
A Promising Intracellular Protein-Degradation Strategy: TRIMbody-Away Technique Based on Nanobody Fragment

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Expression and Purification of TRIM21 and TRIMbody
2.3. Size Exclusion Chromatography (SEC)
2.4. Binding ELISA
2.5. Cell Culture and Transient Transfection
2.6. Generation of Cell Lines Stably Expressing EGFP and TRIMbody
2.7. Flow Cytometry Analysis
2.8. Induction of αEGFP TRIMbody Expression with Doxycycline
2.9. Laser Scanning Confocal Microscope and Live Cell Imaging
2.10. Immunostaining
2.11. Protein Extraction and Western Blot Assay
2.12. RNA Isolation/cDNA Synthesis and Quantitative Real-Time PCR (qRT-PCR) Assay
2.13. Statistical Analysis
3. Results
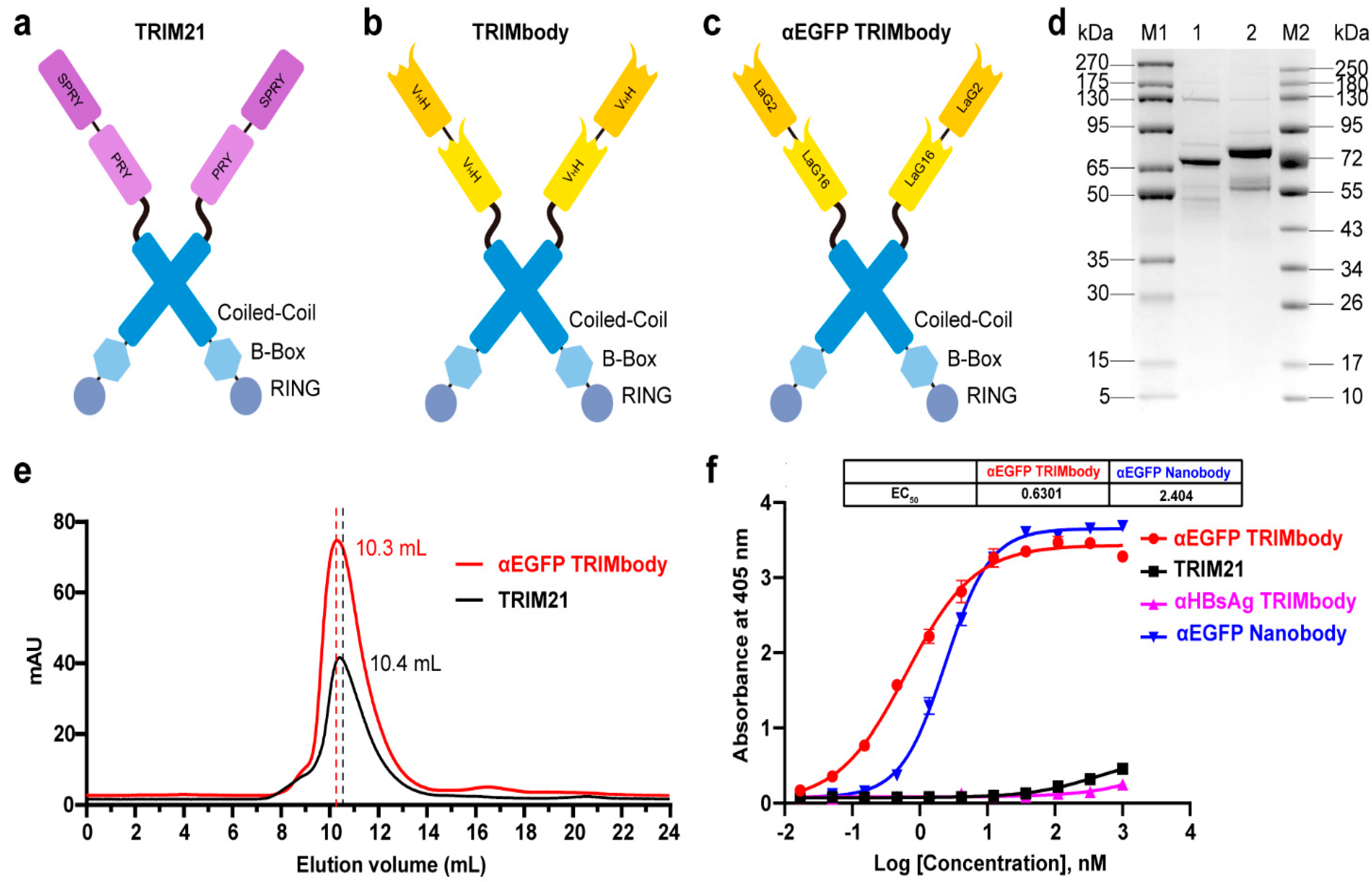
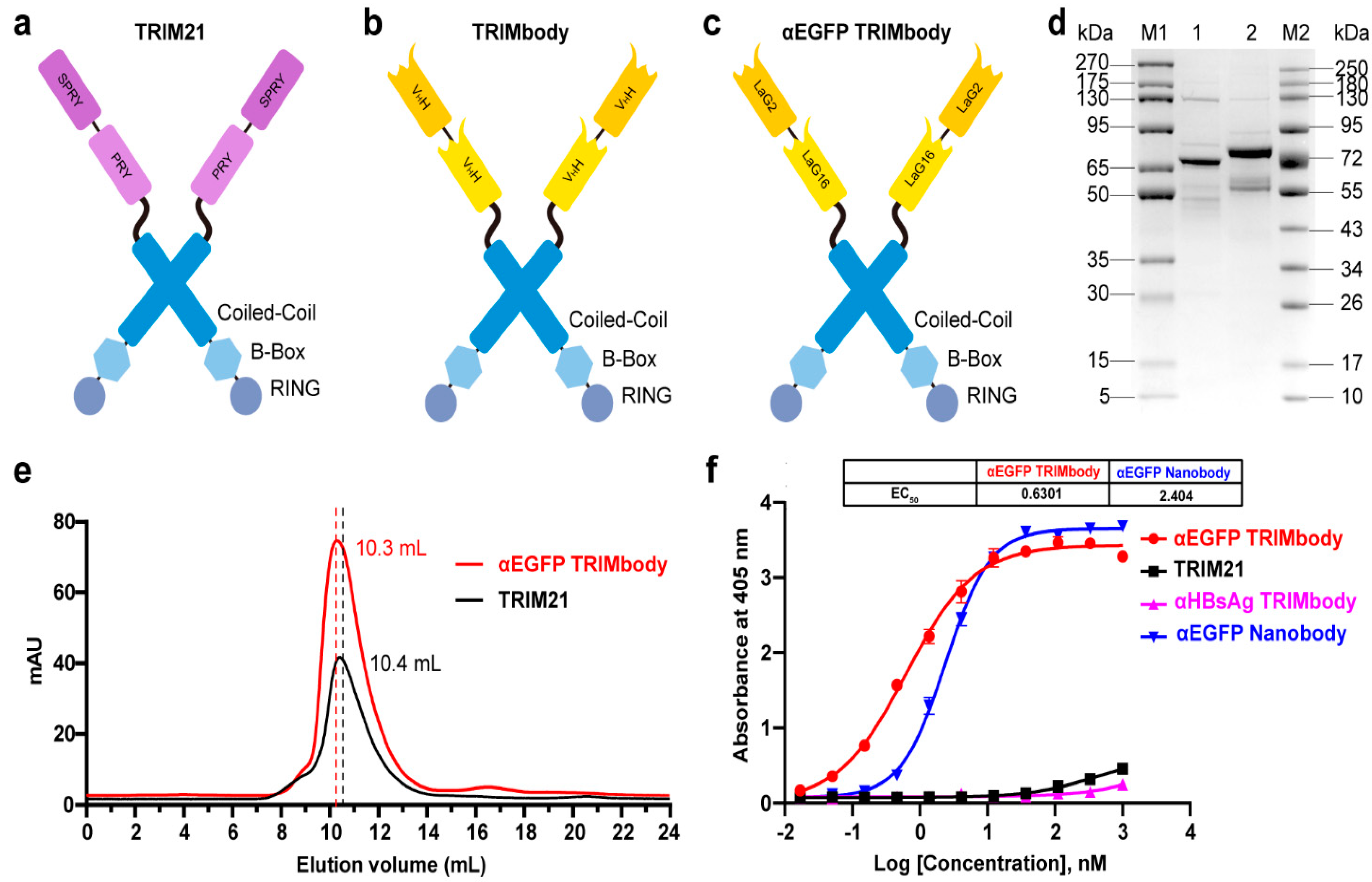
3.1. αEGFP TRIMbody has High Binding Activity to EGFP Protein In Vitro
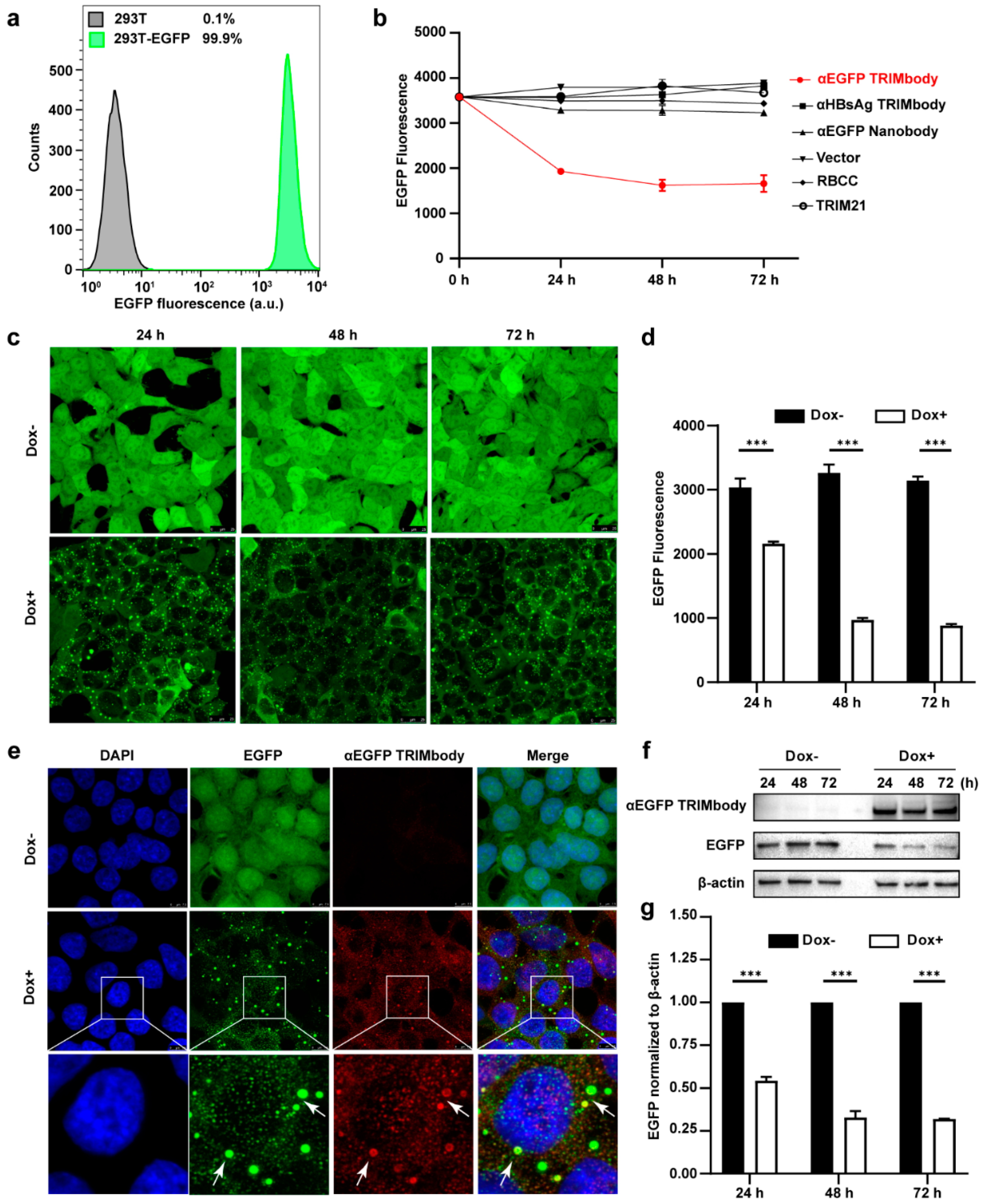
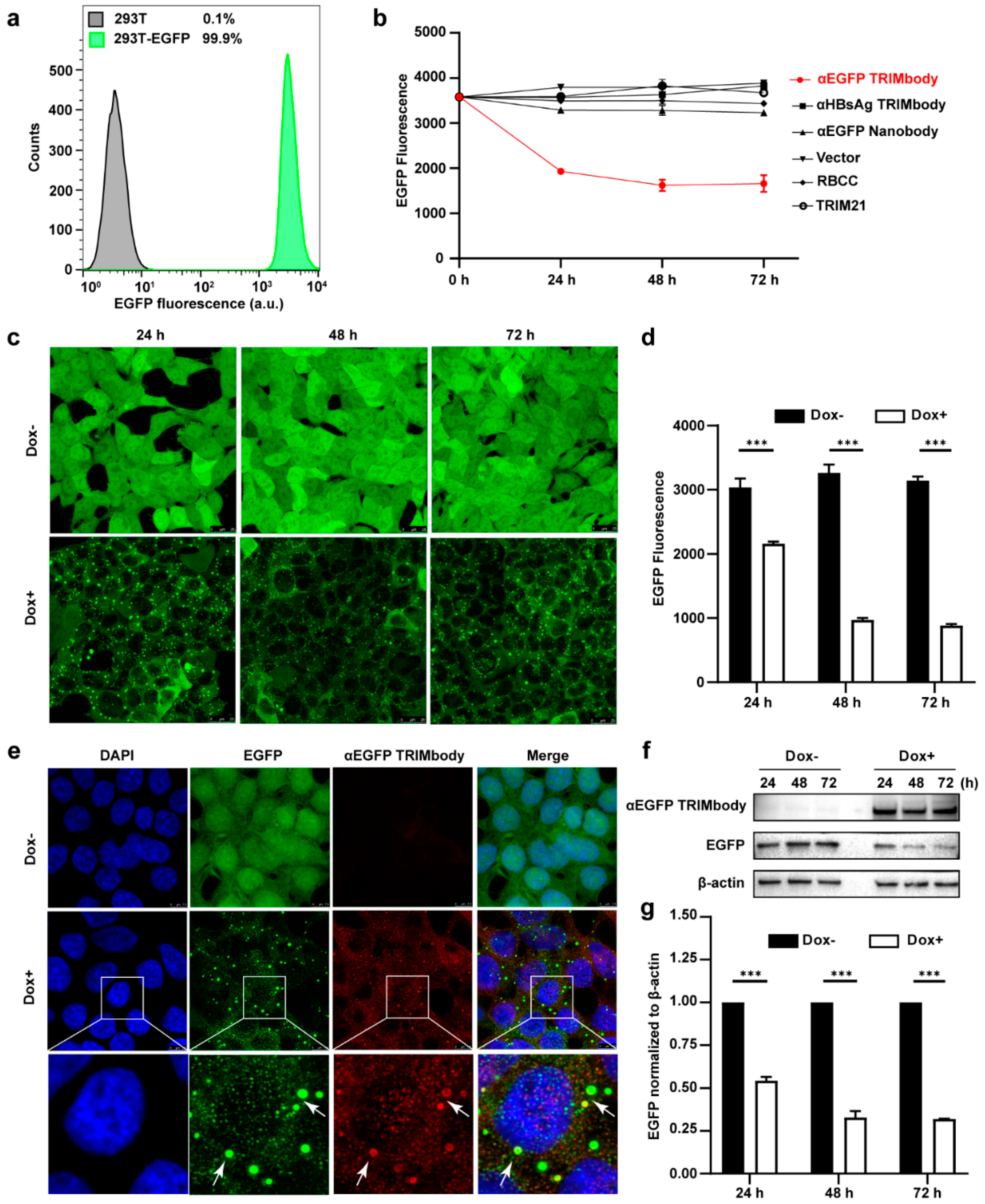
3.2. Degradation of Intracellular EGFP by Inducible Expression of αEGFP TRIMbody
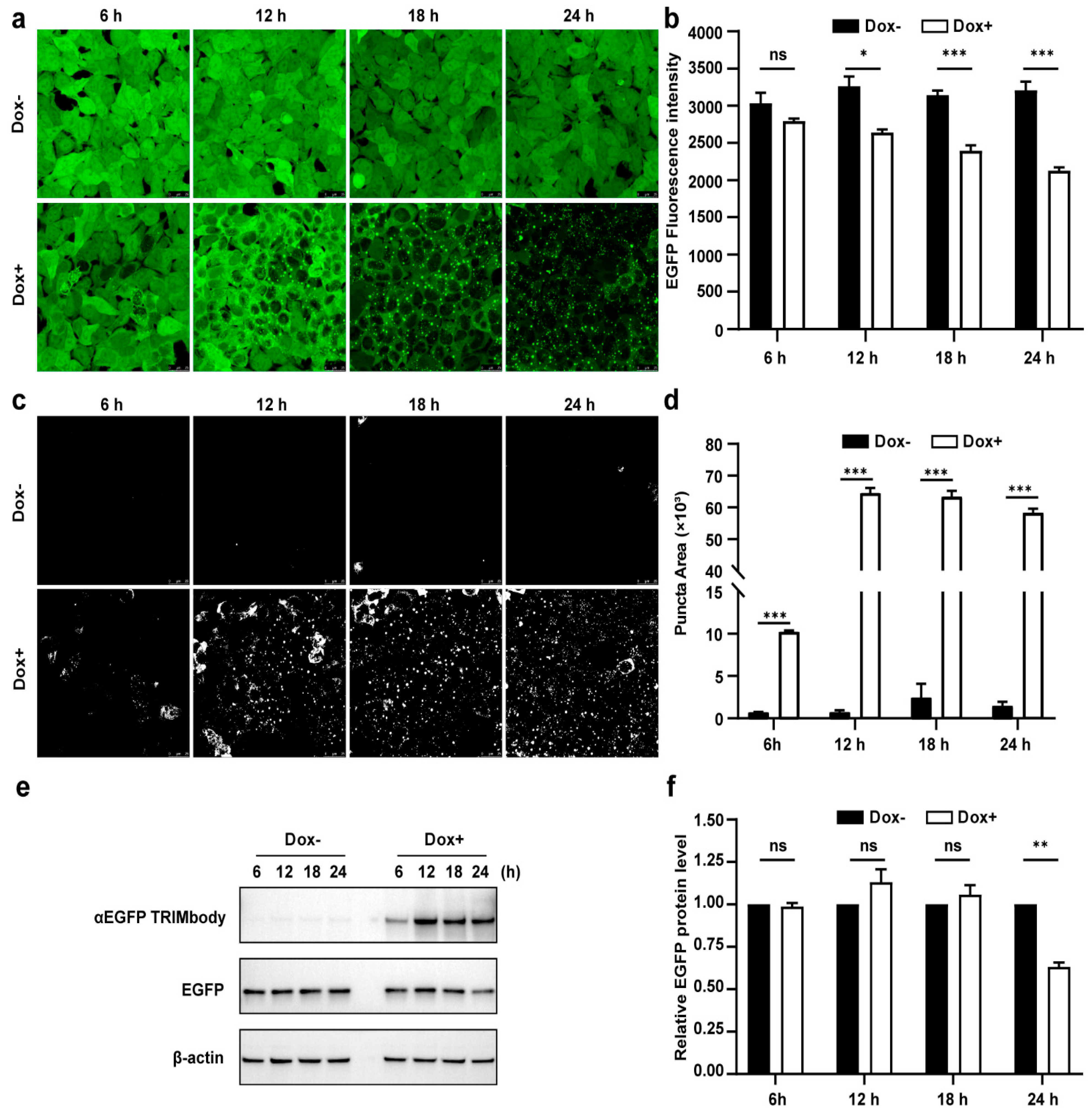
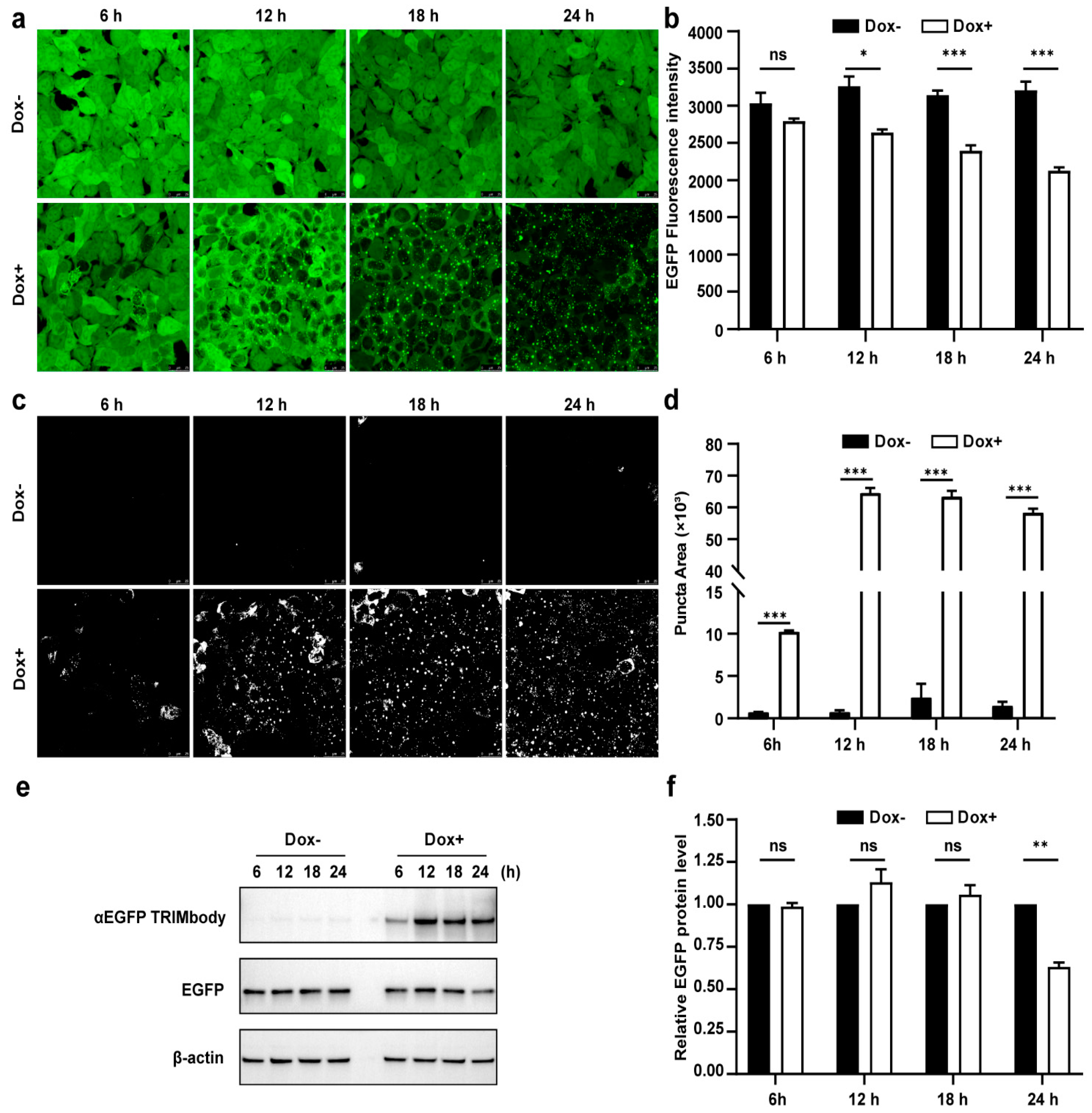
3.3. Dynamic Examination of EGFP Degradation and Accompanying Fluorescent Puncta within 24 h after αEGFP TRIMbody Induction
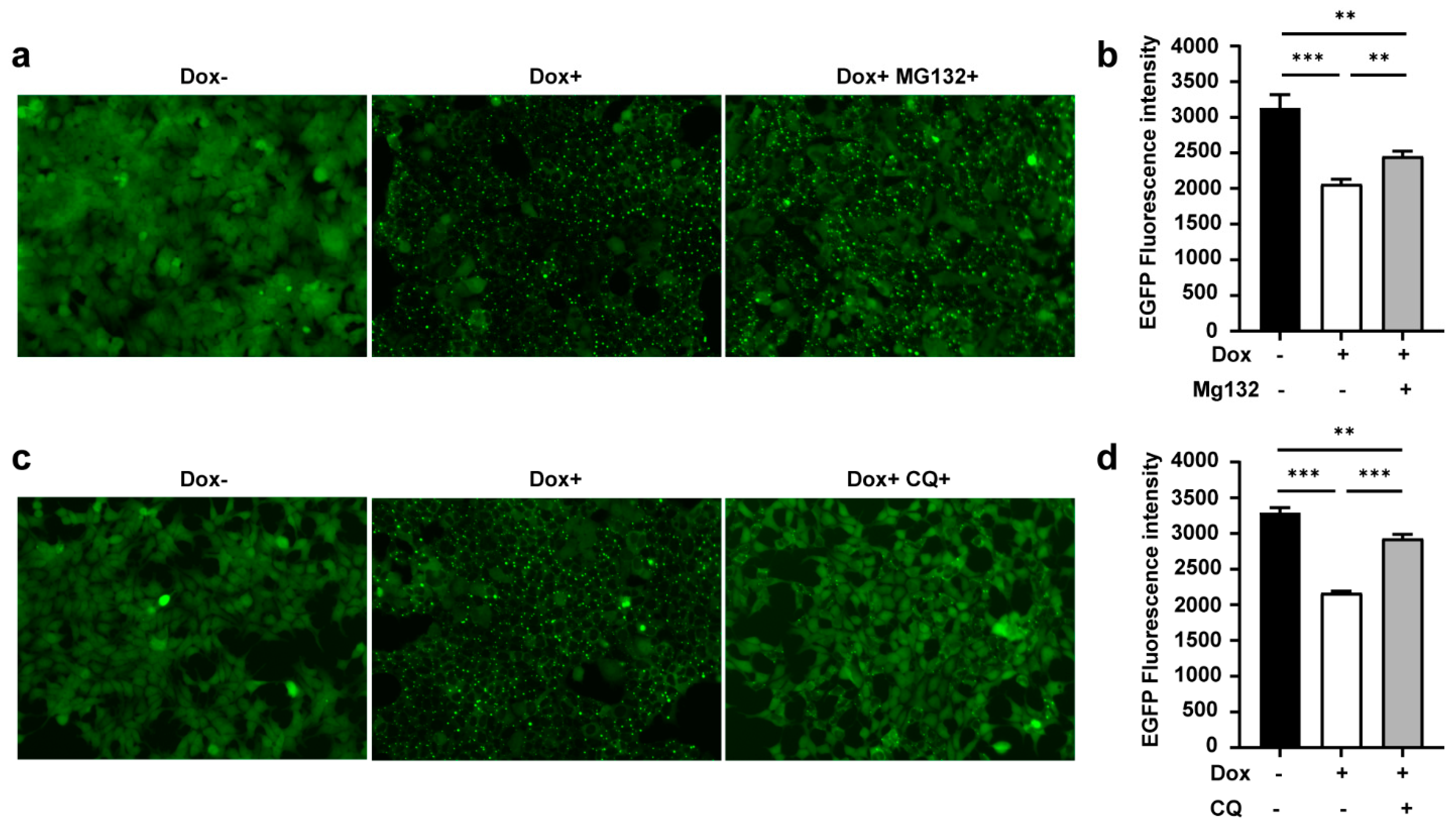
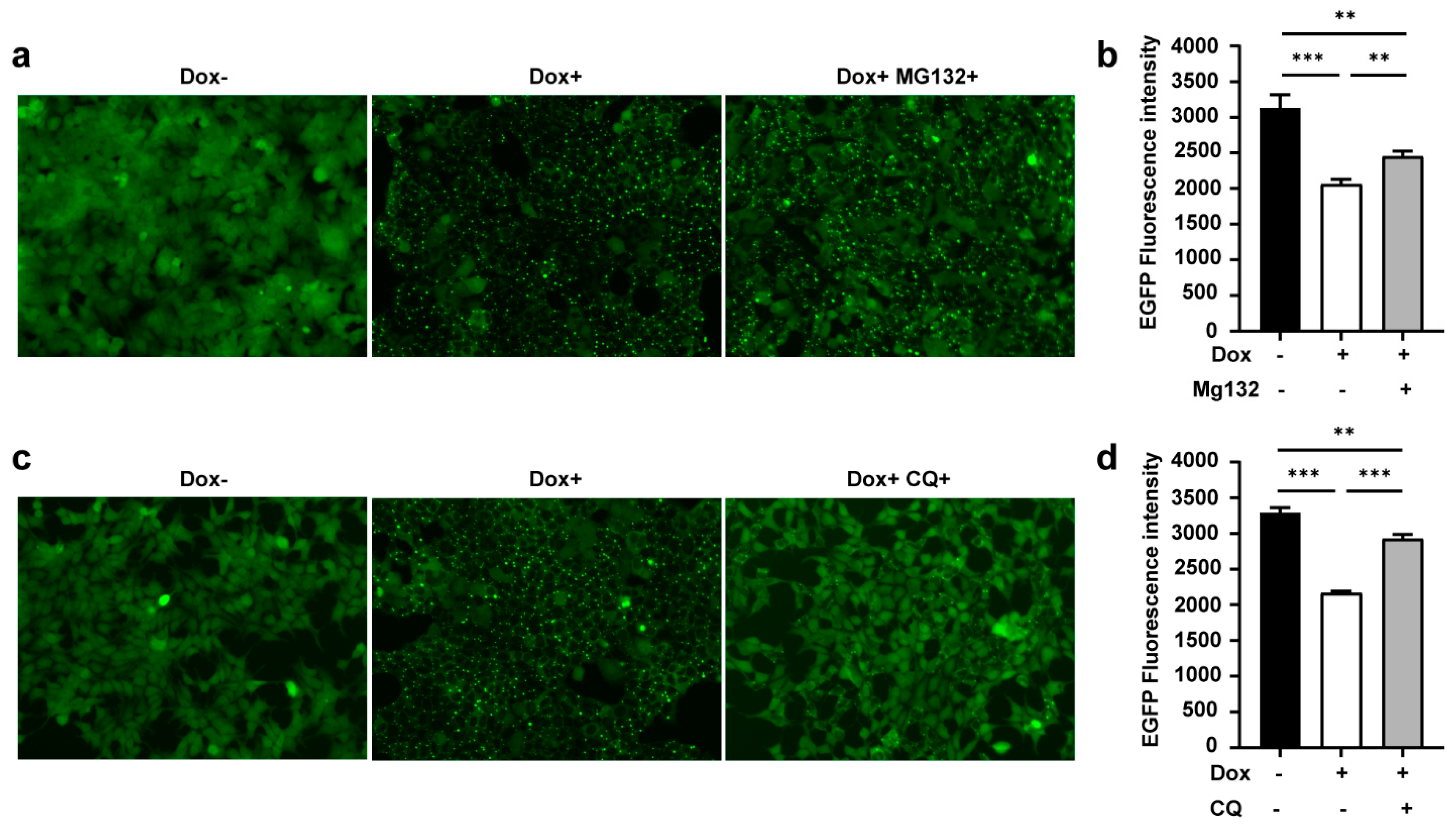
3.4. TRIMbody Induces Intracellular Protein Degradation through the Proteasome and Lysosomal Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bondeson, D.P.; Crews, C.M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röth, S.; Fulcher, L.J.; Sapkota, G.P. Advances in targeted degradation of endogenous proteins. Cell. Mol. Life Sci. 2019, 76, 2761–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Nikan, M.; Tanowitz, M.; Dwyer, C.A.; Jackson, M.; Gaus, H.J.; Swayze, E.E.; Rigo, F.; Seth, P.P.; Prakash, T.P. Targeted Delivery of Antisense Oligonucleotides Using Neurotensin Peptides. J. Med. Chem. 2020, 63, 8471–8484. [Google Scholar] [CrossRef]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N. Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Ottis, P.; Palladino, C.; Thienger, P.; Britschgi, A.; Heichinger, C.; Berrera, M.; Julien-Laferriere, A.; Roudnicky, F.; Kam-Thong, T.; Bischoff, J.R.; et al. Cellular Resistance Mechanisms to Targeted Protein Degradation Converge Toward Impairment of the Engaged Ubiquitin Transfer Pathway. ACS Chem. Biol. 2019, 14, 2215–2223. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Q.; Kao, Y.R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Ohoka, N.; Ujikawa, O.; Shimokawa, K.; Sameshima, T.; Shibata, N.; Hattori, T.; Nara, H.; Cho, N.; Naito, M. Different Degradation Mechanisms of Inhibitor of Apoptosis Proteins (IAPs) by the Specific and Nongenetic IAP-Dependent Protein Eraser (SNIPER). Chem. Pharm. Bull. 2019, 67, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Clift, D.; So, C.; McEwan, W.A.; James, L.C.; Schuh, M. Acute and rapid degradation of endogenous proteins by Trim-Away. Nat. Protoc. 2018, 13, 2149–2175. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Santos, A.F.; Mukadam, A.S.; Osswald, M.; Jacques, D.A.; Dickson, C.F.; McLaughlin, S.H.; Johnson, C.M.; Kiss, L.; Luptak, J.; et al. Target-induced clustering activates Trim-Away of pathogens and proteins. Nat. Struct. Mol. Biol. 2021, 28, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Khan, R.; Khan, A.; Ali, A.; Idrees, M. The interplay between viruses and TRIM family proteins. Rev. Med. Virol. 2019, 29, e2028. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; García-Sastre, A.; Versteeg, G.A. TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 2014, 426, 1265–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhang, Z.; Xu, G. TRIM proteins in neuroblastoma. Biosci. Rep. 2019, 39, BSR20192050. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Koliopoulos, M.G.; Rittinger, K. Structural determinants of TRIM protein function. Biochem. Soc. Trans. 2017, 45, 183–191. [Google Scholar] [CrossRef]
- Zhang, J.R.; Li, X.X.; Hu, W.N.; Li, C.Y. Emerging Role of TRIM Family Proteins in Cardiovascular Disease. Cardiology 2020, 145, 390–400. [Google Scholar] [CrossRef]
- Tomar, D.; Singh, R. TRIM family proteins: Emerging class of RING E3 ligases as regulator of NF-κB pathway. Biol. Cell 2015, 107, 22–40. [Google Scholar] [CrossRef]
- Imam, S.; Talley, S.; Nelson, R.S.; Dharan, A.; O’Connor, C.; Hope, T.J.; Campbell, E.M. TRIM5α Degradation via Autophagy Is Not Required for Retroviral Restriction. J. Virol. 2016, 90, 3400–3410. [Google Scholar] [CrossRef] [Green Version]
- Foss, S.; Bottermann, M.; Jonsson, A.; Sandlie, I.; James, L.C.; Andersen, J.T. TRIM21-From Intracellular Immunity to Therapy. Front. Immunol. 2019, 10, 2049. [Google Scholar] [CrossRef]
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Vunjak, M.; Versteeg, G.A. TRIM proteins. Curr. Biol. 2019, 29, R42–R44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, L.M.; Meroni, G. TRIM family: Pleiotropy and diversification through homomultimer and heteromultimer formation. IUBMB Life 2012, 64, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Li, N.L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-Terminal Tail of TRIM56 Dictates Antiviral Restriction of Influenza A and B Viruses by Impeding Viral RNA Synthesis. J. Virol. 2016, 90, 4369–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepke, L.; Gack, M.U.; Sparrer, K.M. The antiviral activities of TRIM proteins. Curr. Opin. Microbiol. 2021, 59, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.F.M.; Shen, L.; Tatham, M.H.; Dickerson, D.; Prescott, A.R.; Abidi, N.; Xirodimas, D.P.; Hay, R.T. Antibody RING-Mediated Destruction of Endogenous Proteins. Mol. Cell 2020, 79, 155–166.e9. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, S.; Ying, T. Single-Domain Antibodies as Therapeutics against Human Viral Diseases. Front. Immunol. 2017, 8, 1802. [Google Scholar] [CrossRef]
- Ding, Y.; Fei, Y.; Lu, B. Emerging New Concepts of Degrader Technologies. Trends Pharmacol. Sci. 2020, 41, 464–474. [Google Scholar] [CrossRef]
- Chen, W.; Gong, R.; Ying, T.; Prabakaran, P.; Zhu, Z.; Feng, Y.; Dimitrov, D.S. Discovery of novel candidate therapeutics and diagnostics based on engineered human antibody domains. Curr. Drug Discov. Technol. 2014, 11, 28–40. [Google Scholar] [CrossRef]
- Wu, Y.; Li, C.; Xia, S.; Tian, X.; Kong, Y.; Wang, Z.; Gu, C.; Zhang, R.; Tu, C.; Xie, Y.; et al. Identification of Human Single-Domain Antibodies against SARS-CoV-2. Cell Host Microbe 2020, 27, 891–898.e5. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Isenberg, D.A. TRIM21 and the Function of Antibodies inside Cells. Trends Immunol. 2017, 38, 916–926. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- de Taeye, S.W.; Rispens, T.; Vidarsson, G. The Ligands for Human IgG and Their Effector Functions. Antibodies 2019, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridy, P.C.; Li, Y.; Keegan, S.; Thompson, M.K.; Nudelman, I.; Scheid, J.F.; Oeffinger, M.; Nussenzweig, M.C.; Fenyö, D.; Chait, B.T.; et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 2014, 11, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Li, H.; Guo, M.; Wang, J.; Xu, Y.; Zou, X.; Deng, R.; Li, G.; Zhu, H. TRIM21 Promotes Innate Immune Response to RNA Viral Infection through Lys27-Linked Polyubiquitination of MAVS. J. Virol. 2018, 92, e00321-18. [Google Scholar] [CrossRef] [Green Version]
- Manocha, G.D.; Mishra, R.; Sharma, N.; Kumawat, K.L.; Basu, A.; Singh, S.K. Regulatory role of TRIM21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells. J. Neuroinflamm. 2014, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R., 3rd; Hetzer, M.W. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzijl, N.; DeGroot, J.; Thorpe, S.R.; Bank, R.A.; Shaw, J.N.; Lyons, T.J.; Bijlsma, J.W.; Lafeber, F.P.; Baynes, J.W.; TeKoppele, J.M. Effect of collagen turnover on the accumulation of advanced glycation end products. J. Biol. Chem. 2000, 275, 39027–39031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razafsky, D.; Ward, C.; Potter, C.; Zhu, W.; Xue, Y.; Kefalov, V.J.; Fong, L.G.; Young, S.G.; Hodzic, D. Lamin B1 and lamin B2 are long-lived proteins with distinct functions in retinal development. Mol. Biol. Cell 2016, 27, 1928–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savas, J.N.; Toyama, B.H.; Xu, T.; Yates, J.R., 3rd; Hetzer, M.W. Extremely long-lived nuclear pore proteins in the rat brain. Science 2012, 335, 942. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ma, Z.; Wang, H.; Niu, K.; Cao, Y.; Sun, L.; Geng, Y.; Yang, B.; Gao, F.; Chen, Z.; et al. Ubiquitylome study identifies increased histone 2A ubiquitylation as an evolutionarily conserved aging biomarker. Nat. Commun. 2019, 10, 2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, G.; Kong, Y.; Li, Y.; Huang, A.; Wang, C.; Zhou, S.; Yang, Z.; Wu, Y.; Ren, J.; Ying, T. A Promising Intracellular Protein-Degradation Strategy: TRIMbody-Away Technique Based on Nanobody Fragment. Biomolecules 2021, 11, 1512. https://doi.org/10.3390/biom11101512
Chen G, Kong Y, Li Y, Huang A, Wang C, Zhou S, Yang Z, Wu Y, Ren J, Ying T. A Promising Intracellular Protein-Degradation Strategy: TRIMbody-Away Technique Based on Nanobody Fragment. Biomolecules. 2021; 11(10):1512. https://doi.org/10.3390/biom11101512
Chicago/Turabian StyleChen, Gang, Yu Kong, You Li, Ailing Huang, Chunyu Wang, Shanshan Zhou, Zhenlin Yang, Yanling Wu, Jianke Ren, and Tianlei Ying. 2021. "A Promising Intracellular Protein-Degradation Strategy: TRIMbody-Away Technique Based on Nanobody Fragment" Biomolecules 11, no. 10: 1512. https://doi.org/10.3390/biom11101512
APA StyleChen, G., Kong, Y., Li, Y., Huang, A., Wang, C., Zhou, S., Yang, Z., Wu, Y., Ren, J., & Ying, T. (2021). A Promising Intracellular Protein-Degradation Strategy: TRIMbody-Away Technique Based on Nanobody Fragment. Biomolecules, 11(10), 1512. https://doi.org/10.3390/biom11101512