
Design of Inhibitors of the Intrinsically Disordered Protein NUPR1: Balance between Drug Affinity and Target Function

 ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chemistry: General Methods
2.3. Protein Expression and Purification
2.4. Computational Modelling of ZZW-115-Derived Compounds
2.5. Organic Synthesis of ZZW-115-Derived Compounds
2.6. Cell Production and Viability Assays
2.7. Isothermal Titration Calorimetry (ITC)
2.8. Immunofluorescence Staining
2.9. Genotoxicity Evaluation for Compounds Combination
2.10. Statistics
3. Results
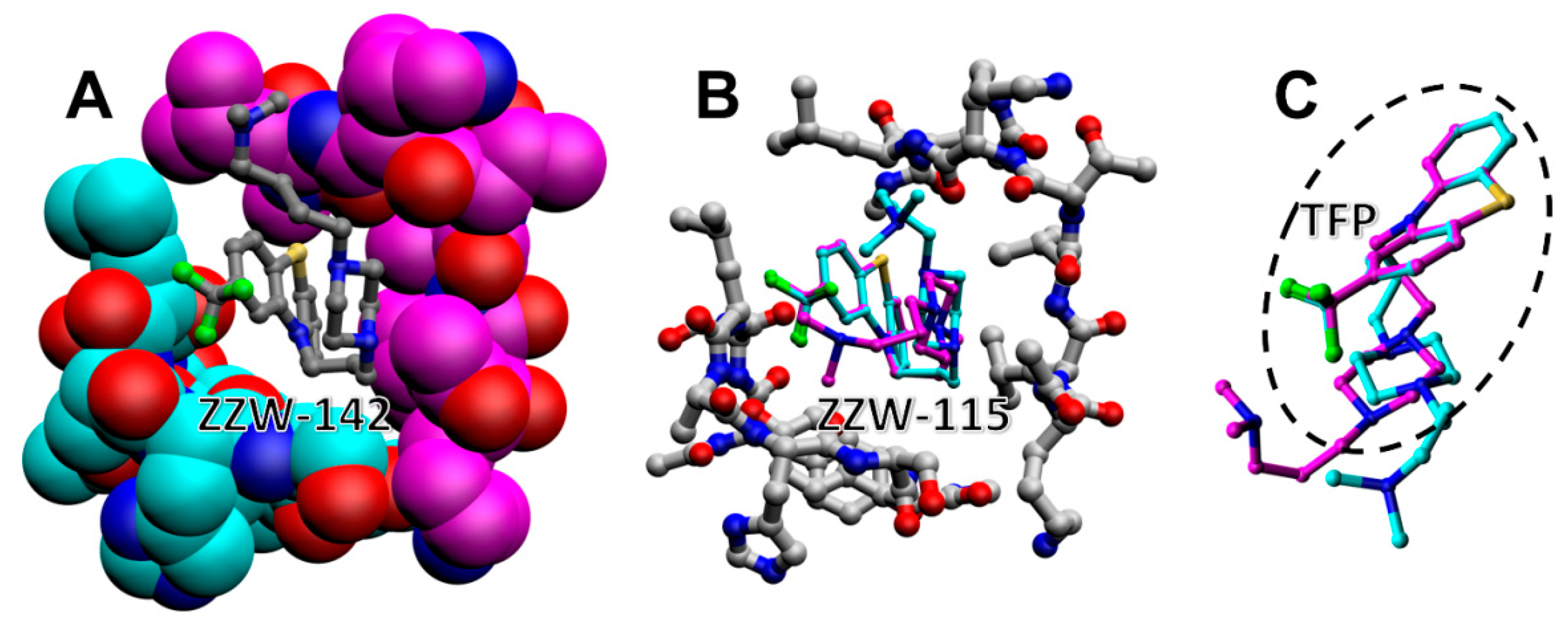
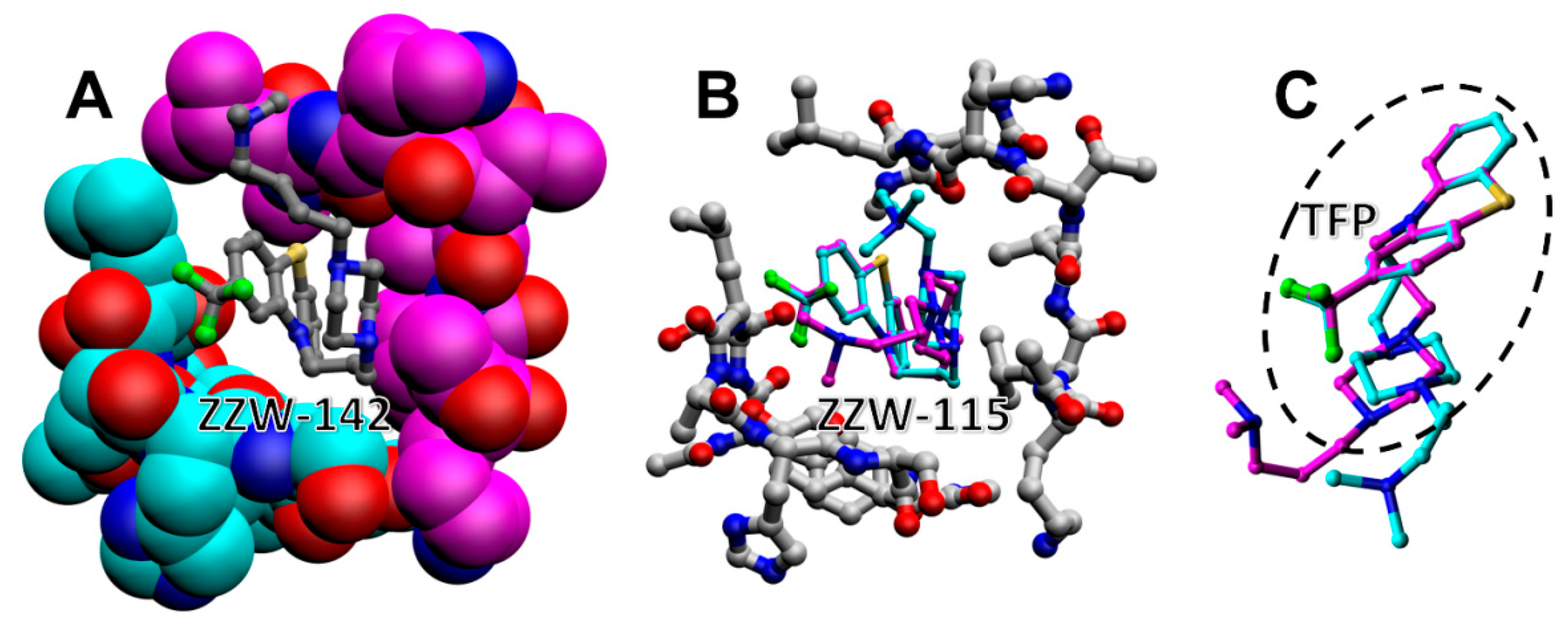
3.1. Docking of Compounds to a Simulated Binding Pocket of NUPR1
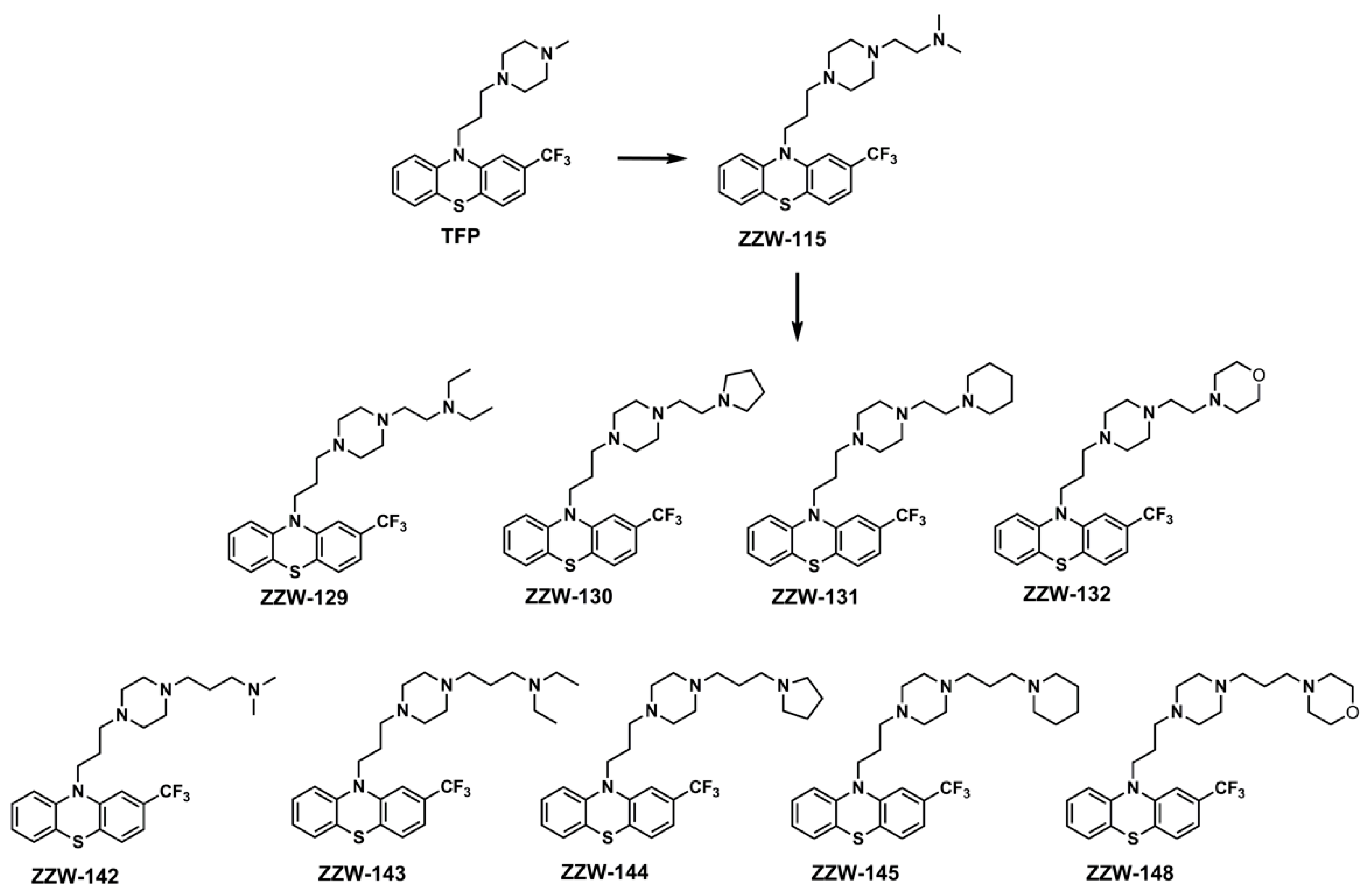
3.2. Synthesis of the ZZW-115-Derived Compounds
3.3. Isothermal Titration Calorimetry in the Presence of ZZW-115-Derived Compounds
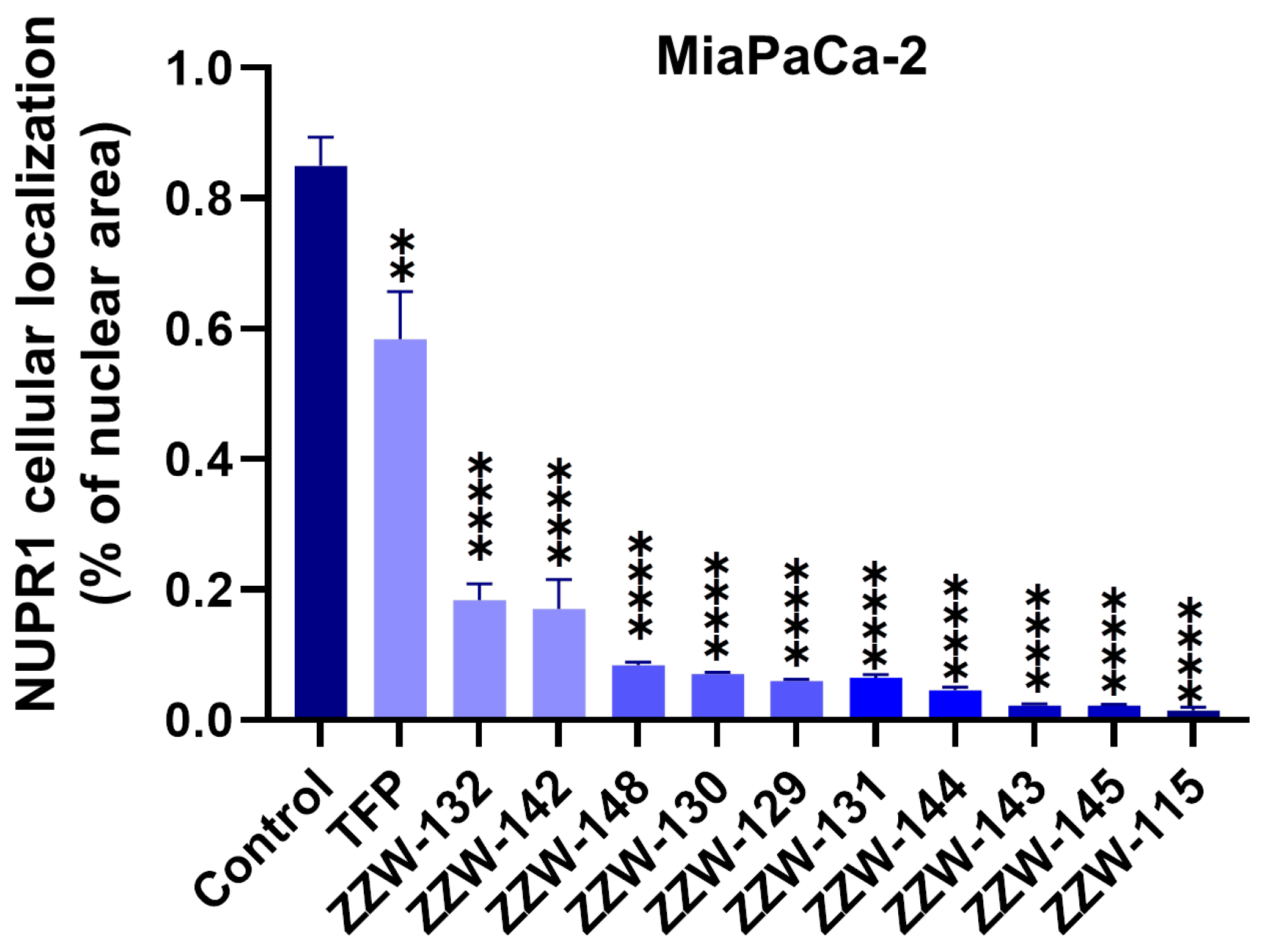
3.4. Effects of ZZW-115-Derived Compounds on Nuclear Translocation of NUPR1
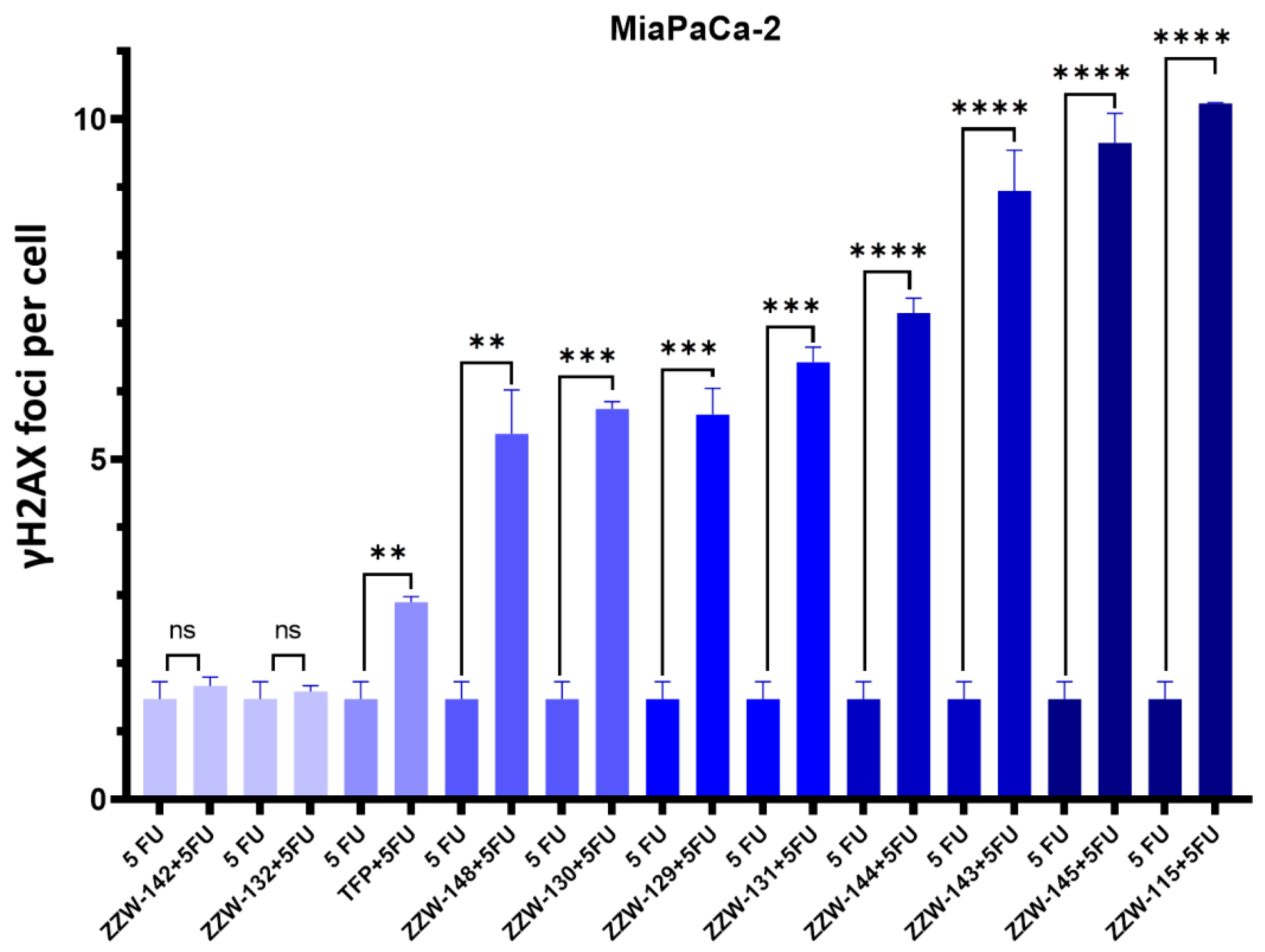
3.5. Treatment with ZZW-115-Derived Compounds Sensitizes Cancer Cells to Genotoxic-induced DNA Damage
3.6. Effects of ZZW-115-Derived Compounds on Pancreatic Cancer Cell Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically Disordered Proteins: Regulation and Disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional Anthology of Intrinsic Disorder. 1. Biological Processes and Functions of Proteins with Long Disordered Regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Biol. 2018, 430, 2309–2320. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight Regulation of Unstructured Proteins: From Transcript Synthesis to Protein Degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int. J. Mol. Sci. 2017, 18, 2761. [Google Scholar] [CrossRef] [Green Version]
- Mallo, G.V.; Fiedler, F.; Calvo, E.L.; Ortiz, E.M.; Vasseur, S.; Keim, V.; Morisset, J.; Iovanna, J.L. Cloning and Expression of the Rat P8 CDNA, a New Gene Activated in Pancreas during the Acute Phase of Pancreatitis, Pancreatic Development, and Regeneration, and Which Promotes Cellular Growth. J. Biol. Chem. 1997, 272, 32360–32369. [Google Scholar] [CrossRef] [Green Version]
- Cano, C.E.; Hamidi, T.; Sandi, M.J.; Iovanna, J.L. Nupr1: The Swiss-Knife of Cancer. J. Cell Physiol. 2011, 226, 1439–1443. [Google Scholar] [CrossRef]
- Goruppi, S.; Iovanna, J.L. Stress-Inducible Protein P8 Is Involved in Several Physiological and Pathological Processes. J. Biol. Chem. 2010, 285, 1577–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, U.R.; Samant, R.S.; Fodstad, O.; Shevde, L.A. Emerging Role of Nuclear Protein 1 (NUPR1) in Cancer Biology. Cancer Metastasis Rev. 2009, 28, 225–232. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Xia, Y.; Peng, L.; Velázquez-Campoy, A.; Abián, O.; Lan, W.; Lomberk, G.; Urrutia, R.; Rizzuti, B.; Soubeyran, P.; et al. Targeting the Stress-Induced Protein NUPR1 to Treat Pancreatic Adenocarcinoma. Cells 2019, 8, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malicet, C.; Giroux, V.; Vasseur, S.; Dagorn, J.C.; Neira, J.L.; Iovanna, J.L. Regulation of Apoptosis by the P8/Prothymosin Alpha Complex. Proc Natl Acad Sci USA 2006, 103, 2671–2676. [Google Scholar] [CrossRef] [Green Version]
- Aguado-Llera, D.; Hamidi, T.; Doménech, R.; Pantoja-Uceda, D.; Gironella, M.; Santoro, J.; Velázquez-Campoy, A.; Neira, J.L.; Iovanna, J.L. Deciphering the Binding between Nupr1 and MSL1 and Their DNA-Repairing Activity. PLoS ONE 2013, 8, e78101. [Google Scholar] [CrossRef] [PubMed]
- Encinar, J.A.; Mallo, G.V.; Mizyrycki, C.; Giono, L.; Gonzalez-Ros, J.M.; Rico, M.; Cánepa, E.; Moreno, S.; Neira, J.L.; Iovanna, J.L. Human P8 Is a HMG-I/Y-like Protein with DNA Binding Activity Enhanced by Phosphorylation. J. Biol. Chem. 2001, 276, 2742–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santofimia-Castaño, P.; Rizzuti, B.; Pey, Á.L.; Soubeyran, P.; Vidal, M.; Urrutia, R.; Iovanna, J.L.; Neira, J.L. Intrinsically Disordered Chromatin Protein NUPR1 Binds to the C-Terminal Region of Polycomb RING1B. Proc. Natl. Acad. Sci. USA 2017, 114, E6332–E6341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandi, M.J.; Hamidi, T.; Malicet, C.; Cano, C.; Loncle, C.; Pierres, A.; Dagorn, J.C.; Iovanna, J.L. P8 Expression Controls Pancreatic Cancer Cell Migration, Invasion, Adhesion, and Tumorigenesis. J. Cell Physiol. 2011, 226, 3442–3451. [Google Scholar] [CrossRef]
- Vasseur, S.; Hoffmeister, A.; Garcia, S.; Bagnis, C.; Dagorn, J.-C.; Iovanna, J.L. P8 Is Critical for Tumour Development Induced by RasV12 Mutated Protein and E1A Oncogene. EMBO Rep. 2002, 3, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Emma, M.R.; Iovanna, J.L.; Bachvarov, D.; Puleio, R.; Loria, G.R.; Augello, G.; Candido, S.; Libra, M.; Gulino, A.; Cancila, V.; et al. NUPR1, a New Target in Liver Cancer: Implication in Controlling Cell Growth, Migration, Invasion and Sorafenib Resistance. Cell Death Dis. 2016, 7, e2269. [Google Scholar] [CrossRef]
- Kim, K.-S.; Jin, D.-I.; Yoon, S.; Baek, S.-Y.; Kim, B.-S.; Oh, S.-O. Expression and Roles of NUPR1 in Cholangiocarcinoma Cells. Anat. Cell Biol. 2012, 45, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Wang, W.; Hu, J.; Feng, K.; Pan, Y.; Zhang, L.; Feng, Y. Lentivirus-Mediated RNAi Knockdown of NUPR1 Inhibits Human Nonsmall Cell Lung Cancer Growth in Vitro and in Vivo. Anat. Rec. (Hoboken) 2012, 295, 2114–2121. [Google Scholar] [CrossRef]
- Li, J.; Ren, S.; Liu, Y.; Lian, Z.; Dong, B.; Yao, Y.; Xu, Y. Knockdown of NUPR1 Inhibits the Proliferation of Glioblastoma Cells via ERK1/2, P38 MAPK and Caspase-3. J. Neurooncol. 2017, 132, 15–26. [Google Scholar] [CrossRef]
- Zhou, C.; Xu, J.; Lin, J.; Lin, R.; Chen, K.; Kong, J.; Shui, X. Long Noncoding RNA FEZF1-AS1 Promotes Osteosarcoma Progression by Regulating the MiR-4443/NUPR1 Axis. Oncol. Res. 2018, 26, 1335–1343. [Google Scholar] [CrossRef]
- Zeng, C.; Li, X.; Li, A.; Yi, B.; Peng, X.; Huang, X.; Chen, J. Knockdown of NUPR1 Inhibits the Growth of U266 and RPMI8226 Multiple Myeloma Cell Lines via Activating PTEN and Caspase Activation--dependent Apoptosis. Oncol. Rep. 2018, 40, 1487–1494. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velázquez-Campoy, A.; Iovanna, J.L.; Neira, J.L. Designing and Repurposing Drugs to Target Intrinsically Disordered Proteins for Cancer Treatment: Using NUPR1 as a Paradigm. Mol. Cell Oncol. 2019, 6, e1612678. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velázquez-Campoy, A.; Abián, O.; Rizzuti, B.; et al. Ligand-Based Design Identifies a Potent NUPR1 Inhibitor Exerting Anticancer Activity via Necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.; Santofimia-Castaño, P.; Swayden, M.; Xia, Y.; Zhou, Z.; Audebert, S.; Camoin, L.; Huang, C.; Peng, L.; Jiménez-Alesanco, A.; et al. ZZW-115-Dependent Inhibition of NUPR1 Nuclear Translocation Sensitizes Cancer Cells to Genotoxic Agents. JCI Insight 2020, 5, e138117. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational Methods in Drug Discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [Green Version]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of Computer-Aided Drug Design in Modern Drug Discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Rizzuti, B.; Grande, F. Virtual screening in drug discovery: A precious tool for a still-demanding challenge. In Protein Homeostasis Diseases; Elsevier BV: Amsterdam, The Netherlands, 2020; pp. 309–327. [Google Scholar]
- Gill, S.C.; von Hippel, P.H. Calculation of Protein Extinction Coefficients from Amino Acid Sequence Data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, A.; Rizzuti, B.; Guzzi, R. Stereoselective and Domain-Specific Effects of Ibuprofen on the Thermal Stability of Human Serum Albumin. Eur. J. Pharm. Sci. 2018, 112, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; Rizzuti, B.; Iovanna, J.L. Determinants of the pKa Values of Ionizable Residues in an Intrinsically Disordered Protein. Arch. Biochem. Biophys. 2016, 598, 18–27. [Google Scholar] [CrossRef]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenograft. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, F.S.; Deramaudt, T.B.; Brunner, T.B.; Boretti, M.I.; Gooch, K.J.; Stoffers, D.A.; Bernhard, E.J.; Rustgi, A.K. Successful Growth and Characterization of Mouse Pancreatic Ductal Cells: Functional Properties of the Ki-RAS(G12V) Oncogene. Gastroenterology 2004, 127, 250–260. [Google Scholar] [CrossRef]
- Duconseil, P.; Gilabert, M.; Gayet, O.; Loncle, C.; Moutardier, V.; Turrini, O.; Calvo, E.; Ewald, J.; Giovannini, M.; Gasmi, M.; et al. Transcriptomic Analysis Predicts Survival and Sensitivity to Anticancer Drugs of Patients with a Pancreatic Adenocarcinoma. Am. J. Pathol. 2015, 185, 1022–1032. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Abián, O.; Velázquez-Campoy, A.; Iovanna, J.L.; Neira, J.L. Amphipathic Helical Peptides Hamper Protein-Protein Interactions of the Intrinsically Disordered Chromatin Nuclear Protein 1 (NUPR1). BBA Gen. Subj. 2018, 1862, 1283–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein-Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Ruan, H.; Sun, Q.; Zhang, W.; Liu, Y.; Lai, L. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velázquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell Mol. Life Sci. 2020, 77, 1695–1707. [Google Scholar] [CrossRef] [Green Version]
- Bumbak, F.; Thomas, T.; Noonan-Williams, B.J.; Vaid, T.M.; Yan, F.; Whitehead, A.R.; Bruell, S.; Kocan, M.; Tan, X.; Johnson, M.A.; et al. Conformational Changes in Tyrosine 11 of Neurotensin Are Required to Activate the Neurotensin Receptor 1. ACS Pharmacol. Transl. Sci. 2020, 3, 690–705. [Google Scholar] [CrossRef]
- Loening, N.M.; Saravanan, S.; Jespersen, N.E.; Jara, K.; Barbar, E. Interplay of Disorder and Sequence Specificity in the Formation of Stable Dynein-Dynactin Complexes. Biophys. J. 2020, 119, 950–965. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.T.; Sormanni, P.; Vendruscolo, M. Targeting Disordered Proteins with Small Molecules Using Entropy. Trends Biochem. Sci. 2015, 40, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Heller, G.T.; Bonomi, M.; Vendruscolo, M. Structural Ensemble Modulation upon Small-Molecule Binding to Disordered Proteins. J. Mol. Biol. 2018, 430, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Crespo, A.; Fernández, A. Induced Disorder in Protein-Ligand Complexes as a Drug-Design Strategy. Mol. Pharm. 2008, 5, 430–437. [Google Scholar] [CrossRef]
- Diehl, C.; Engström, O.; Delaine, T.; Håkansson, M.; Genheden, S.; Modig, K.; Leffler, H.; Ryde, U.; Nilsson, U.J.; Akke, M. Protein Flexibility and Conformational Entropy in Ligand Design Targeting the Carbohydrate Recognition Domain of Galectin-3. J. Am. Chem. Soc. 2010, 132, 14577–14589. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Kang, L.-W.; Velazquez-Campoy, A.; Kiso, Y.; Amzel, L.M.; Freire, E. A Structural and Thermodynamic Escape Mechanism from a Drug Resistant Mutation of the HIV-1 Protease. Proteins 2004, 55, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; He, S.; Nowak, R.P.; Wang, J.; Zimmerman, M.W.; Fu, C.; Durbin, A.D.; Martel, M.W.; Prutsch, N.; Gray, N.S.; et al. Allosteric Activators of Protein Phosphatase 2A Display Broad Antitumor Activity Mediated by Dephosphorylation of MYBL2. Cell 2020, 181, 702–715.e20. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ka (M–1) × 105 | Kd (µM) (=1/Ka) a | ΔG (kcal mol–1) a | ΔH (kcal mol–1) a | −TΔS (kcal mol–1) a | N a |
|---|---|---|---|---|---|---|
| ZZW-129 | 3.1 ± 0.3 | 3.2 ± 0.3 | −7.5 ± 0.1 | −0.7 ± 0.4 | −6.8 ± 0.4 | 1.2 ± 0.1 |
| ZZW-130 | 6.4 ± 0.4 | 1.6 ± 0.1 | −7.9 ± 0.1 | 0.3 ± 0.3 | −8.2 ± 0.3 | 1.4 ± 0.1 |
| ZZW-131 | 4.7 ± 0.3 | 2.1 ± 0.1 | −7.7 ± 0.1 | 0.5 ± 0.3 | −8.2 ± 0.3 | 0.8 ± 0.1 |
| ZZW-132 | 5.8 ± 0.4 | 1.7 ± 0.1 | −7.9 ± 0.1 | 0.3 ± 0.3 | −8.2 ± 0.3 | 1.2 ± 0.1 |
| ZZW-142 | 4.9 ± 0.3 | 2.0 ± 0.2 | −7.8 ± 0.1 | 0.3 ± 0.3 | −8.1 ± 0.3 | 1.3 ± 0.1 |
| ZZW-143 | 0.5 ± 0.1 | 20 ± 4 | −6.4 ± 0.1 | 0.8 ± 0.4 | −7.2 ± 0.4 | 1.2 ± 0.1 |
| ZZW-144 | 0.85 ± 0.8 | 12 ± 1 | −6.7 ± 0.1 | 0.5 ± 0.5 | −7.2 ± 0.5 | 1.2 ± 0.1 |
| ZZW-145 | 0.82 ± 0.9 | 11 ± 1 | −6.7 ± 0.1 | 0.7 ± 0.5 | −7.4 ± 0.5 | 1.2 ± 0.1 |
| ZZW-148 | 1.0 ± 0.1 | 9.6 ± 1.0 | −6.8 ± 0.1 | 0.7 ± 0.5 | −7.5 ± 0.5 | 1.2 ± 0.1 |
| ZZW-115 b | 4.7 ± 0.4 | 2.1 ± 0.2 | −7.7 ± 0.1 | −0.4 ± 0.3 | −7.3 ± 0.3 | 0.9 ± 0.1 |
| TFP b | 1.9 ± 0.2 | 5.2 ± 0.6 | −7.2 ± 0.1 | −1.1 ± 0.4 | −6.1 ± 0.4 | 1.0 ± 0.1 |
| PDAC001T | PDAC012T | PDAC021T | PDAC081T | PDAC082T | PDAC087T | PDAC088T | PDAC089T | PDAC115T | MiaPaCa-2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ZZW-129 | 1.94 | 2.77 | 3.41 | 0.86 | 3.56 | 4.28 | 7.28 | 7.90 | 2.46 | 0.33 |
| ZZW-130 | 2.45 | 4.18 | 3.81 | 1.12 | 4.33 | 4.95 | 6.75 | 6.03 | 2.91 | 0.35 |
| ZZW-131 | 3.98 | 5.25 | 6.47 | 0.97 | 6.49 | 4.43 | 8.11 | 10.59 | 5.75 | 0.30 |
| ZZW-132 | 12.47 | 12.21 | 16.50 | 1.91 | 21.10 | 16.55 | 23.63 | 29.10 | 16.06 | 1.04 |
| ZZW-142 | 11.93 | 12.78 | 17.01 | 2.27 | 22.97 | 16.55 | 23.92 | 22.46 | 14.70 | 0.95 |
| ZZW-143 | 2.31 | 2.18 | 2.57 | 0.97 | 3.55 | 2.22 | 5.94 | 6.12 | 2.50 | 0.29 |
| ZZW-144 | 2.51 | 2.51 | 3.69 | 0.98 | 3.04 | 3.57 | 5.80 | 8.38 | 3.22 | 0.26 |
| ZZW-145 | 2.36 | 2.36 | 2.33 | 0.78 | 2.33 | 2.37 | 5.00 | 6.22 | 2.44 | 0.19 |
| ZZW-148 | 3.75 | 3.70 | 4.35 | 1.11 | 4.10 | 6.57 | 10.73 | 14.77 | 4.37 | 0.47 |
| ZZW-115 | 1.84 | 2.44 | 2.54 | 0.84 | 2.36 | 3.72 | 5.29 | 4.72 | 2.21 | 0.32 |
| TFP | 12.48 | 20.39 | 23.51 | 2.48 | 21.47 | 7.80 | 15.90 | 8.09 | 10.12 | 9.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzuti, B.; Lan, W.; Santofimia-Castaño, P.; Zhou, Z.; Velázquez-Campoy, A.; Abián, O.; Peng, L.; Neira, J.L.; Xia, Y.; Iovanna, J.L. Design of Inhibitors of the Intrinsically Disordered Protein NUPR1: Balance between Drug Affinity and Target Function. Biomolecules 2021, 11, 1453. https://doi.org/10.3390/biom11101453
Rizzuti B, Lan W, Santofimia-Castaño P, Zhou Z, Velázquez-Campoy A, Abián O, Peng L, Neira JL, Xia Y, Iovanna JL. Design of Inhibitors of the Intrinsically Disordered Protein NUPR1: Balance between Drug Affinity and Target Function. Biomolecules. 2021; 11(10):1453. https://doi.org/10.3390/biom11101453
Chicago/Turabian StyleRizzuti, Bruno, Wenjun Lan, Patricia Santofimia-Castaño, Zhengwei Zhou, Adrián Velázquez-Campoy, Olga Abián, Ling Peng, José L. Neira, Yi Xia, and Juan L. Iovanna. 2021. "Design of Inhibitors of the Intrinsically Disordered Protein NUPR1: Balance between Drug Affinity and Target Function" Biomolecules 11, no. 10: 1453. https://doi.org/10.3390/biom11101453
APA StyleRizzuti, B., Lan, W., Santofimia-Castaño, P., Zhou, Z., Velázquez-Campoy, A., Abián, O., Peng, L., Neira, J. L., Xia, Y., & Iovanna, J. L. (2021). Design of Inhibitors of the Intrinsically Disordered Protein NUPR1: Balance between Drug Affinity and Target Function. Biomolecules, 11(10), 1453. https://doi.org/10.3390/biom11101453