
Activation of Aldehyde Dehydrogenase 2 Ameliorates Glucolipotoxicity of Pancreatic Beta Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Measurement of GSIS
2.4. Islet Isolation and Perifusion Study
2.5. Intracellular ATP Assay
2.6. Cellular Oxygen Consumption Rate (OCR)
2.7. Cell Viability Assay
2.8. Apoptosis Detection with 7-AAD
2.9. Detection of Mitochondrial and Intracellular ROS
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
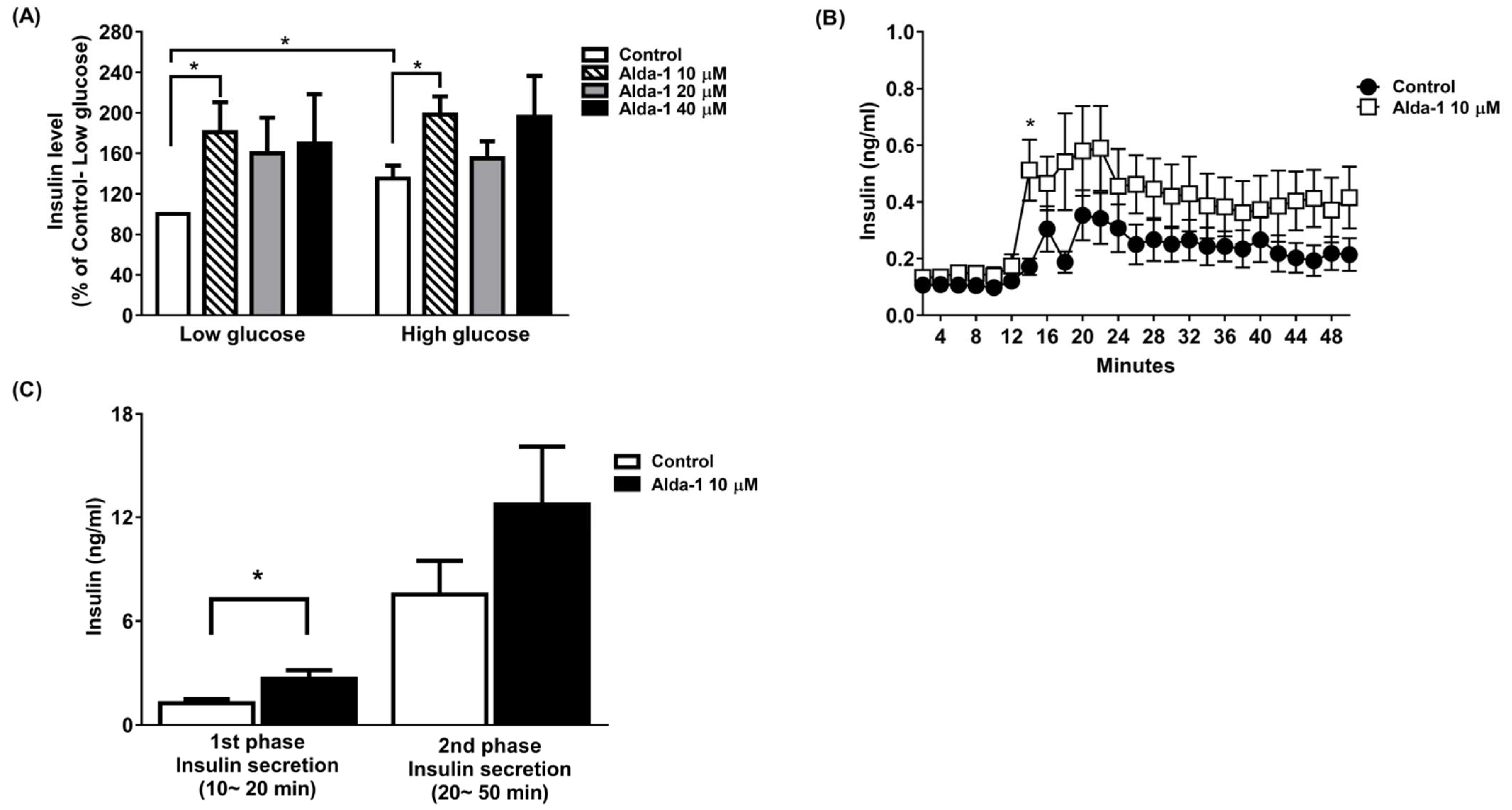
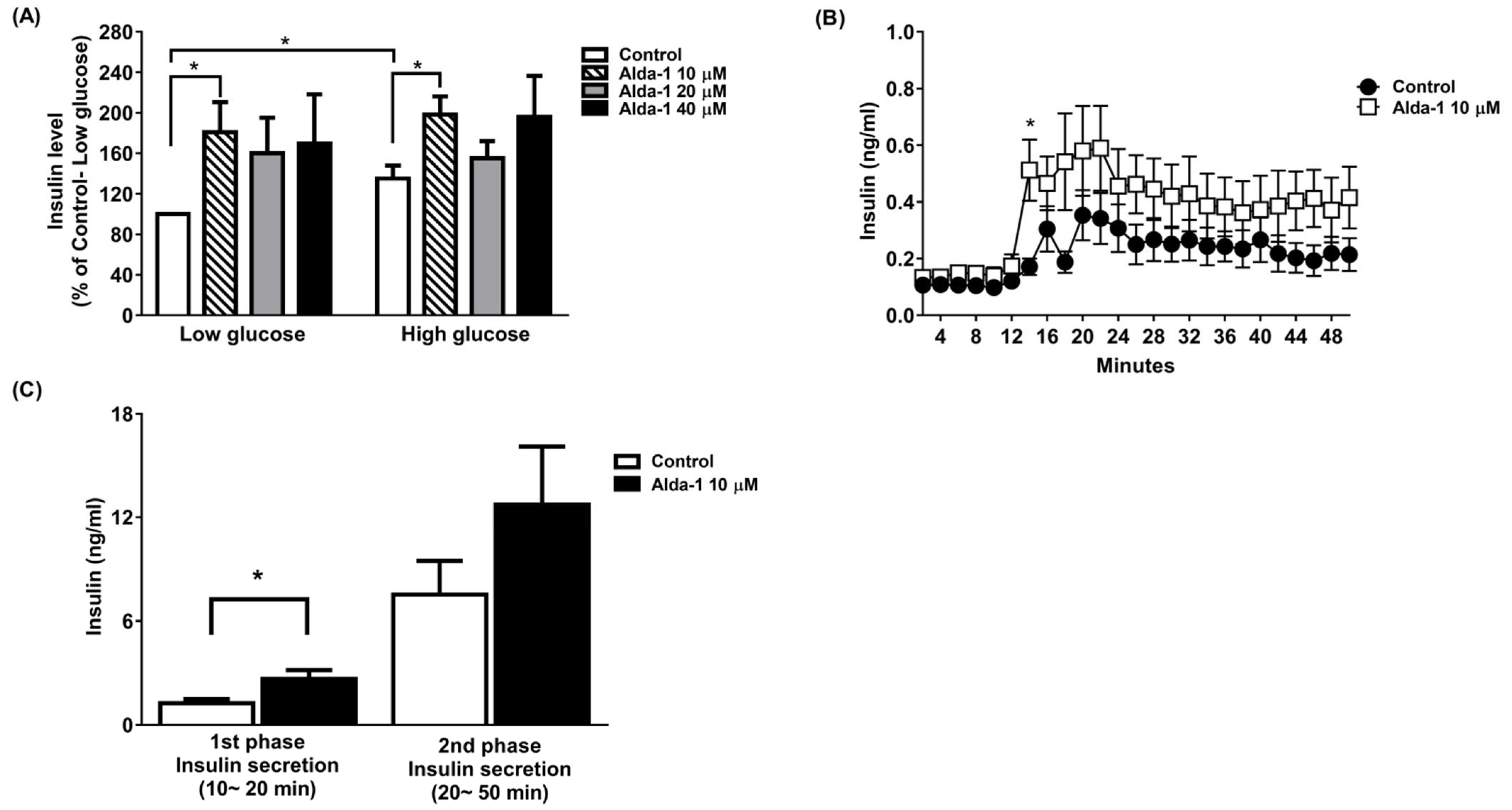
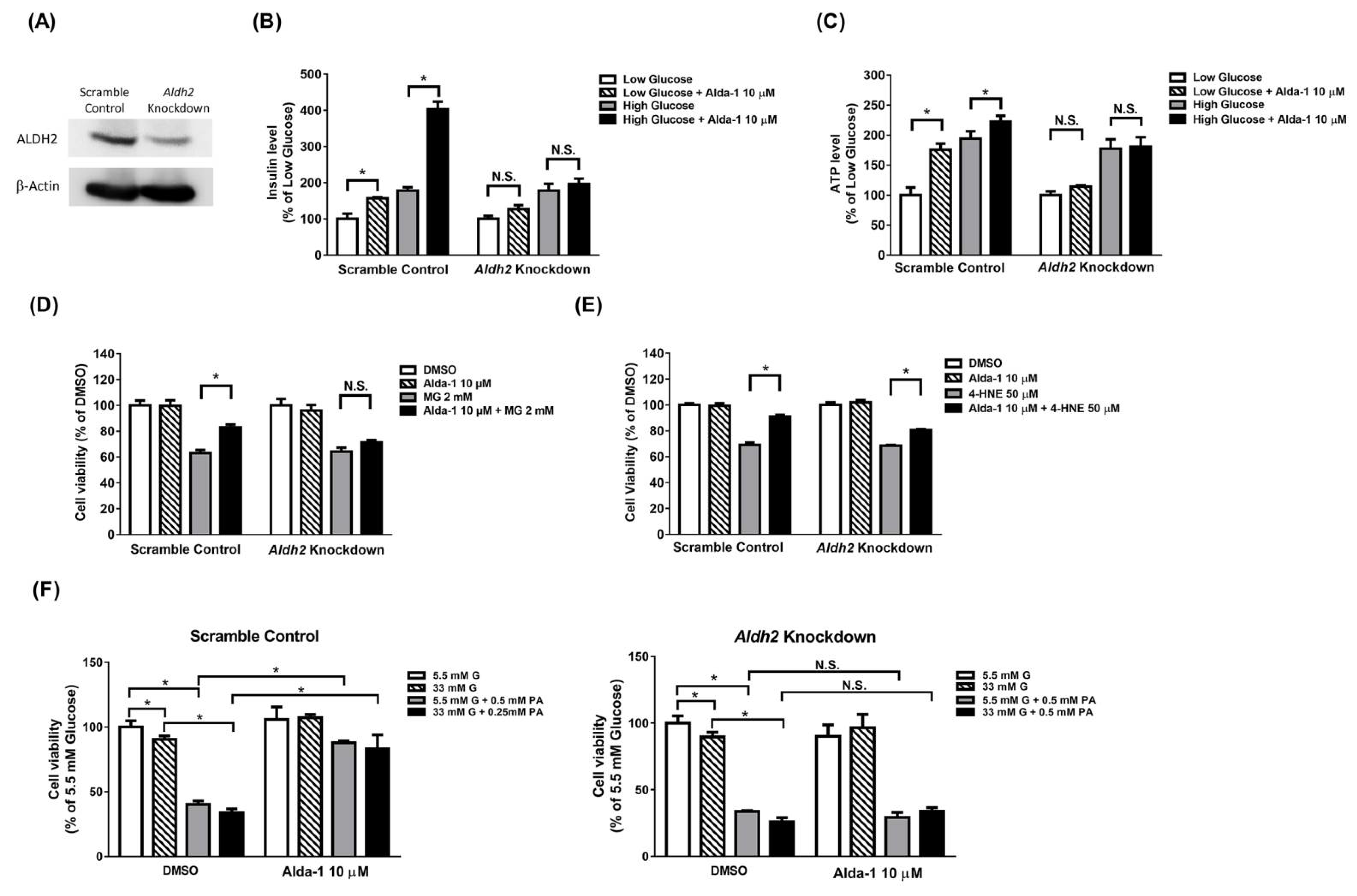
3.1. ALDH2 Activator Enhanced GSIS
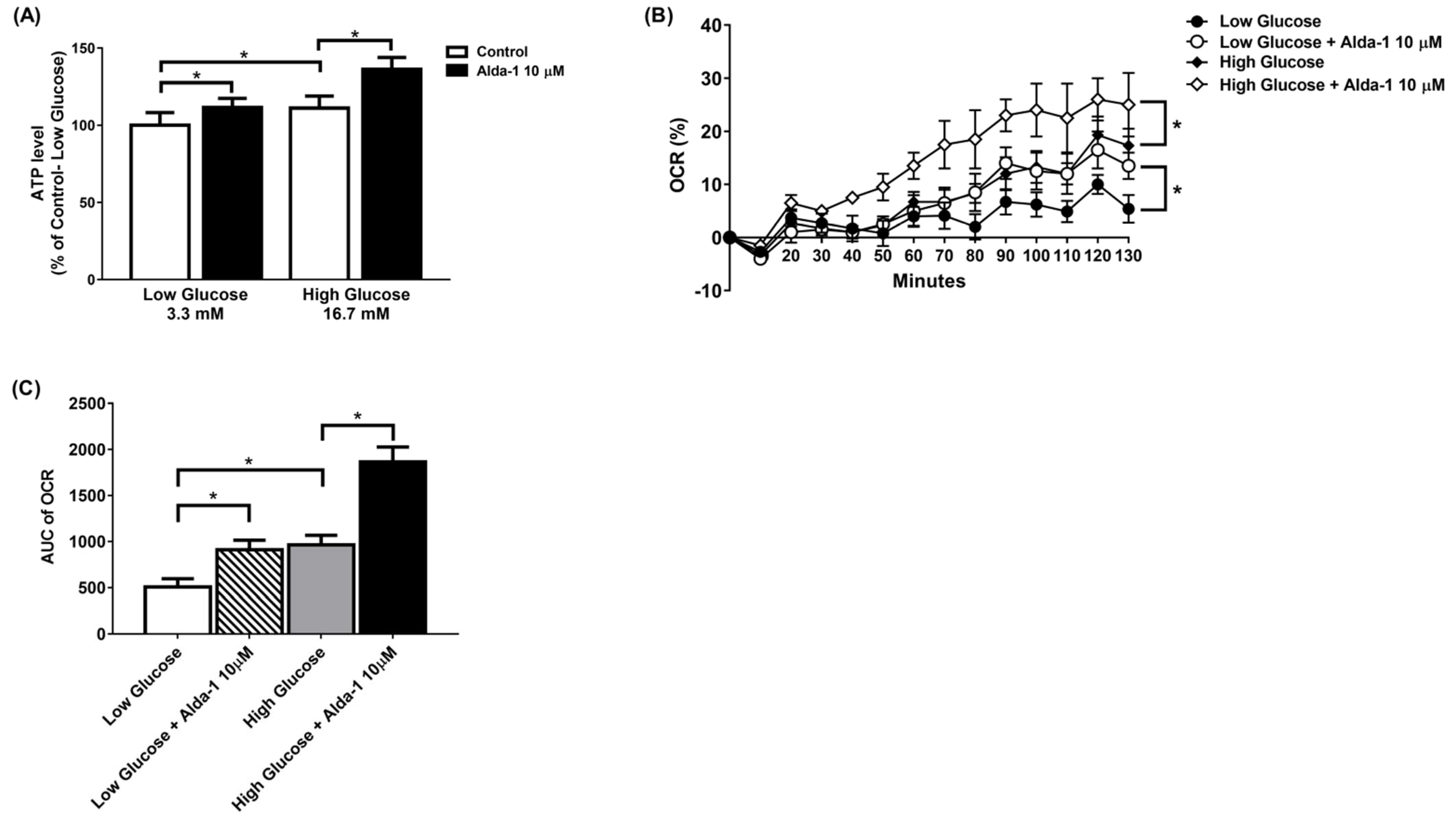
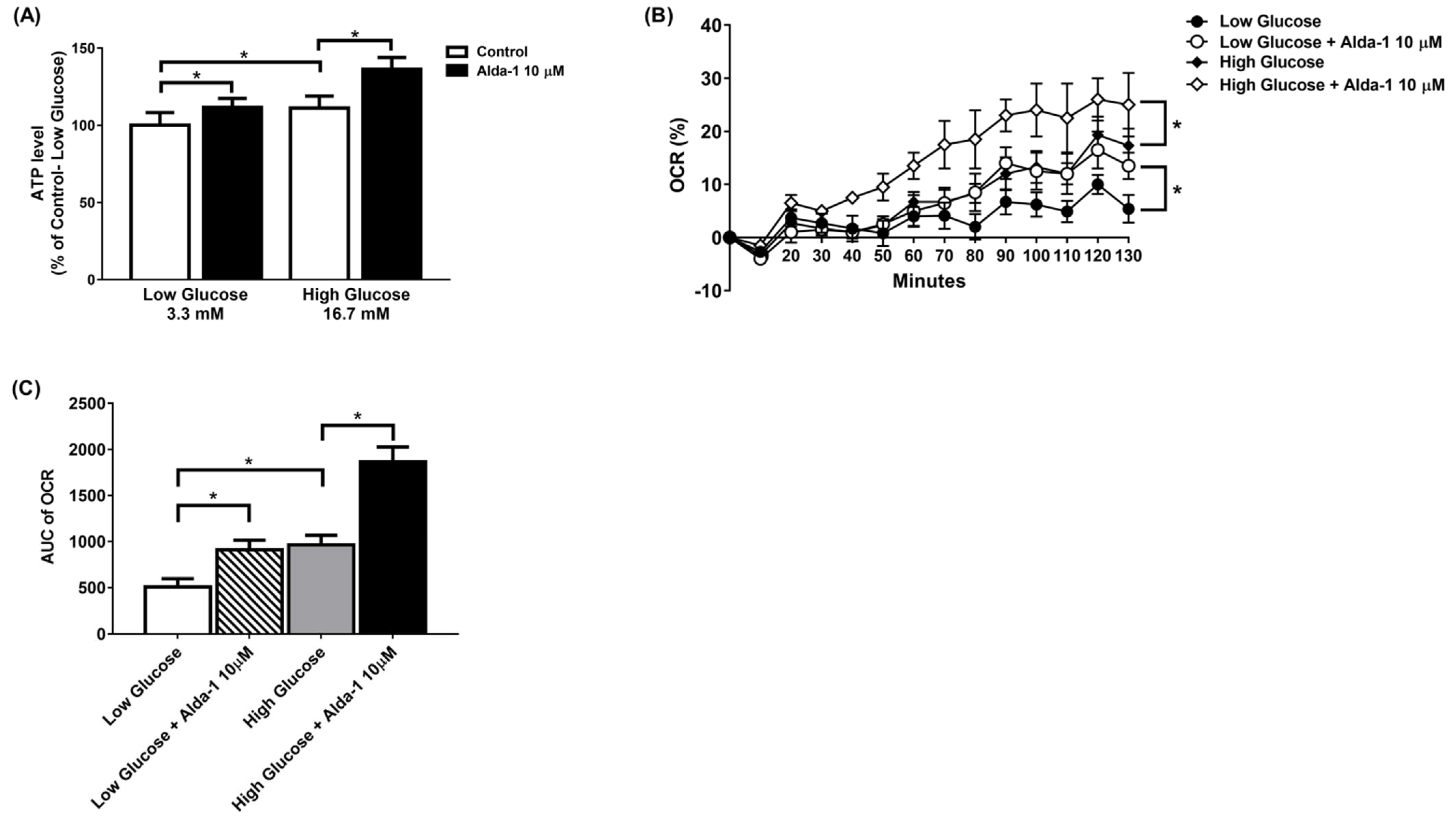
3.2. ALDH2 Activator Improved Mitochondrial Function of Pancreatic Beta Cells
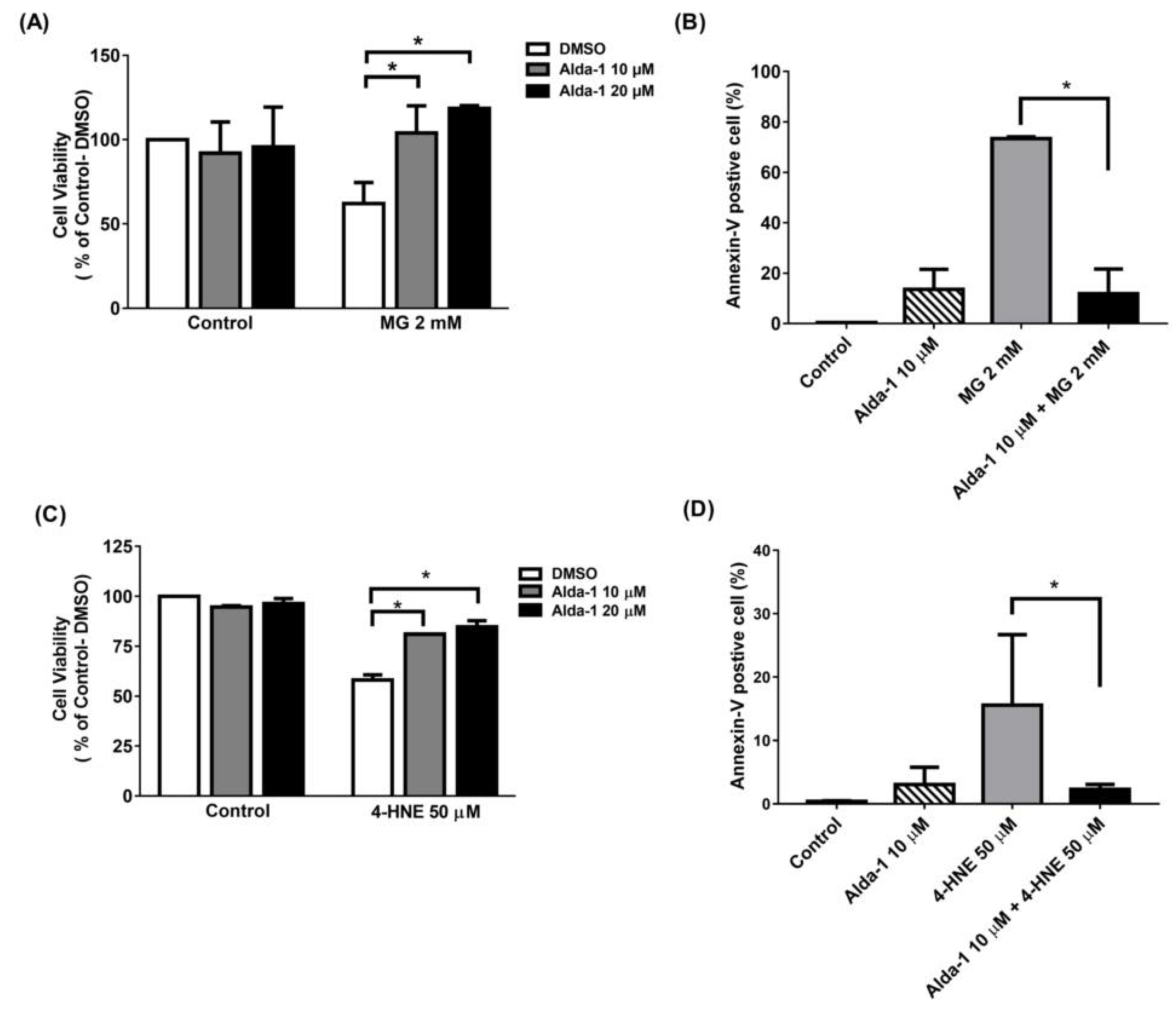
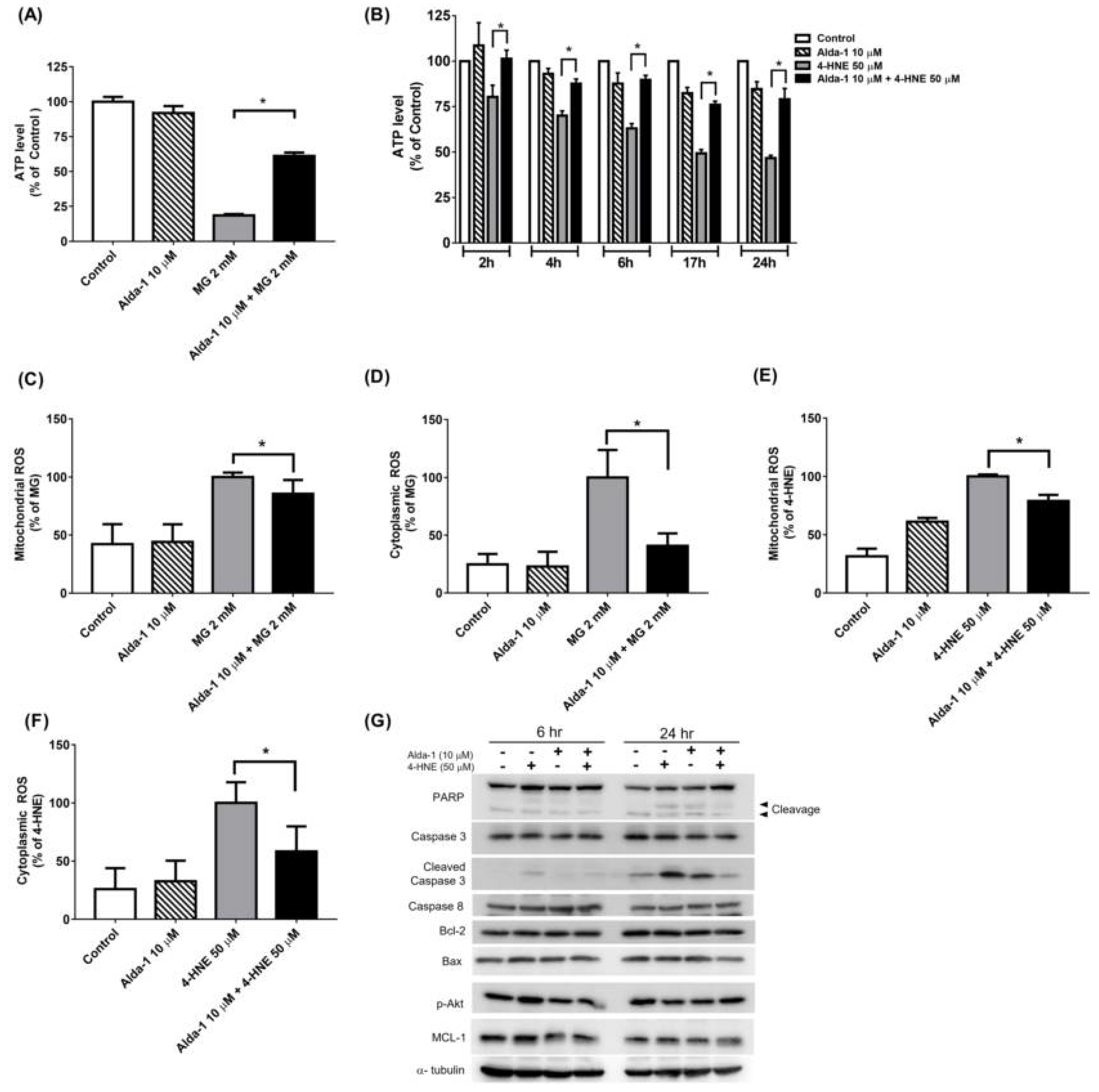
3.3. Alda-1 Rescued MIN6 Cells from MG- and 4-HNE- Induced Beta Cell Death and Apoptosis
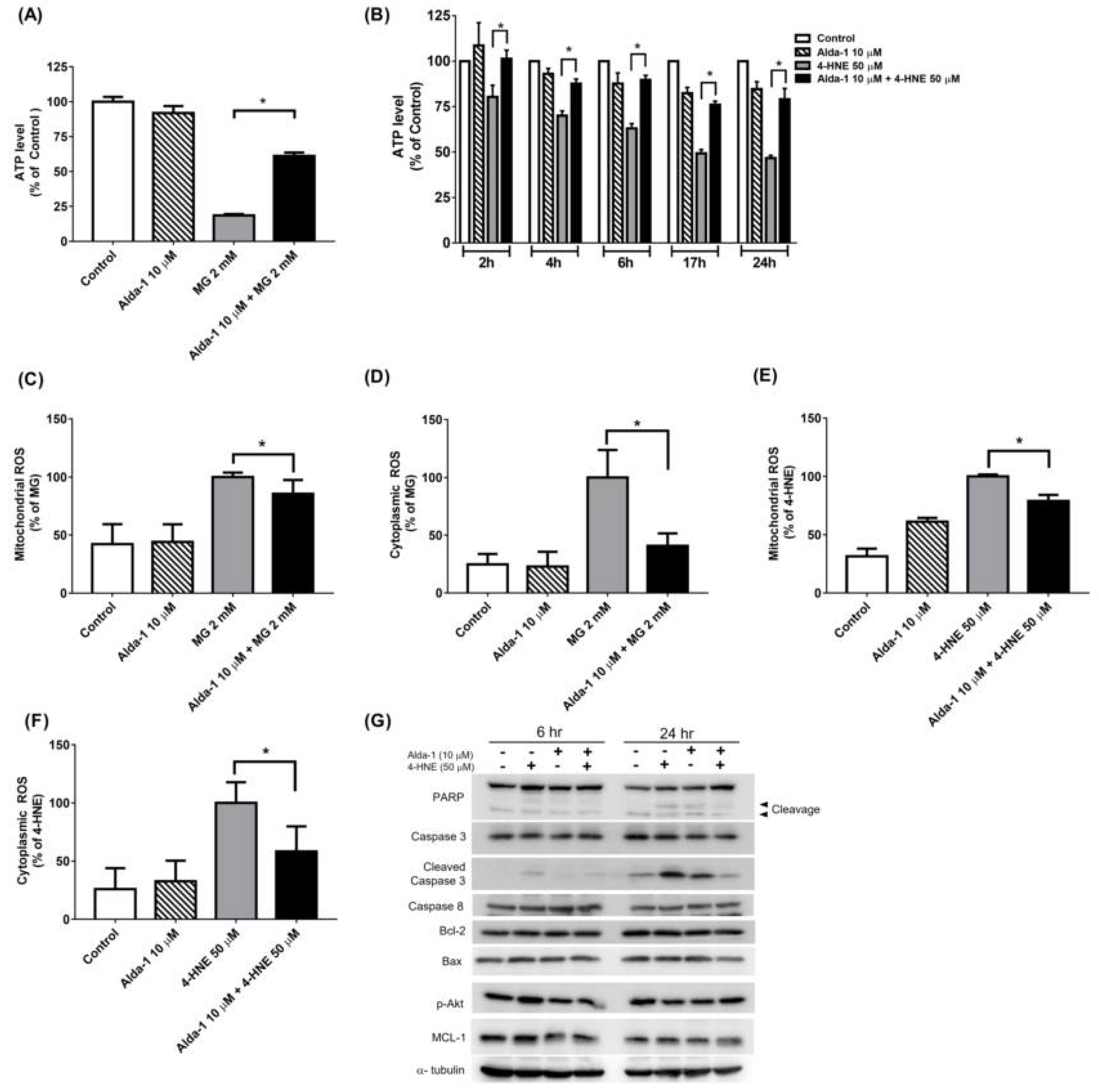
3.4. Alda-1 Rescued MG- and 4-HNE-Induced Mitochondrial Dysfunction in Beta Cells
3.5. Alda-1 Ameliorated MG- and 4-HNE-Increased Oxidative Stress in Mitochondria and Cytoplasm of Beta Cells
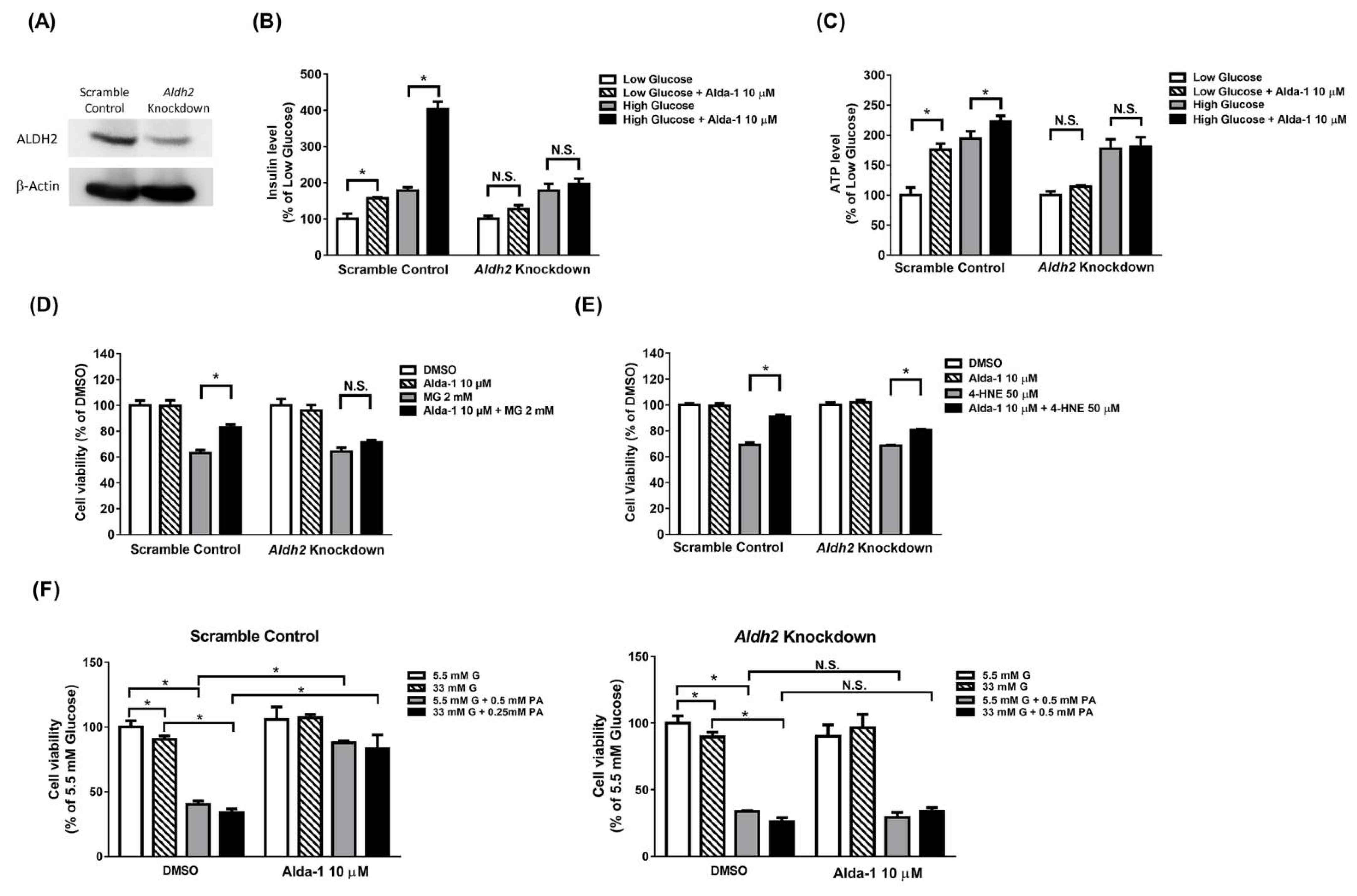
3.6. Potentiation Effect on Insulin Secretion and Mitochondrial Function of ALDH2 Activator Was Abolished in Aldh2 Knockdown MIN6 Cells
3.7. Beta Cell Death Evoked by MG and 4-HNE Cannot Be Fully Prevented by ALDH2 Activator in Aldh2 Knockdown MIN6 Cells
3.8. Beta Cell Death Induced by Either Hyperglycemia or Palmitate or Both Cannot Be Fully Prevented by ALDH2 Activator in Aldh2-Knockdown MIN6 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Brien, P.J.; Siraki, A.G.; Shangari, N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005, 35, 609–662. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53 (Suppl. 1), S119–S124. [Google Scholar] [CrossRef] [Green Version]
- Kohnke, R.; Mei, J.; Park, M.; York, D.A.; Erlanson-Albertsson, C. Fatty acids and glucose in high concentration down-regulates ATP synthase beta-subunit protein expression in INS-1 cells. Nutr. Neurosci. 2007, 10, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J. Type 2 diabetes-a matter of beta-cell life and death? Science 2005, 307, 380–384. [Google Scholar] [CrossRef]
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.; Sourris, K.C.; Penfold, S.A.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes 2008, 57, 460–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganjac, M.; Tirosh, O.; Cohen, G.; Sasson, S.; Zarkovic, N. Reactive aldehydes--second messengers of free radicals in diabetes mellitus. Free Radic. Res. 2013, 47 (Suppl. 1), 39–48. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.; Riahi, Y.; Shamni, O.; Guichardant, M.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Role of lipid peroxidation and PPAR-delta in amplifying glucose-stimulated insulin secretion. Diabetes 2011, 60, 2830–2842. [Google Scholar] [CrossRef] [Green Version]
- Sheader, E.A.; Benson, R.S.; Best, L. Cytotoxic action of methylglyoxal on insulin-secreting cells. Biochem. Pharmacol. 2001, 61, 1381–1386. [Google Scholar] [CrossRef]
- Chen, C.H.; Cruz, L.A.; Mochly-Rosen, D. Pharmacological recruitment of aldehyde dehydrogenase 3A1 (ALDH3A1) to assist ALDH2 in acetaldehyde and ethanol metabolism in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, 3074–3079. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Gilbert, S.A.; Li, Q.; Ren, J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J. Mol. Cell. Cardiol. 2009, 47, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.H.; Liao, P.R.; Guo, C.J.; Chen, C.H.; Mochly-Rosen, D.; Chuang, L.M. PKC-ALDH2 Pathway Plays a Novel Role in Adipocyte Differentiation. PLoS ONE 2016, 11, e0161993. [Google Scholar] [CrossRef]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, K.M.; Campos, J.C.; Bechara, L.R.; Queliconi, B.; Lima, V.M.; Disatnik, M.H.; Magno, P.; Chen, C.H.; Brum, P.C.; Kowaltowski, A.J.; et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc. Res. 2014, 103, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Hu, Q.; Fu, Z.; Wang, R.; Xiong, Y.; Zhang, Y.; Liu, Z.; Wang, Y.; Ye, Q. Increased Expression of Aldehyde Dehydrogenase 2 Reduces Renal Cell Apoptosis during Ischemia/Reperfusion Injury after Hypothermic Machine Perfusion. Artif. Organs 2016, 40, 596–603. [Google Scholar] [CrossRef]
- Li, D.S.; Yuan, Y.H.; Tu, H.J.; Liang, Q.L.; Dai, L.J. A protocol for islet isolation from mouse pancreas. Nat. Protoc. 2009, 4, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Nunemaker, C.S.; Wasserman, D.H.; McGuinness, O.P.; Sweet, I.R.; Teague, J.C.; Satin, L.S. Insulin secretion in the conscious mouse is biphasic and pulsatile. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E523–E529. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003, 289, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rorsman, P.; Renstrom, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045. [Google Scholar] [CrossRef] [PubMed]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, J. ALDH2 in alcoholic heart diseases: Molecular mechanism and clinical implications. Pharmacol. Ther. 2011, 132, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.U.; Van Wassenhove, L.D.; Logas, K.R.; Minhas, P.S.; Andreasson, K.I.; Weinberg, K.I.; Chen, C.H.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 activity and aldehydic load contribute to neuroinflammation and Alzheimer’s disease related pathology. Acta Neuropathol. Commun. 2019, 7, 190. [Google Scholar] [CrossRef] [Green Version]
- Kharroubi, I.; Ladriere, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: Role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 2004, 145, 5087–5096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Stein, D.T.; Stevenson, B.E.; Chester, M.W.; Basit, M.; Daniels, M.B.; Turley, S.D.; McGarry, J.D. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J. Clin. Investig. 1997, 100, 398–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 2011, 60, 200–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, G.; Shamni, O.; Avrahami, Y.; Cohen, O.; Broner, E.C.; Filippov-Levy, N.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Beta cell response to nutrient overload involves phospholipid remodelling and lipid peroxidation. Diabetologia 2015, 58, 1333–1343. [Google Scholar] [CrossRef]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Perez-Miller, S.; Younus, H.; Vanam, R.; Chen, C.H.; Mochly-Rosen, D.; Hurley, T.D. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat. Struct. Mol. Biol. 2010, 17, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, B.; Fan, X.; Wang, W.; Xu, T.; Wei, S.; Zheng, W.; Yuan, Q.; Gao, L.; Yin, X.; et al. Aldehyde Dehydrogenase 2 Protects against Post-Cardiac Arrest Myocardial Dysfunction through a Novel Mechanism of Suppressing Mitochondrial Reactive Oxygen Species Production. Front. Pharmacol. 2020, 11, 373. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Brouwers, O.; Stehouwer, C.D. Modulation of insulin action by advanced glycation end products: A new player in the field. Horm. Metab. Res. 2008, 40, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.J.; Tseng, H.C.; Liu, M.W.; Chang, Y.C.; Hsieh, M.L.; Chuang, L.M. Glucagon-like peptide-1 prevents methylglyoxal-induced apoptosis of beta cells through improving mitochondrial function and suppressing prolonged AMPK activation. Sci. Rep. 2016, 6, 23403. [Google Scholar] [CrossRef] [PubMed]
- Shangari, N.; O’Brien, P.J. The cytotoxic mechanism of glyoxal involves oxidative stress. Biochem. Pharmacol. 2004, 68, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- De Arriba, S.G.; Stuchbury, G.; Yarin, J.; Burnell, J.; Loske, C.; Munch, G. Methylglyoxal impairs glucose metabolism and leads to energy depletion in neuronal cells--Protection by carbonyl scavengers. Neurobiol. Aging 2007, 28, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, J.; Wu, L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem. Pharmacol. 2009, 77, 1709–1716. [Google Scholar] [CrossRef]
- Robertson, R.P. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J. Biol. Chem. 2004, 279, 42351–42354. [Google Scholar] [CrossRef] [Green Version]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in beta-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Delghingaro-Augusto, V.; Nolan, C.J.; Turner, N.; Hallahan, N.; Andrikopoulos, S.; Gunton, J.E. High passage MIN6 cells have impaired insulin secretion with impaired glucose and lipid oxidation. PLoS ONE 2012, 7, e40868. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, L.; Gammell, P.; McKiernan, E.; Ryan, E.; Jeppesen, P.B.; Rani, S.; Clynes, M. Phenotypic and global gene expression profile changes between low passage and high passage MIN-6 cells. J. Endocrinol. 2006, 191, 665–676. [Google Scholar] [CrossRef]
- Matthews, D.R.; Cull, C.A.; Stratton, I.M.; Holman, R.R.; Turner, R.C. UKPDS 26: Sulphonylurea failure in non-insulin-dependent diabetic patients over six years. UK Prospective Diabetes Study (UKPDS) Group. Diabet. Med. 1998, 15, 297–303. [Google Scholar] [CrossRef]
- Donath, M.Y.; Halban, P.A. Decreased beta-cell mass in diabetes: Significance, mechanisms and therapeutic implications. Diabetologia 2004, 47, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Harrity, T.; Farrelly, D.; Tieman, A.; Chu, C.; Kunselman, L.; Gu, L.; Ponticiello, R.; Cap, M.; Qu, F.; Shao, C.; et al. Muraglitazar, a novel dual (alpha/gamma) peroxisome proliferator-activated receptor activator, improves diabetes and other metabolic abnormalities and preserves beta-cell function in db/db mice. Diabetes 2006, 55, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leahy, J.L.; Hirsch, I.B.; Peterson, K.A.; Schneider, D. Targeting beta-cell function early in the course of therapy for type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2010, 95, 4206–4216. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-M.; Hee, S.-W.; Chou, S.-Y.; Liu, M.-W.; Chen, C.-H.; Mochly-Rosen, D.; Chang, T.-J.; Chuang, L.-M. Activation of Aldehyde Dehydrogenase 2 Ameliorates Glucolipotoxicity of Pancreatic Beta Cells. Biomolecules 2021, 11, 1474. https://doi.org/10.3390/biom11101474
Chen S-M, Hee S-W, Chou S-Y, Liu M-W, Chen C-H, Mochly-Rosen D, Chang T-J, Chuang L-M. Activation of Aldehyde Dehydrogenase 2 Ameliorates Glucolipotoxicity of Pancreatic Beta Cells. Biomolecules. 2021; 11(10):1474. https://doi.org/10.3390/biom11101474
Chicago/Turabian StyleChen, Shiau-Mei, Siow-Wey Hee, Shih-Yun Chou, Meng-Wei Liu, Che-Hong Chen, Daria Mochly-Rosen, Tien-Jyun Chang, and Lee-Ming Chuang. 2021. "Activation of Aldehyde Dehydrogenase 2 Ameliorates Glucolipotoxicity of Pancreatic Beta Cells" Biomolecules 11, no. 10: 1474. https://doi.org/10.3390/biom11101474
APA StyleChen, S.-M., Hee, S.-W., Chou, S.-Y., Liu, M.-W., Chen, C.-H., Mochly-Rosen, D., Chang, T.-J., & Chuang, L.-M. (2021). Activation of Aldehyde Dehydrogenase 2 Ameliorates Glucolipotoxicity of Pancreatic Beta Cells. Biomolecules, 11(10), 1474. https://doi.org/10.3390/biom11101474