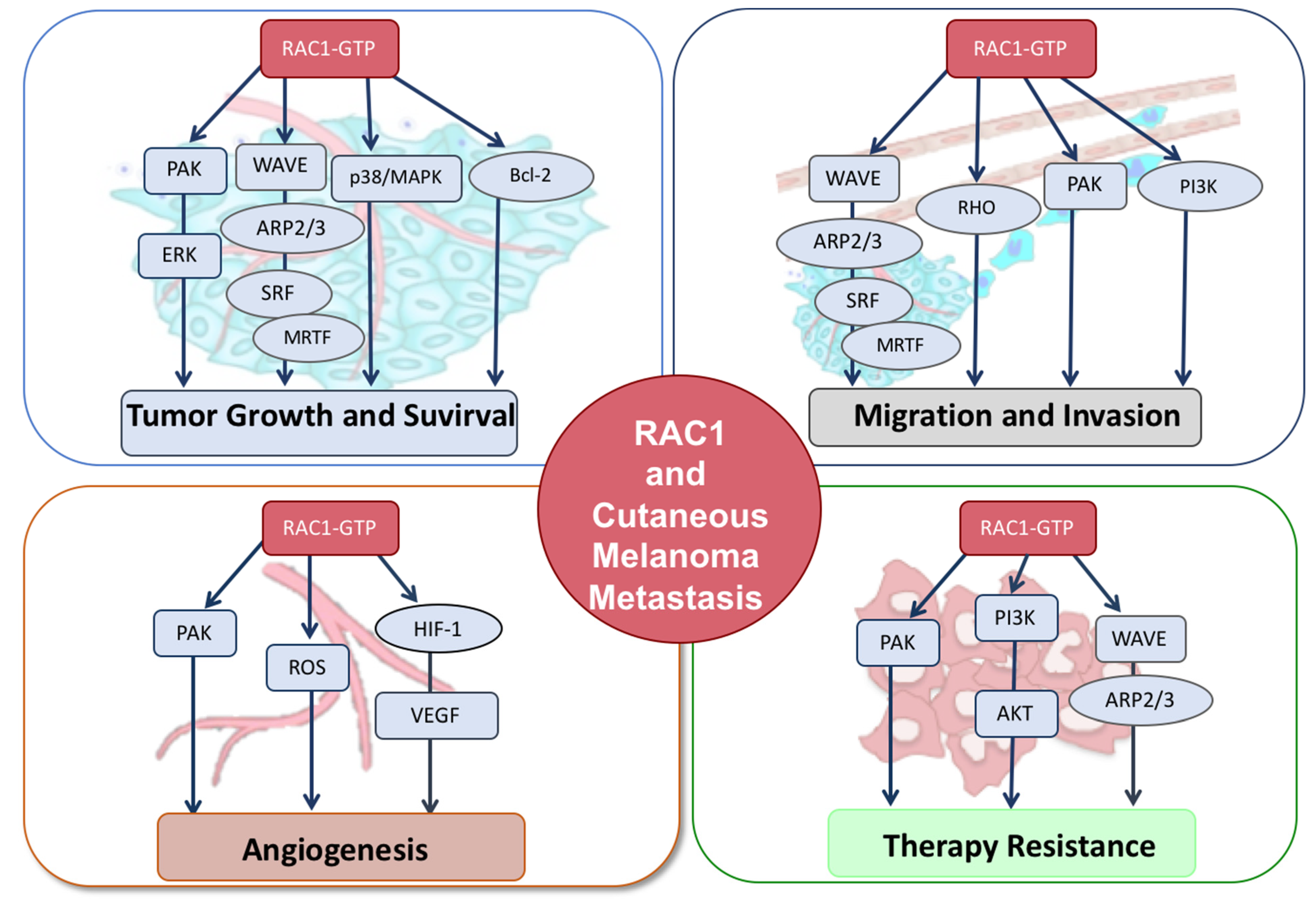
2. RAC1 Pathway Activation in Melanoma Formation
3. RAC1 Signaling in Tumor Cell Migration and Invasion
4. RAC1 Signaling in Angiogenesis
5. RAC1 Targeting Therapies and Therapy Resistance
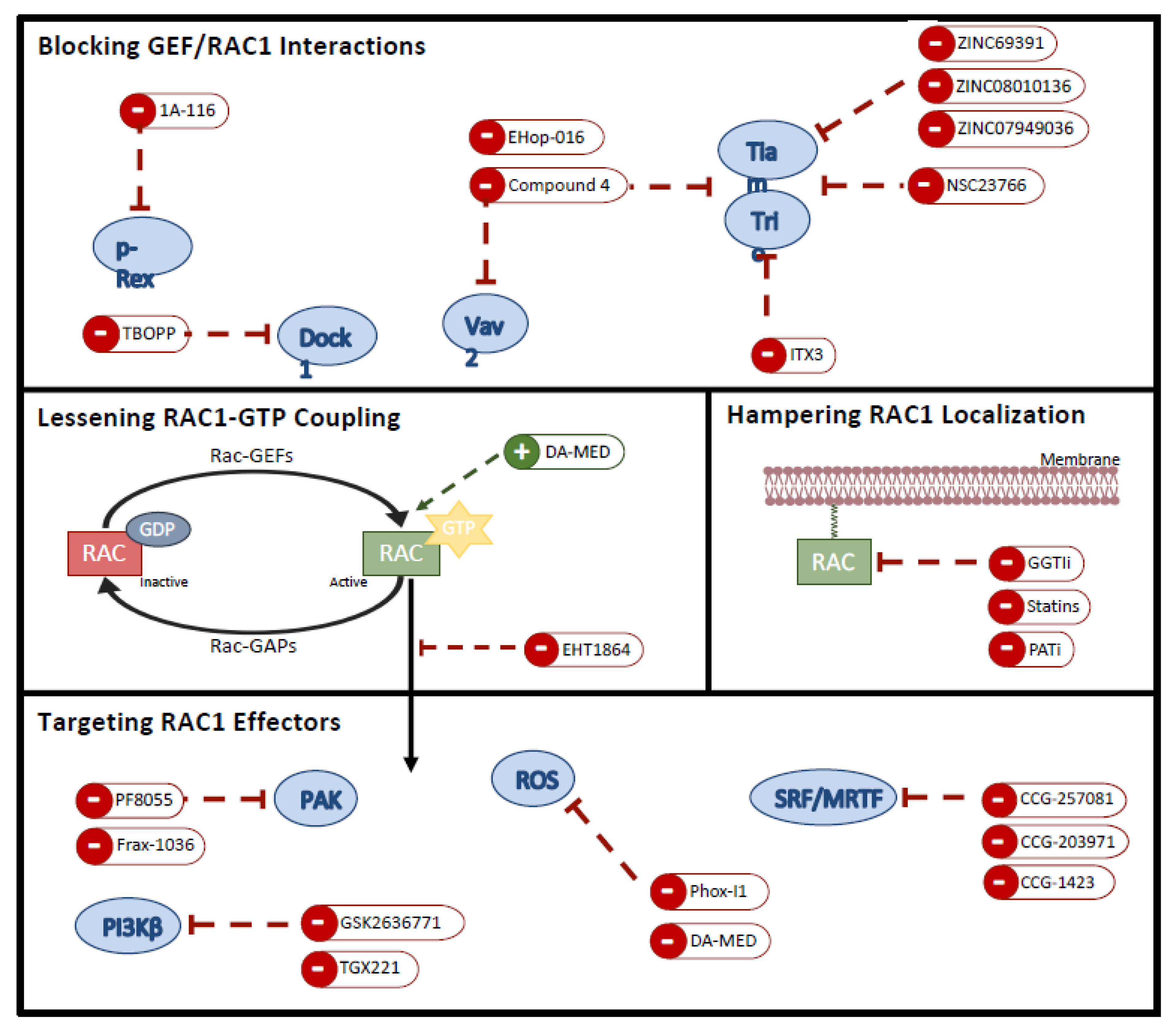
5.1. Preventing RAC1 Localization
5.2. Hampering Nucleotide Coupling
5.3. Blocking GEF/RAC1 Interactions
5.4. Targeting RAC1 Effectors
5.5. Therapy Resistance
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Greene, F.L.; Sobin, L.H. The staging of cancer: A retrospective and prospective appraisal. CA Cancer J. Clin. 2008, 58, 180–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol. Cancer Ther. 2013, 12, 1925–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshfegh, Y.; Bravo-Cordero, J.J.; Miskolci, V.; Condeelis, J.; Hodgson, L. A Trio-Rac1-Pak1 signalling axis drives invadopodia disassembly. Nat. Cell Biol. 2014, 16, 574–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, M.F. Rho GTPases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases 2018, 9, 203–215. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef]
- Mar, V.J.; Wong, S.Q.; Logan, A.; Nguyen, T.; Cebon, J.; Kelly, J.W.; Wolfe, R.; Dobrovic, A.; McLean, C.; McArthur, G.A. Clinical and pathological associations of the activating RAC1 P29S mutation in primary cutaneous melanoma. Pigment Cell Melanoma Res. 2014, 27, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Watson, I.R.; Li, L.; Cabeceiras, P.K.; Mahdavi, M.; Gutschner, T.; Genovese, G.; Wang, G.; Fang, Z.; Tepper, J.M.; Stemke-Hale, K.; et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014, 74, 4845–4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, H.L.; Rosenbaum, S.; Purwin, T.J.; Davies, M.A.; Aplin, A.E. RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015, 28, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, C.R.; Li, A.; Faller, W.; Ozanne, B.; Welch, H.; Machesky, L.M.; Sansom, O.J. A Rac1-independent role for P-Rex1 in melanoblasts. J. Investig. Dermatol. 2015, 135, 314–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [Green Version]
- Wertheimer, E.; Gutierrez-Uzquiza, A.; Rosemblit, C.; Lopez-Haber, C.; Sosa, M.S.; Kazanietz, M.G. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell Signal 2012, 24, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Mack, N.A.; Whalley, H.J.; Castillo-Lluva, S.; Malliri, A. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle 2011, 10, 1571–1581. [Google Scholar] [CrossRef] [Green Version]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.; Monboisse, J. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef] [Green Version]
- Revach, O.Y.; Winograd-Katz, S.E.; Samuels, Y.; Geiger, B. The involvement of mutant Rac1 in the formation of invadopodia in cultured melanoma cells. Exp. Cell Res. 2016, 343, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [Green Version]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Araiza-Olivera, D.; Feng, Y.; Semenova, G.; Prudnikova, T.Y.; Rhodes, J.; Chernoff, J. Suppression of RAC1-driven malignant melanoma by group A PAK inhibitors. Oncogene 2018, 37, 944–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Lerida, I.; Pellinen, T.; Sanchez, S.A.; Guadamillas, M.C.; Wang, Y.; Mirtti, T.; Calvo, E.; Del Pozo, M.A. Rac1 nucleocytoplasmic shuttling drives nuclear shape changes and tumor invasion. Dev. Cell 2015, 32, 318–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Duijn, T.J.; Anthony, E.C.; Hensbergen, P.J.; Deelder, A.M.; Hordijk, P.L. Rac1 recruits the adapter protein CMS/CD2AP to cell-cell contacts. J. Biol. Chem. 2010, 285, 20137–20146. [Google Scholar] [CrossRef] [Green Version]
- Nouri, K.; Fansa, E.K.; Amin, E.; Dvorsky, R.; Gremer, L.; Willbold, D.; Schmitt, L.; Timson, D.J.; Ahmadian, M.R. IQGAP1 Interaction with RHO Family Proteins Revisited: KINETIC AND EQUILIBRIUM EVIDENCE FOR MULTIPLE DISTINCT BINDING SITES. J. Biol. Chem. 2016, 291, 26364–26376. [Google Scholar] [CrossRef] [Green Version]
- Nohata, N.; Uchida, Y.; Stratman, A.N.; Adams, R.H.; Zheng, Y.; Weinstein, B.M.; Mukouyama, Y.S.; Gutkind, J.S. Temporal-specific roles of Rac1 during vascular development and retinal angiogenesis. Dev. Biol. 2016, 411, 183–194. [Google Scholar] [CrossRef]
- Goel, H.L.; Pursell, B.; Shultz, L.D.; Greiner, D.L.; Brekken, R.A.; Vander Kooi, C.W.; Mercurio, A.M. P-Rex1 Promotes Resistance to VEGF/VEGFR-Targeted Therapy in Prostate Cancer. Cell Rep. 2016, 14, 2193–2208. [Google Scholar] [CrossRef] [Green Version]
- Low, I.C.; Loh, T.; Huang, Y.; Virshup, D.M.; Pervaiz, S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56δ stabilizes its antiapoptotic activity. Blood 2014, 124, 2223–2234. [Google Scholar] [CrossRef]
- Chong, S.J.F.; Lai, J.X.H.; Qu, J.; Hirpara, J.; Kang, J.; Swaminathan, K.; Loh, T.; Kumar, A.; Vali, S.; Abbasi, T.; et al. A feedforward relationship between active Rac1 and phosphorylated Bcl-2 is critical for sustaining Bcl-2 phosphorylation and promoting cancer progression. Cancer Lett. 2019, 457, 151–167. [Google Scholar] [CrossRef]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Bahirwani, R.; Ghabril, M.; Forde, K.A.; Chatrath, H.; Wolf, K.M.; Uribe, L.; Reddy, K.R.; Fuchs, B.; Chalasani, N. Factors that predict short-term intensive care unit mortality in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 2013, 11, 1194–1200.e2. [Google Scholar] [CrossRef] [Green Version]
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1. Cancer Cell 2019, 36, 68–83.e69. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Fu, M.; Wang, L.; Liu, J.; Li, Y.; Brakebusch, C.; Mei, Q. p21-activated kinase 1 (PAK1) can promote ERK activation in a kinase-independent manner. J. Biol. Chem. 2013, 288, 20093–20099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larribère, L.; Cakrapradipta Wibowo, Y.; Patil, N.; Abba, M.; Tundidor, I.; Aguiñón Olivares, R.G.; Allgayer, H.; Utikal, J. NF1-RAC1 axis regulates migration of the melanocytic lineage. Transl. Oncol. 2020, 13, 100858. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Kurisu, S.; Suetsugu, S.; Yamazaki, D.; Yamaguchi, H.; Takenawa, T. Rac-WAVE2 signaling is involved in the invasive and metastatic phenotypes of murine melanoma cells. Oncogene 2005, 24, 1309–1319. [Google Scholar] [CrossRef] [Green Version]
- Clayton, N.S.; Ridley, A.J. Targeting Rho GTPase Signaling Networks in Cancer. Front. Cell Dev. Biol. 2020, 8, 222. [Google Scholar] [CrossRef]
- Melendez, J.; Memtsa, M.; Stavroulis, A.; Fakokunde, A.; Yoong, W. The best way to determine the best way to undertake a hysterectomy. BJOG 2009, 116, 1539–1540. [Google Scholar] [CrossRef]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef]
- Wang, J.; Hirose, H.; Du, G.; Chong, K.; Kiyohara, E.; Witz, I.P.; Hoon, D.S.B. P-REX1 amplification promotes progression of cutaneous melanoma via the PAK1/P38/MMP-2 pathway. Cancer Lett. 2017, 407, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kempers, L.; Driessen, A.J.M.; van Rijssel, J.; Nolte, M.A.; van Buul, J.D. The RhoGEF Trio: A Protein with a Wide Range of Functions in the Vascular Endothelium. Int. J. Mol. Sci. 2021, 22, 10168. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.S.; Dean, K.M.; Isogai, T.; Kasitinon, S.Y.; Murali, V.S.; Roudot, P.; Groisman, A.; Reed, D.K.; Welf, E.S.; Han, S.J.; et al. Enhanced Dendritic Actin Network Formation in Extended Lamellipodia Drives Proliferation in Growth-Challenged Rac1. Dev. Cell 2019, 49, 444–460.e9. [Google Scholar] [CrossRef] [PubMed]
- Mani, T.; Hennigan, R.F.; Foster, L.A.; Conrady, D.G.; Herr, A.B.; Ip, W. FERM domain phosphoinositide binding targets merlin to the membrane and is essential for its growth-suppressive function. Mol. Cell Biol. 2011, 31, 1983–1996. [Google Scholar] [CrossRef] [Green Version]
- Colón-Bolea, P.; García-Gómez, R.; Shackleton, S.; Crespo, P.; Bustelo, X.R.; Casar, B. RAC1 induces nuclear alterations through the LINC complex to enhance melanoma invasiveness. Mol. Biol. Cell 2020, 31, 2768–2778. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Dey, N.; Crosswell, H.E.; De, P.; Parsons, R.; Peng, Q.; Su, J.D.; Durden, D.L. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 2008, 68, 1862–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, L.S.; Leng, J.; Schwartz, M.A.; Bokoch, G.M. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell 1998, 9, 1863–1871. [Google Scholar] [CrossRef] [Green Version]
- Soga, N.; Connolly, J.O.; Chellaiah, M.; Kawamura, J.; Hruska, K.A. Rac regulates vascular endothelial growth factor stimulated motility. Cell Commun. Adhes. 2001, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Palmby, T.R.; Gavard, J.; Amornphimoltham, P.; Zheng, Y.; Gutkind, J.S. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008, 22, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rigamonti, D.; Badr, A.; Zhang, J. Ccm1 regulates microvascular morphogenesis during angiogenesis. J. Vasc. Res. 2011, 48, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nelson, W.J. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J. Cell Biol. 2007, 178, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Connolly, J.O.; Simpson, N.; Hewlett, L.; Hall, A. Rac regulates endothelial morphogenesis and capillary assembly. Mol. Biol. Cell 2002, 13, 2474–2485. [Google Scholar] [CrossRef] [Green Version]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Bryan, B.A.; D’Amore, P.A. What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell Mol. Life Sci. 2007, 64, 2053–2065. [Google Scholar] [CrossRef]
- Turcotte, S.; Desrosiers, R.R.; Béliveau, R. HIF-1alpha mRNA and protein upregulation involves Rho GTPase expression during hypoxia in renal cell carcinoma. J. Cell Sci. 2003, 116, 2247–2260. [Google Scholar] [CrossRef] [Green Version]
- Draper, J.M.; Smith, C.D. Palmitoyl acyltransferase assays and inhibitors (Review). Mol. Membr. Biol. 2009, 26, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavda, B.; Arnott, J.A.; Planey, S.L. Targeting protein palmitoylation: Selective inhibitors and implications in disease. Expert Opin. Drug Discov. 2014, 9, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzaneh Behelgardi, M.; Zahri, S.; Gholami Shahvir, Z.; Mashayekhi, F.; Mirzanejad, L.; Asghari, S.M. Targeting signaling pathways of VEGFR1 and VEGFR2 as a potential target in the treatment of breast cancer. Mol. Biol. Rep. 2020, 47, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; van der Meel, R.; Symons, M.H.; Fens, M.H.; Pieters, E.; Wilschut, K.J.; Storm, G.; Jarzabek, M.; Gallagher, W.M.; Schiffelers, R.M.; et al. Examining the role of Rac1 in tumor angiogenesis and growth: A clinically relevant RNAi-mediated approach. Angiogenesis 2011, 14, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol. Cell Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef]
- Yamaoka-Tojo, M.; Ushio-Fukai, M.; Hilenski, L.; Dikalov, S.I.; Chen, Y.E.; Tojo, T.; Fukai, T.; Fujimoto, M.; Patrushev, N.A.; Wang, N.; et al. IQGAP1, a novel vascular endothelial growth factor receptor binding protein, is involved in reactive oxygen species--dependent endothelial migration and proliferation. Circ. Res. 2004, 95, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ramshekar, A.; Kunz, E.; Sacks, D.B.; Hartnett, M.E. IQGAP1 causes choroidal neovascularization by sustaining VEGFR2-mediated Rac1 activation. Angiogenesis 2020, 23, 685–698. [Google Scholar] [CrossRef]
- Bu, F.; Min, J.W.; Munshi, Y.; Lai, Y.J.; Qi, L.; Urayama, A.; McCullough, L.D.; Li, J. Activation of endothelial ras-related C3 botulinum toxin substrate 1 (Rac1) improves post-stroke recovery and angiogenesis via activating Pak1 in mice. Exp. Neurol. 2019, 322, 113059. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Cannon, A.C.; Uribe-Alvarez, C.; Chernoff, J. RAC1 as a Therapeutic Target in Malignant Melanoma. Trends Cancer 2020, 6, 478–488. [Google Scholar] [CrossRef]
- Cardama, G.A.; Comin, M.J.; Hornos, L.; Gonzalez, N.; Defelipe, L.; Turjanski, A.G.; Alonso, D.F.; Gomez, D.E.; Menna, P.L. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med. Chem. 2014, 14, 840–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomino, T.; Tajiri, H.; Tatsuguchi, T.; Shirai, T.; Oisaki, K.; Matsunaga, S.; Sanematsu, F.; Sakata, D.; Yoshizumi, T.; Maehara, Y.; et al. DOCK1 inhibition suppresses cancer cell invasion and macropinocytosis induced by self-activating Rac1. Biochem. Biophys. Res. Commun. 2018, 497, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernández, E.; Humphries-Bickley, T.; De la Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; De La Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A.; et al. The Rac Inhibitor EHop-016 Inhibits Mammary Tumor Growth and Metastasis in a Nude Mouse Model. Transl. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef]
- Ferri, N.; Contini, A.; Bernini, S.K.; Corsini, A. Role of small GTPase protein Rac1 in cardiovascular diseases: Development of new selective pharmacological inhibitors. J. Cardiovasc. Pharmacol. 2013, 62, 425–435. [Google Scholar] [CrossRef]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouquier, N.; Vignal, E.; Charrasse, S.; Weill, M.; Schmidt, S.; Léonetti, J.P.; Blangy, A.; Fort, P. A cell active chemical GEF inhibitor selectively targets the Trio/RhoG/Rac1 signaling pathway. Chem. Biol. 2009, 16, 657–666. [Google Scholar] [CrossRef]
- Ferri, N.; Corsini, A.; Bottino, P.; Clerici, F.; Contini, A. Virtual screening approach for the identification of new Rac1 inhibitors. J. Med. Chem. 2009, 52, 4087–4090. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, W.; Sun, M.; Lu, C.; Shen, Y. Deacetylmycoepoxydiene is an agonist of Rac1, and simultaneously induces autophagy and apoptosis. Appl. Microbiol. Biotechnol. 2018, 102, 5965–5975. [Google Scholar] [CrossRef]
- Dütting, S.; Heidenreich, J.; Cherpokova, D.; Amin, E.; Zhang, S.C.; Ahmadian, M.R.; Brakebusch, C.; Nieswandt, B. Critical off-target effects of the widely used Rac1 inhibitors NSC23766 and EHT1864 in mouse platelets. J. Thromb. Haemost. 2015, 13, 827–838. [Google Scholar] [CrossRef]
- Abdrabou, A.; Wang, Z. Post-Translational Modification and Subcellular Distribution of Rac1: An Update. Cells 2018, 7, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.; Yang, F.; Wise, C.E.; Meng, F.; Priester, S.; Munshi, M.K.; Guerrier, M.; Dostal, D.E.; Glaser, S.S. Simvastatin stimulates apoptosis in cholangiocarcinoma by inhibition of Rac1 activity. Dig. Liver Dis. 2011, 43, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Lerida, I.; Sanchez-Perales, S.; Calvo, M.; Rentero, C.; Zheng, Y.; Enrich, C.; Del Pozo, M.A. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 2012, 31, 534–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Alvarez, C.; Guerrero-Rodríguez, S.L.; Rhodes, J.; Cannon, A.; Chernoff, J.; Araiza-Olivera, D. Targeting effector pathways in RAC1. Small GTPases 2021, 12, 273–281. [Google Scholar] [CrossRef]
- Lu, J.; Chan, L.; Fiji, H.D.G.; Dahl, R.; Kwon, O.; Tamanoi, F. In vivo antitumor effect of a novel inhibitor of protein geranylgeranyltransferase-I. Mol. Cancer Ther. 2009, 8, 1218–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimonjic, D.B.; Chan, L.N.; Tripathi, V.; Lu, J.; Kwon, O.; Popescu, N.C.; Lowy, D.R.; Tamanoi, F. In vitro and in vivo effects of geranylgeranyltransferase I inhibitor P61A6 on non-small cell lung cancer cells. BMC Cancer 2013, 13, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, O.M.; Ibrahim, M.X.; Jonsson, I.M.; Karlsson, C.; Liu, M.; Sjogren, A.K.; Olofsson, F.J.; Brisslert, M.; Andersson, S.; Ohlsson, C.; et al. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J. Clin. Investig. 2011, 121, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochocki, J.D.; Distefano, M.D. Prenyltransferase Inhibitors: Treating Human Ailments from Cancer to Parasitic Infections. Medchemcomm 2013, 4, 476–492. [Google Scholar] [CrossRef] [Green Version]
- Ferri, N.; Colombo, G.; Ferrandi, C.; Raines, E.W.; Levkau, B.; Corsini, A. Simvastatin reduces MMP1 expression in human smooth muscle cells cultured on polymerized collagen by inhibiting Rac1 activation. Arterioscler Thromb. Vasc. Biol. 2007, 27, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Fukumoto, Y.; Nochioka, K.; Minami, T.; Kudo, S.; Shiba, N.; Takai, Y.; Williams, C.L.; Liao, J.K.; Shimokawa, H. Statins exert the pleiotropic effects through small GTP-binding protein dissociation stimulator upregulation with a resultant Rac1 degradation. Arterioscler Thromb. Vasc. Biol. 2013, 33, 1591–1600. [Google Scholar] [CrossRef] [Green Version]
- Michaelson, D.; Silletti, J.; Murphy, G.; D’Eustachio, P.; Rush, M.; Philips, M.R. Differential localization of Rho GTPases in live cells: Regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 2001, 152, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.; Malliri, A. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases 2017, 8, 139–163. [Google Scholar] [CrossRef] [Green Version]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Hernandez-O’Farrill, E.; Borrero-Garcia, L.D.; Forestier-Roman, I.; Gerena, Y.; Blanco, M.; Rivera-Robles, M.J.; Rodriguez-Medina, J.R.; Cubano, L.A.; et al. Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in Metastatic Cancer. Mol. Cancer Ther. 2017, 16, 805–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, J.; Murray, L.J.; Ndubaku, C.O.; O’Brien, T.; Blackwood, E.; Wang, W.; Aliagas, I.; Gazzard, L.; Crawford, J.J.; Drobnick, J.; et al. Chemically Diverse Group I p21-Activated Kinase (PAK) Inhibitors Impart Acute Cardiovascular Toxicity with a Narrow Therapeutic Window. J. Med. Chem. 2016, 59, 5520–5541. [Google Scholar] [CrossRef] [PubMed]
- Bosco, E.E.; Kumar, S.; Marchioni, F.; Biesiada, J.; Kordos, M.; Szczur, K.; Meller, J.; Seibel, W.; Mizrahi, A.; Pick, E.; et al. Rational design of small molecule inhibitors targeting the Rac GTPase-p67(phox) signaling axis in inflammation. Chem. Biol. 2012, 19, 228–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisabeth, E.M.; Kahl, D.; Gopallawa, I.; Haynes, S.E.; Misek, S.A.; Campbell, P.L.; Dexheimer, T.S.; Khanna, D.; Fox, D.A.; Jin, X.; et al. Identification of Pirin as a Molecular Target of the CCG-1423/CCG-203971 Series of Antifibrotic and Antimetastatic Compounds. ACS Pharmacol. Transl. Sci. 2019, 2, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Licciulli, S.; Luise, C.; Scafetta, G.; Capra, M.; Giardina, G.; Nuciforo, P.; Bosari, S.; Viale, G.; Mazzarol, G.; Tonelli, C.; et al. Pirin inhibits cellular senescence in melanocytic cells. Am. J. Pathol. 2011, 178, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, I.; Simizu, S.; Okumura, H.; Takagi, S.; Osada, H. A small-molecule inhibitor shows that pirin regulates migration of melanoma cells. Nat. Chem. Biol. 2010, 6, 667–673. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Watanabe, B.; Nakagawa, Y.; Minami, S.; Morita, T. RPEL proteins are the molecular targets for CCG-1423, an inhibitor of Rho signaling. PLoS ONE 2014, 9, e89016. [Google Scholar] [CrossRef]
- Lundquist, M.R.; Storaska, A.J.; Liu, T.C.; Larsen, S.D.; Evans, T.; Neubig, R.R.; Jaffrey, S.R. Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell 2014, 156, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchings, K.M.; Lisabeth, E.M.; Rajeswaran, W.; Wilson, M.W.; Sorenson, R.J.; Campbell, P.L.; Ruth, J.H.; Amin, A.; Tsou, P.S.; Leipprandt, J.R.; et al. Pharmacokinetic optimitzation of CCG-203971: Novel inhibitors of the Rho/MRTF/SRF transcriptional pathway as potential antifibrotic therapeutics for systemic scleroderma. Bioorg. Med. Chem. Lett. 2017, 27, 1744–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef] [PubMed]

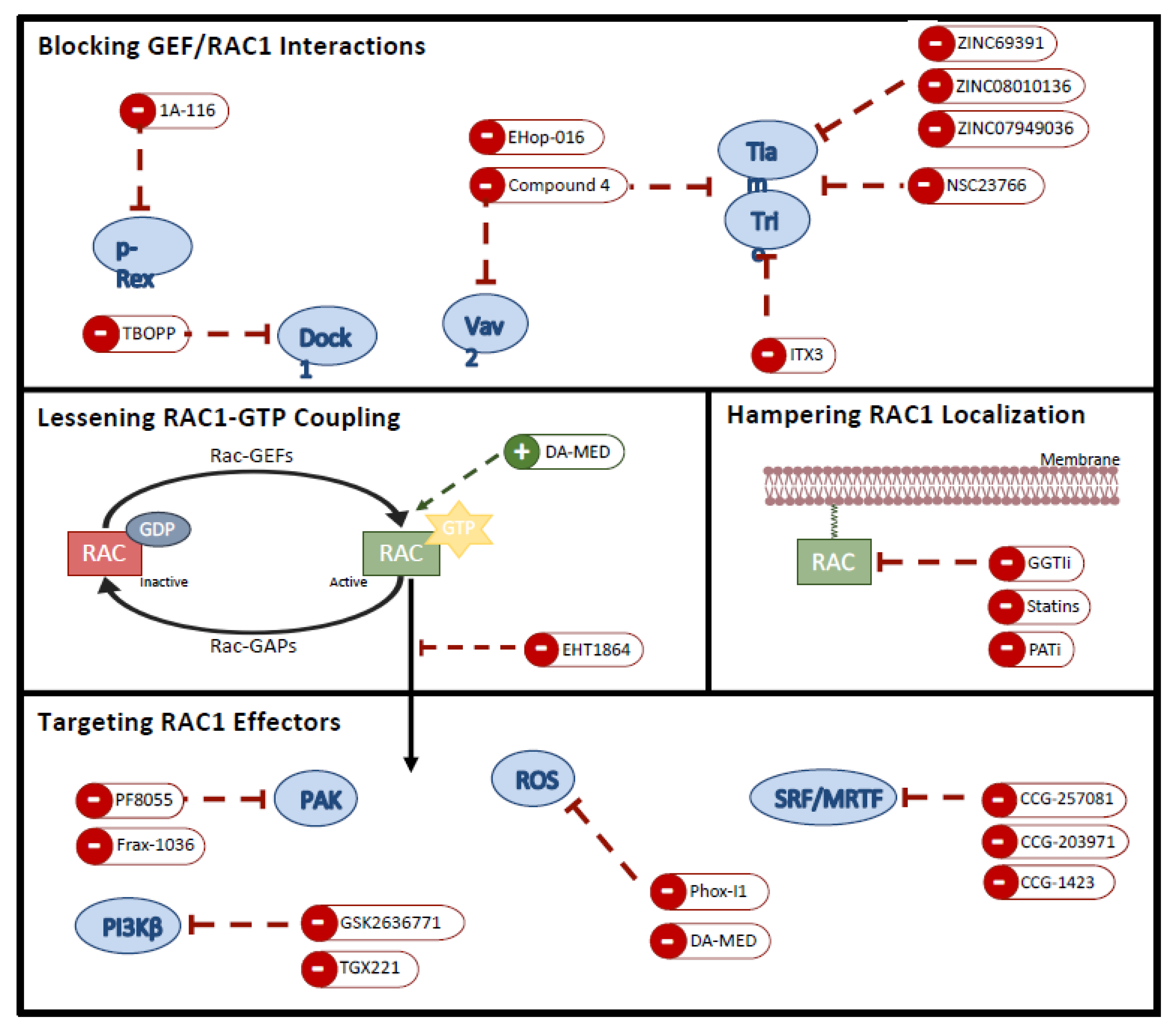

{kind=link}
{kind=link}
| Compound Name | Structure | Target | Mechanism of Action | References |
|---|---|---|---|---|
| Blocking GEF/RAC1 Interactions | ||||
| 1A-116 | 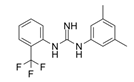 | p-REX | Blocks RAC-p-REX1 interaction, reducing intracellular RAC1-GTP levels. | [73] |
| TBOPP |  | DOCK1 | Binds to the DOCK1 DHR-2 domain, inhibiting DOCK1-mediated RAC activation. | [74] |
| EHop-016 | 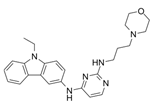 | VAV2 | Prevents RAC1-VAV2 association, inhibiting activity of RAC downstream effector PAK1. It also targets CDC42. | [75,76] |
| Compound 4 | 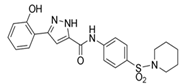 | VAV2, TIAM1, TRIO | Impedes RAC1 binding to TIAM1, TRIO and VAV2. | [77,78] |
| NSC23766 | 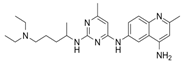 | TIAM1, TRIO | Inhibits RAC1 binding and activation by the RAC-specific GEFs TRIO or TIAM1. | [79] |
| ITX3 | 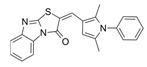 | TRIO-N | Inhibits TRIO N-terminal GEF domain, reducing RAC1 and RHOG activation. | [80] |
| ZINC69391 | 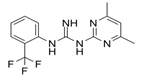 | TIAM1 | Interferes RAC1-TIAM1 interaction, reducing RAC1-GTP levels. | [73] |
| ZINC08010136 | 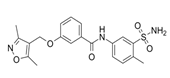 | TIAM1 | Disrupts RAC1-TIAM1 complex, decreasing active RAC1 cytoplasmic levels without affecting RHOA and CDC42. It is four times more effective than NSC23766. | [81] |
| ZINC07949036 | 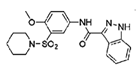 | TIAM1 | Blocks RAC1-TIAM1 interaction without affecting RHOA and CDC42 activation. | [81] |
| Lessening RAC1-GTP Coupling | ||||
| DA-MED | 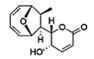 | RAC1 | RAC1 agonist. It has been shown to induce ROS production. | [82] |
| EHT-1864 |  | RAC1 | Inhibits RAC family GTPases. Blocking its activation by direct binding to RAC1, RAC1b, RAC2 and RAC3. | [83] |
| Hampering RAC1 Localization | ||||
| GGTI-2418 | 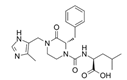 | Geranylgeranyl transferase 1 | Inhibits GGTase I, in charge of lipid modification required for RAC function. | [84] |
| Simvastatin |  | HMG-CoA reductase | Inhibits isoprenoid synthesis, reducing RAC1 membrane association and activity. | [85] |
| PATi | PATs | Inhibits Palmitoyl Acyltransferases (PATs), interfering with RAC1 localization. | [86] | |
| Targeting RAC1 Effectors | ||||
| PF8055 | PAK | PAK inhibitor | [23] | |
| FRAX1036 | 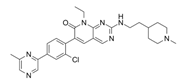 | PAK | PAK inhibitor | [25] |
| GSK2636771 | 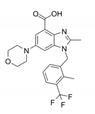 | PI3Kβ | Inhibits p110β catalytic subunit of PI3K, impairing AKT phosphorylation by RAC1. | [15] |
| TGX-221 |  | PI3Kβ | Inhibits p110β catalytic subunit of PI3K, impairing AKT phosphorylation by RAC1. | [87] |
| Phox-I1 | 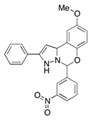 | NOX2 | Inhibits ROS production in neutrophils, by targeting the p67phox interaction site with RAC1 GTPase. | [87] |
| CCG-257081 | 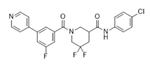 | SRF/MRTF | Inhibits SRF/MRTF pathway. | [34] |
| CCG-203971 |  | SRF/MRTF | Inhibits SRF/MRTF pathway. | [34] |
| CCG-1423 | 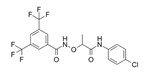 | SRF/MRTF | Inhibits SRF/MRTF pathway. | [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colón-Bolea, P.; García-Gómez, R.; Casar, B. RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomolecules 2021, 11, 1554. https://doi.org/10.3390/biom11111554
Colón-Bolea P, García-Gómez R, Casar B. RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomolecules. 2021; 11(11):1554. https://doi.org/10.3390/biom11111554
Chicago/Turabian StyleColón-Bolea, Paula, Rocío García-Gómez, and Berta Casar. 2021. "RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma" Biomolecules 11, no. 11: 1554. https://doi.org/10.3390/biom11111554
APA StyleColón-Bolea, P., García-Gómez, R., & Casar, B. (2021). RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomolecules, 11(11), 1554. https://doi.org/10.3390/biom11111554