Development of Possible Next Line of Systemic Therapies for Gemcitabine-Resistant Biliary Tract Cancers: A Perspective from Clinical Trials


 and
and
Abstract
1. Introduction
2. Chemotherapy
3. Targeted Therapy
3.1. FGFR2 Fusions
3.2. IDH1/2 Mutations and PARP
4. EGFR/HER2
5. RAF/MEK
6. c-MET
7. PI3K/AKT/mTOR
8. Anti-Angiogenesis Agents
9. NTRK
10. Immune Checkpoint Inhibitors
11. Complementary Agents
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tariq, N.U.; McNamara, M.G.; Valle, J.W. Biliary tract cancers: Current knowledge, clinical candidates and future challenges. Cancer Manag. Res. 2019, 11, 2623–2642. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.M.; Vogel, A. Epidemiology and Risk Factors of Cholangiocarcinoma. Visc. Med. 2016, 32, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Bragazzi, M.C.; Cardinale, V.; Carpino, G.; Venere, R.; Semeraro, R.; Gentile, R.; Gaudio, E.; Alvaro, D. Cholangiocarcinoma: Epidemiology and risk factors. Transl. Gastrointest. Cancer 2011, 1, 21–32. [Google Scholar]
- Pellino, A.; Loupakis, F.; Cadamuro, M.; Dadduzio, V.; Fassan, M.; Guido, M.; Cillo, U.; Indraccolo, S.; Fabris, L. Precision medicine in cholangiocarcinoma. Transl. Gastroenterol. Hepatol. 2018, 3, 40. [Google Scholar] [CrossRef]
- Lee, T.Y.; Hsu, Y.C.; Yu, S.H.; Lin, J.T.; Wu, M.S.; Wu, C.Y. Effect of Nucleos(t)ide Analogue Therapy on Risk of Intrahepatic Cholangiocarcinoma in Patients With Chronic Hepatitis B. Clin Gastroenterol Hepatol. 2018, 16, 947–954.e94. [Google Scholar] [CrossRef]
- Okusaka, T.; Nakachi, K.; Fukutomi, A.; Mizuno, N.; Ohkawa, S.; Funakoshi, A.; Nagino, M.; Kondo, S.; Nagaoka, S.; Funai, J.; et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: A comparative multicentre study in Japan. Br. J. Cancer 2010, 103, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Morizane, C.; Okusaka, T.; Mizusawa, J.; Katayama, H.; Ueno, M.; Ikeda, M.; Ozaka, M.; Okano, N.; Sugimori, K.; Fukutomi, A.; et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: The FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann. Oncol. 2019, 30, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Morizane, C.; Okusaka, T.; Mizusawa, J.; Takashima, A.; Ueno, M.; Ikeda, M.; Hamamoto, Y.; Ishii, H.; Boku, N.; Furuse, J. Randomized phase II study of gemcitabine plus S-1 versus S-1 in advanced biliary tract cancer: A Japan Clinical Oncology Group trial (JCOG 0805). Cancer Sci. 2013, 104, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Kanai, M.; Kobayashi, S.; Eguchi, H.; Baba, H.; Seo, S.; Taketomi, A.; Takayama, T.; Yamaue, H.; Ishioka, C.; et al. Randomized phase III study of gemcitabine, cisplatin plus S-1 (GCS) versus gemcitabine, cisplatin (GC) for advanced biliary tract cancer (KHBO1401-MITSUBA). Ann. Oncol. 2018, 29, viii205. [Google Scholar] [CrossRef]
- Fornaro, L.; Cereda, S.; Aprile, G.; Di Girolamo, S.; Santini, D.; Silvestris, N.; Lonardi, S.; Leone, F.; Milella, M.; Vivaldi, C.; et al. Multivariate prognostic factors analysis for second-line chemotherapy in advanced biliary tract cancer. Br. J. Cancer. 2014, 110, 2165–2169. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Hubner, R.A.; David Ryder, W.; Valle, J.W. Second-line chemotherapy in advanced biliary cancer: A systematic review. Ann. Oncol. 2014, 25, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Wardell, C.P.; Fujita, M.; Yamada, T.; Simbolo, M.; Fassan, M.; Karlic, R.; Polak, P.; Kim, J.; Hatanaka, Y.; Maejima, K.; et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 2018, 68, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Krook, M.A.; Lenyo, A.; Wilberding, M.; Barker, H.; Dantuono, M.; Bailey, K.M.; Chen, H.-Z.; Reeser, J.W.; Wing, M.R.; Miya, J.; et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol. Cancer Ther. 2020. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Molecular targeted therapies: Ready for prime time in biliary tract cancer. J. Hepatol. 2020, 73, 170–185. [Google Scholar] [CrossRef]
- Rizvi, S.; Gores, G.J. Emerging molecular therapeutic targets for cholangiocarcinoma. J. Hepatol. 2017, 67, 632–644. [Google Scholar] [CrossRef]
- Jusakul, A.; Kongpetch, S.; Teh, B.T. Genetics of Opisthorchis viverrini-related cholangiocarcinoma. Curr. Opin. Gastroenterol. 2015, 31, 258–263. [Google Scholar] [CrossRef]
- Cao, J.; Hu, J.; Liu, S.; Meric-Bernstam, F.; Abdel-Wahab, R.; Xu, J.; Li, Q.; Yan, M.; Feng, Y.; Lin, J.; et al. Intrahepatic Cholangiocarcinoma: Genomic Heterogeneity Between Eastern and Western Patients. JCO Precis. Oncol. 2020, 557–569. [Google Scholar] [CrossRef]
- Ying, J.; Chen, J. Combination versus mono-therapy as salvage treatment for advanced biliary tract cancer: A comprehensive meta-analysis of published data. Crit. Rev. Oncol. Hematol. 2019, 139, 134–142. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. ABC-06 | A randomised phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin/5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. J. Clin. Oncol. 2019, 37, 4003. [Google Scholar] [CrossRef]
- Brieau, B.; Dahan, L.; De Rycke, Y.; Boussaha, T.; Vasseur, P.; Tougeron, D.; Lecomte, T.; Coriat, R.; Bachet, J.B.; Claudez, P.; et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: A large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 2015, 121, 3290–3297. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Ikeda, M.; Okusaka, T.; Nakamori, S.; Ohkawa, S.; Nagakawa, T.; Boku, N.; Yanagimoto, H.; Sato, T.; Furuse, J. A multicenter phase II study of S-1 for gemcitabine-refractory biliary tract cancer. Cancer Chemother Pharm. 2013, 71, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Arima, S.; Shimizu, K.; Okamoto, T.; Toki, M.; Suzuki, Y.; Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Kasuga, A.; et al. A Multicenter Phase II Study of Gemcitabine plus S-1 Chemotherapy for Advanced Biliary Tract Cancer. Anticancer Res. 2017, 37, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.O.; Markussen, A.; Diness, L.V.; Nielsen, D. Efficacy and Safety of Capecitabine, Irinotecan, Gemcitabine, and Bevacizumab as Second-Line Treatment in Advanced Biliary Tract Cancer: A Phase II Study. Oncology 2018, 94, 19–24. [Google Scholar] [CrossRef]
- Belkouz, A.; de Vos-Geelen, J.; Mathôt, R.A.A.; Eskens, F.A.L.M.; van Gulik, T.M.; van Oijen, M.G.H.; Punt, C.J.A.; Wilmink, J.W.; Klümpen, H.-J. Efficacy and safety of FOLFIRINOX as salvage treatment in advanced biliary tract cancer: An open-label, single arm, phase 2 trial. Br. J. Cancer 2020, 122, 634–639. [Google Scholar] [CrossRef]
- Prager, G.W.; Kornek, G.; Scheithauer, W.; Steger, G.G.; Zielinski, C.; Unseld, M. Nab-paclitaxel as second-line treatment in advanced biliary cancer. J. Clin. Oncol. 2015, 33, 463. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, H.; Zhao, Y.; Pan, Q.; Mao, A.; Zhu, W.; Zhang, N.; Lin, Z.; Zhou, J.; Wang, Y.; et al. Comprehensive molecular profiling of intrahepatic cholangiocarcinoma in the Chinese population and therapeutic experience. J. Transl. Med. 2020, 18, 273. [Google Scholar] [CrossRef]
- Graham, R.P.; Barr Fritcher, E.G.; Pestova, E.; Schulz, J.; Sitailo, L.A.; Vasmatzis, G.; Murphy, S.J.; McWilliams, R.R.; Hart, S.N.; Halling, K.C.; et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum. Pathol. 2014, 45, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Borad, M.J.; Kelley, R.K.; Wang, Y.; Abdel-Wahab, R.; Meric-Bernstam, F.; Baggerly, K.A.; Kaseb, A.O.; Al-shamsi, H.O.; Ahn, D.H.; et al. Cholangiocarcinoma With FGFR Genetic Aberrations: A Unique Clinical Phenotype. JCO Precis. Oncol. 2018, 1–12. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration: FDA Grants Accelerated Approval to Pemigatinib for Cholangiocarcinoma with an FGFR2 Rearrangement or Fusion. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pemigatinib-cholangiocarcinoma-fgfr2-rearrangement-or-fusion (accessed on 11 May 2020).
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef]
- Javle, M.; Kelley, R.K.; Roychowdhury, S.; Weiss, K.H.; Abou-Alfa, G.K.; Macarulla, T.; Sadeghi, S.; Waldschmidt, D.; Zhu, A.X.; Goyal, L.; et al. Updated results from a phase II study of infigratinib (BGJ398), a selective pan-FGFR kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma containing FGFR2 fusions. Ann. Oncol. 2018, 29, viii720. [Google Scholar] [CrossRef]
- Mazzaferro, V.; El-Rayes, B.F.; Droz Dit Busset, M.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br. J. Cancer 2019, 120, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Shi, L.; Liu, L.Y.; Fece de la Cruz, F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion–Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Meric-Bernstam, F.; Arkenau, H.T.; Bahleda, R.; Kelley, R.K.; Hierro, C.; Ahn, D.; Zhu, A.; Javle, M.; Winkler, R.; et al. Efficacy of TAS-120, an irreversible fibroblast growth factor receptor inhibitor (FGFRi), in patients with cholangiocarcinoma and FGFR pathway alterations previously treated with chemotherapy and other FGFRi’s. Ann. Oncol. 2018, 29, ix49–ix50. [Google Scholar] [CrossRef]
- Bahleda, R.; Italiano, A.; Hierro, C.; Mita, A.; Cervantes, A.; Chan, N.; Awad, M.; Calvo, E.; Moreno, V.; Govindan, R.; et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 4888. [Google Scholar] [CrossRef]
- Vogel, A.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. FIGHT-202: A phase II study of pemigatinib in patients (pts) with previously treated locally advanced or metastatic cholangiocarcinoma (CCA). Ann. Oncol. 2019, 30, v876. [Google Scholar] [CrossRef]
- Liu, S.; Quarles, L.D. How fibroblast growth factor 23 works. J. Am. Soc. Nephrol. 2007, 18, 1637–1647. [Google Scholar] [CrossRef]
- Wöhrle, S.; Bonny, O.; Beluch, N.; Gaulis, S.; Stamm, C.; Scheibler, M.; Müller, M.; Kinzel, B.; Thuery, A.; Brueggen, J.; et al. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J. Bone Min. Res. 2011, 26, 2486–2497. [Google Scholar] [CrossRef]
- Wöhrle, S.; Henninger, C.; Bonny, O.; Thuery, A.; Beluch, N.; Hynes, N.E.; Guagnano, V.; Sellers, W.R.; Hofmann, F.; Kneissel, M.; et al. Pharmacological inhibition of fibroblast growth factor (FGF) receptor signaling ameliorates FGF23-mediated hypophosphatemic rickets. J. Bone Min. Res. 2013, 28, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Raineri, S.; Mellor, J. IDH1: Linking Metabolism and Epigenetics. Front. Genet. 2018, 9, 493. [Google Scholar] [CrossRef]
- Zhao, D.Y.; Lim, K.H. Current biologics for treatment of biliary tract cancers. J. Gastrointest Oncol. 2017, 8, 430–440. [Google Scholar] [CrossRef]
- Boscoe, A.N.; Rolland, C.; Kelley, R.K. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: A systematic literature review. J. Gastrointest Oncol. 2019, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Weil, M.K.; Chen, A.P. PARP inhibitor treatment in ovarian and breast cancer. Curr. Probl. Cancer 2011, 35, 7–50. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 185–195. [Google Scholar] [CrossRef]
- Chae, H.; Kim, D.; Yoo, C.; Kim, K.-P.; Jeong, J.H.; Chang, H.-M.; Lee, S.S.; Park, D.H.; Song, T.J.; Hwang, S.; et al. Therapeutic relevance of targeted sequencing in management of patients with advanced biliary tract cancer: DNA damage repair gene mutations as a predictive biomarker. Eur. J. Cancer 2019, 120, 31–39. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Raitses-Gurevich, M.; Kelley, R.K.; Bocobo, A.G.; Borgida, A.; Shroff, R.T.; Holter, S.; Gallinger, S.; Ahn, D.H.; Aderka, D.; et al. Overall Survival and Clinical Characteristics of BRCA-Associated Cholangiocarcinoma: A Multicenter Retrospective Study. Oncologist 2017, 22, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr Treat Options Oncol. 2016, 17, 58. [Google Scholar] [CrossRef]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushmann, M.d.; Salem, M.E.; Battaglin, F.; et al. Molecular profile of BRCA-mutated biliary tract cancers. ESMO Open 2020, 5, e000682. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, J.M.; Reuter, V.P.; Koche, R.P.; Thompson, C.B. 2-hydroxyglutarate inhibits MyoD-mediated differentiation by preventing H3K9 demethylation. Proc. Natl. Acad. Sci. USA 2019, 116, 12851–12856. [Google Scholar] [CrossRef]
- Pignochino, Y.; Sarotto, I.; Peraldo-Neia, C.; Penachioni, J.Y.; Cavalloni, G.; Migliardi, G.; Casorzo, L.; Chiorino, G.; Risio, M.; Bardelli, A.; et al. Targeting EGFR/HER2 pathways enhances the antiproliferative effect of gemcitabine in biliary tract and gallbladder carcinomas. BMC Cancer 2010, 10, 631. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.H.; Lindebjerg, J.; Ploen, J.; Hansen, T.F.; Jakobsen, A. Phase II marker-driven trial of panitumumab and chemotherapy in KRAS wild-type biliary tract cancer. Ann. Oncol. 2012, 23, 2341–2346. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase II study of erlotinib in patients with advanced biliary cancer. J. Clin. Oncol. 2006, 24, 3069–3074. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; Ramasubbaiah, R.; Yu, M.; Picus, J.; Bufill, J.A.; Tong, Y.; Coleman, N.; Johnston, E.L.; Currie, C.; Loehrer, P.J. Phase II Trial of Erlotinib and Docetaxel in Advanced and Refractory Hepatocellular and Biliary Cancers: Hoosier Oncology Group GI06-101. Oncology 2012, 17, 13-e23. [Google Scholar] [CrossRef]
- Gwak, G.Y.; Yoon, J.H.; Shin, C.M.; Ahn, Y.J.; Chung, J.K.; Kim, Y.A.; Kim, T.Y.; Lee, H.S. Detection of response-predicting mutations in the kinase domain of the epidermal growth factor receptor gene in cholangiocarcinomas. J. Cancer Res. Clin. Oncol. 2005, 131, 649–652. [Google Scholar] [CrossRef]
- Leone, F.; Cavalloni, G.; Pignochino, Y.; Sarotto, I.; Ferraris, R.; Piacibello, W.; Venesio, T.; Capussotti, L.; Risio, M.; Aglietta, M. Somatic Mutations of Epidermal Growth Factor Receptor in Bile Duct and Gallbladder Carcinoma. Clin. Cancer Res. 2006, 12, 1680. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor–Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C.; Schiff, R. HER2: Biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Bang, Y.-J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Javle, M.; Churi, C.; Kang, H.C.; Shroff, R.; Janku, F.; Surapaneni, R.; Zuo, M.; Barrera, C.; Alshamsi, H.; Krishnan, S.; et al. HER2/neu-directed therapy for biliary tract cancer. J. Hematol. Oncol. 2015, 8, 58. [Google Scholar] [CrossRef]
- Javle, M.M.; Hainsworth, J.D.; Swanton, C.; Burris, H.A.; Kurzrock, R.; Sweeney, C.; Meric-Bernstam, F.; Spigel, D.R.; Bose, R.; Guo, S.; et al. Pertuzumab + trastuzumab for HER2-positive metastatic biliary cancer: Preliminary data from MyPathway. J. Clin. Oncol. 2017, 35, 402. [Google Scholar] [CrossRef]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef]
- Harding, J.; Cleary, J.; Shapiro, G.; Braña, I.; Moreno, V.; Quinn, D.; Borad, M.; Loi, S.; Spanggaard, I.; Stemmer, S.; et al. Treating HER2-mutant advanced biliary tract cancer with neratinib: Benefits of HER2-directed targeted therapy in the phase 2 SUMMIT ‘basket’ trial. Ann. Oncol. 2019, 30, iv127. [Google Scholar] [CrossRef]
- Tan, A.C.; Oh, D.-Y.; Chao, Y.; Hsieh, C.-Y.; Chang, W.-L.; Isanto, F.; Chen, Y.-C.; McHale, M.; Lindmark, B.; Ng, M.C.H. Efficacy and safety of varlitinib, a reversible pan-HER tyrosine kinase inhibitor, in combination with platinum-based regimens in biliary tract cancers: A pooled analysis from three phase I studies. J. Clin. Oncol. 2019, 37, 331. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Belani, C.P.; Singh, D.A.; Tanaka, M.; Lenz, H.J.; Yen, Y.; Kindler, H.L.; Iqbal, S.; Longmate, J.; Mack, P.C.; et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother. Pharmacol. 2009, 64, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.M.; Oh, D.-Y.; Ikeda, M.; Yong, W.-P.; McIntyre, N.; Lindmark, B.; McHale, M. Results from TreeTopp: A randomized phase II study of the efficacy and safety of varlitinib plus capecitabine versus placebo in second-line (2L) advanced or metastatic biliary tract cancer (BTC). J. Clin. Oncol. 2020, 38, 4597. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018, 9, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Tsurutani, J.; Iwata, H.; Krop, I.; Jänne, P.A.; Doi, T.; Takahashi, S.; Park, H.; Redfern, C.; Tamura, K.; Wise-Draper, T.M.; et al. Targeting HER2 with Trastuzumab Deruxtecan: A Dose-Expansion, Phase I Study in Multiple Advanced Solid Tumors. Cancer Discov. 2020, 10, 688. [Google Scholar] [CrossRef] [PubMed]
- Ohba, A.; Morizane, C.; Ueno, M.; Kobayashi, S.; Kawamoto, Y.; Komatsu, Y.; Ikeda, M.; Sasaki, M.; Okano, N.; Furuse, J.; et al. Multicenter phase II study of trastuzumab deruxtecan (DS-8201) for HER2-positive unresectable or recurrent biliary tract cancer: HERB trial. J. Clin. Oncol. 2020, 38, TPS4654. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Schaider, H.; Sturm, R.A. The evolving universe of BRAF mutations in melanoma. Br. J. Dermatol. 2017, 177, 893. [Google Scholar] [CrossRef] [PubMed]
- Goeppert, B.; Frauenschuh, L.; Renner, M.; Roessler, S.; Stenzinger, A.; Klauschen, F.; Warth, A.; Vogel, M.N.; Mehrabi, A.; Hafezi, M.; et al. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod. Pathol. 2014, 27, 1028–1034. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Phelps, M.A.; Li, X.; Saji, M.; Goff, L.; Kauh, J.S.W.; O’Neil, B.H.; Balsom, S.; Balint, C.; Liersemann, R.; et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 2357–2363. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; McDonough, S.; El-Khoueiry, A.B.; Bekaii-Saab, T.S.; Stein, S.M.; Sahai, V.; Keogh, G.P.; Kim, E.J.; Baron, A.D.; Siegel, A.B.; et al. Randomised phase II trial (SWOG S1310) of single agent MEK inhibitor trametinib Versus 5-fluorouracil or capecitabine in refractory advanced biliary cancer. Eur. J. Cancer 2020, 130, 219–227. [Google Scholar] [CrossRef]
- Finn, R.S.; Ahn, D.H.; Javle, M.M.; Tan, B.R.; Weekes, C.D.; Bendell, J.C.; Patnaik, A.; Khan, G.N.; Laheru, D.; Chavira, R.; et al. Phase 1b investigation of the MEK inhibitor binimetinib in patients with advanced or metastatic biliary tract cancer. Investig. New Drugs 2018, 36, 1037–1043. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, K.-H.; Kim, J.-W.; Suh, K.J.; Nam, A.-R.; Bang, J.-H.; Bang, Y.-J.; Oh, D.-Y. Enhanced antitumor effect of binimetinib in combination with capecitabine for biliary tract cancer patients with mutations in the RAS/RAF/MEK/ERK pathway: Phase Ib study. Br. J. Cancer 2019, 121, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Lassen, U.N.; Elez, E.; Italiano, A.; Curigliano, G.; De Braud, F.G.; Prager, G.; Greil, R.; Stein, A.; Fasolo, A.; et al. Efficacy and safety of dabrafenib (D) and trametinib (T) in patients (pts) with BRAF V600E–mutated biliary tract cancer (BTC): A cohort of the ROAR basket trial. J. Clin. Oncol. 2019, 37, 187. [Google Scholar] [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutated biliary tract cancer (ROAR): A phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 2020. [Google Scholar] [CrossRef]
- Blumenschein, G.R., Jr.; Mills, G.B.; Gonzalez-Angulo, A.M. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 3287–3296. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Ojima, H.; Iwasaki, M.; Shimizu, H.; Kokubu, A.; Hiraoka, N.; Kosuge, T.; Yoshikawa, D.; Kono, T.; Furukawa, H.; et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br. J. Cancer 2011, 105, 131–138. [Google Scholar] [CrossRef]
- Goyal, L.; Zheng, H.; Yurgelun, M.B.; Abrams, T.A.; Allen, J.N.; Cleary, J.M.; Knowles, M.; Regan, E.; Reardon, A.; Khachatryan, A.; et al. A phase 2 and biomarker study of cabozantinib in patients with advanced cholangiocarcinoma. Cancer 2017, 123, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Saleh, M.; Bendell, J.; Infante, J.R.; Jones, S.; Kurkjian, C.D.; Moore, K.M.; Kazakin, J.; Abbadessa, G.; Wang, Y.; et al. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann. Oncol. 2014, 25, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Corti, F.; Nichetti, F.; Raimondi, A.; Niger, M.; Prinzi, N.; Torchio, M.; Tamborini, E.; Perrone, F.; Pruneri, G.; Di Bartolomeo, M.; et al. Targeting the PI3K/AKT/mTOR pathway in biliary tract cancers: A review of current evidences and future perspectives. Cancer Treat Rev. 2019, 72, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Roa, I.; de Toro, G.; Fernández, F.; Game, A.; Muñoz, S.; de Aretxabala, X.; Javle, M. Inactivation of tumor suppressor gene pten in early and advanced gallbladder cancer. Diagn. Pathol. 2015, 10, 148. [Google Scholar] [CrossRef]
- Turkes, F.; Carmichael, J.; Cunningham, D.; Starling, N. Contemporary Tailored Oncology Treatment of Biliary Tract Cancers. Gastroenterol. Res. Pract. 2019, 2019, 7698786. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kunnimalaiyaan, S.; Kunnimalaiyaan, M.; Gamblin, T.C. Inhibition of the AKT pathway in cholangiocarcinoma by MK2206 reduces cellular viability via induction of apoptosis. Cancer Cell Int. 2015, 15, 13. [Google Scholar] [CrossRef]
- RIZZO, A.; RICCI, A.D.; TOBER, N.; NIGRO, M.C.; MOSCA, M.; PALLONI, A.; ABBATI, F.; FREGA, G.; DE LORENZO, S.; TAVOLARI, S.; et al. Second-line Treatment in Advanced Biliary Tract Cancer: Today and Tomorrow. Anticancer Res. 2020, 40, 3013–3030. [Google Scholar] [CrossRef]
- Wu, C.-E.; Chen, M.-H.; Yeh, C.-N. mTOR Inhibitors in Advanced Biliary Tract Cancers. Int. J. Mol. Sci. 2019, 20, 500. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Li, J.; Wei, L.; Doyle, A.; Marshall, J.L.; Schaaf, L.J.; Phelps, M.A.; Villalona-Calero, M.A.; Bekaii-Saab, T. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci. Rep. 2015, 5, 12122. [Google Scholar] [CrossRef] [PubMed]
- Buzzoni, R.; Pusceddu, S.; Bajetta, E.; De Braud, F.; Platania, M.; Iannacone, C.; Cantore, M.; Mambrini, A.; Bertolini, A.; Alabiso, O.; et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: A phase II ITMO study. Ann. Oncol. 2014, 25, 1597–1603. [Google Scholar] [CrossRef]
- Leyva-Illades, D.; McMillin, M.; Quinn, M.; Demorrow, S. Cholangiocarcinoma pathogenesis: Role of the tumor microenvironment. Transl. Gastrointest. Cancer 2012, 1, 71–80. [Google Scholar] [PubMed]
- Simone, V.; Brunetti, O.; Lupo, L.; Testini, M.; Maiorano, E.; Simone, M.; Longo, V.; Rolfo, C.; Peeters, M.; Scarpa, A.; et al. Targeting Angiogenesis in Biliary Tract Cancers: An Open Option. Int. J. Mol. Sci. 2017, 18, 418. [Google Scholar] [CrossRef] [PubMed]
- Bengala, C.; Bertolini, F.; Malavasi, N.; Boni, C.; Aitini, E.; Dealis, C.; Zironi, S.; Depenni, R.; Fontana, A.; Del Giovane, C.; et al. Sorafenib in patients with advanced biliary tract carcinoma: A phase II trial. Br. J. Cancer 2010, 102, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Seitz, J.-F.; Fartoux, L.; Malka, D.; Lledo, G.; Tijeras-Raballand, A.; De Gramont, A.; Ronot, M.; Bouattour, M.; Dreyer, C.; et al. Sunitinib as second-line treatment in patients with advanced intrahepatic cholangiocarcinoma (SUN-CK phase II trial): Safety, efficacy, and updated translational results. J. Clin. Oncol. 2015, 33, 343. [Google Scholar] [CrossRef]
- Yi, J.H.; Thongprasert, S.; Lee, J.; Doval, D.C.; Park, S.H.; Park, J.O.; Park, Y.S.; Kang, W.K.; Lim, H.Y. A phase II study of sunitinib as a second-line treatment in advanced biliary tract carcinoma: A multicentre, multinational study. Eur. J. Cancer 2012, 48, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Zong, H.; Jin, S.; Zhong, Q.; Jiang, M.; Jin, M.; Li, R.; Jiang, G. A phase II study of anlotinib with cediranib as a second-line treatment for patients with advanced biliary tract cancers (aBTCs). J. Clin. Oncol. 2020, 38, e16712. [Google Scholar] [CrossRef]
- Sun, W.; Patel, A.; Normolle, D.; Patel, K.; Ohr, J.; Lee, J.J.; Bahary, N.; Chu, E.; Streeter, N.; Drummond, S. A phase 2 trial of regorafenib as a single agent in patients with chemotherapy-refractory, advanced, and metastatic biliary tract adenocarcinoma. Cancer 2019, 125, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; Sanoff, H.K.; Poklepovic, A.S.; Soares, H.; Kim, J.; Lyu, J.; Liu, Y.; Nixon, A.B.; Kim, D.W. A multi-institutional phase 2 trial of regorafenib in refractory advanced biliary tract cancer. Cancer 2020, 126, 3464–3470. [Google Scholar] [CrossRef]
- Demols, A.; Borbath, I.; Van den Eynde, M.; Houbiers, G.; Peeters, M.; Marechal, R.; Delaunoit, T.; Goemine, J.C.; Laurent, S.; Holbrechts, S.; et al. Regorafenib after failure of gemcitabine and platinum-based chemotherapy for locally advanced/metastatic biliary tumors: REACHIN, a randomized, double-blind, phase II trial. Ann. Oncol. 2020, 31, 1169–1177. [Google Scholar] [CrossRef]
- Ikeda, M.; Sasaki, T.; Morizane, C.; Mizuno, N.; Nagashima, F.; Shimizu, S.; Hayata, N.; Ikezawa, H.; Suzuki, T.; Nakajima, R.; et al. 722P-A phase 2 study of lenvatinib monotherapy as second-line treatment in unresectable biliary tract cancer: Primary analysis results. Ann. Oncol. 2017, 28, v246. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. Esmo Open 2016, 1, e000023. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Demols, A.; Rocq, L.; Charry, M.; De Nève, N.; Verrellen, A.; Ramadhan, A.; Van Campenhout, C.; De Clercq, S.; Salmon, I.; D’Haene, N. NTRK gene fusions in biliary tract cancers. J. Clin. Oncol. 2020, 38, 574. [Google Scholar] [CrossRef]
- Drilon, A. TRK inhibitors in TRK fusion-positive cancers. Ann. Oncol. 2019, 30, viii23–viii30. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Rolfo, C.D.; De Braud, F.G.; Doebele, R.C.; Drilon, A.E.; Siena, S.; Patel, M.; Cho, B.C.; Liu, S.V.; Ahn, M.-J.; Chiu, C.-H.; et al. Efficacy and safety of entrectinib in patients (pts) with NTRK-fusion positive (NTRK-fp) solid tumors: An updated integrated analysis. J. Clin. Oncol. 2020, 38, 3605. [Google Scholar] [CrossRef]
- Patel, M.; Siena, S.; Demetri, G.; Doebele, R.; Chae, Y.; Conkling, P.; Garrido-Laguna, I.; Longo, F.; Rolfo, C.; Sigal, D.; et al. O-3 Efficacy and safety of entrectinib in NTRK fusion-positive gastrointestinal cancers: Updated integrated analysis of three clinical trials (STARTRK-2, STARTRK-1 and ALKA-372-001). Ann. Oncol. 2020, 31, 232–233. [Google Scholar] [CrossRef]
- Wu, C.; Ma, S. A selective review of robust variable selection with applications in bioinformatics. Brief Bioinform. 2015, 16, 873–883. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, F.; Ren, J.; Li, X.; Jiang, Y.; Ma, S. A Selective Review of Multi-Level Omics Data Integration Using Variable Selection. High-Throughput 2019, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Jindal, V. Immune checkpoint inhibitors in gastrointestinal malignancies. J. Gastrointest. Oncol. 2018, 9, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Lee, J.-C.; Shin, D.W.; Kim, J.; Hwang, J.-H. High PD-L1 expression is associated with therapeutic response to pembrolizumab in patients with advanced biliary tract cancer. Sci. Rep. 2020, 10, 12348. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Bang, Y.J.; Doi, T.; Braud, F.D.; Piha-Paul, S.; Hollebecque, A.; Razak, A.R.A.; Lin, C.C.; Ott, P.A.; He, A.R.; Yuan, S.S.; et al. 525 Safety and efficacy of pembrolizumab (MK-3475) in patients (pts) with advanced biliary tract cancer: Interim results of KEYNOTE-028. Eur. J. Cancer 2015, 51, S112. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Piha-Paul, S.A.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Pembrolizumab (pembro) for advanced biliary adenocarcinoma: Results from the KEYNOTE-028 (KN028) and KEYNOTE-158 (KN158) basket studies. J. Clin. Oncol. 2019, 37, 4079. [Google Scholar] [CrossRef]
- Ueno, M.; Ikeda, M.; Morizane, C.; Kobayashi, S.; Ohno, I.; Kondo, S.; Okano, N.; Kimura, K.; Asada, S.; Namba, Y.; et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: A non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 611–621. [Google Scholar] [CrossRef]
- Kim, R.D.; Chung, V.; Alese, O.B.; El-Rayes, B.F.; Li, D.; Al-Toubah, T.E.; Schell, M.J.; Zhou, J.-M.; Mahipal, A.; Kim, B.H.; et al. A Phase 2 Multi-institutional Study of Nivolumab for Patients With Advanced Refractory Biliary Tract Cancer. JAMA Oncol. 2020, 6, 888–894. [Google Scholar] [CrossRef]
- Klein, O.; Kee, D.; Nagrial, A.; Markman, B.; Underhill, C.; Michael, M.; Jackett, L.; Lum, C.; Behren, A.; Palmer, J.; et al. Evaluation of Combination Nivolumab and Ipilimumab Immunotherapy in Patients With Advanced Biliary Tract Cancers: Subgroup Analysis of a Phase 2 Nonrandomized Clinical Trial. JAMA Oncol. 2020, e202814. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed]
- Ioka, T.; Ueno, M.; Oh, D.-Y.; Fujiwara, Y.; Chen, J.-S.; Doki, Y.; Mizuno, N.; Park, K.; Asagi, A.; Hayama, M.; et al. Evaluation of safety and tolerability of durvalumab (D) with or without tremelimumab (T) in patients (pts) with biliary tract cancer (BTC). J. Clin. Oncol. 2019, 37, 387. [Google Scholar] [CrossRef]
- Hong, T.S.; Goyal, L.; Parikh, A.R.; Yeap, B.Y.; Ulysse, C.A.; Drapek, L.C.; Allen, J.N.; Clark, J.W.; Christopher, B.; Bolton, C.; et al. A pilot study of durvalumab/tremelimumab (durva/treme) and radiation (XRT) for metastatic biliary tract cancer (mBTC): Preliminary safety and efficacy. J. Clin. Oncol. 2020, 38, 547. [Google Scholar] [CrossRef]
- Ciciola, P.; Cascetta, P.; Bianco, C.; Formisano, L.; Bianco, R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. J. Clin. Med. 2020, 9, 675. [Google Scholar] [CrossRef]
- Arkenau, H.-T.; Martin-Liberal, J.; Calvo, E.; Penel, N.; Krebs, M.G.; Herbst, R.S.; Walgren, R.A.; Widau, R.C.; Mi, G.; Jin, J.; et al. Ramucirumab Plus Pembrolizumab in Patients with Previously Treated Advanced or Metastatic Biliary Tract Cancer: Nonrandomized, Open-Label, Phase I Trial (JVDF). Oncology 2018, 23, 1407. [Google Scholar] [CrossRef]
- Lin, J.; Shi, W.; Zhao, S.; Hu, J.; Hou, Z.; Yao, M.; Chrin, G.; Pan, J.; Hu, K.; Zhao, L.; et al. Lenvatinib plus checkpoint inhibitors in patients (pts) with advanced intrahepatic cholangiocarcinoma (ICC): Preliminary data and correlation with next-generation sequencing. J. Clin. Oncol. 2018, 36, 500. [Google Scholar] [CrossRef]
- Yarchoan, M.C.L.; Anders, R.A.; Anne Noonan, A.; Goff, L.W.; Goyal, L.; Lacy, J.; Li, D.; Patel, A.; He, A.R.; Abou-Alfa, G.; et al. A multicenter randomized phase 2 trial of atezolizumab as monotherapy or in combination with cobimetinib in biliary tract cancers (BTCs): A NCI Experimental Therapeutics Clinical Trials Network (ETCTN) study. In Proceedings of the 2020 American Association for Cancer Research Virtual Annual Meeting I, Philadelphia, PA, USA, 27–28 April 2020. Abstract CT043. bit.ly/042BCTMCw. [Google Scholar]
- Block, K.I.; Block, P.B.; Gyllenhaal, C. Integrative Treatment for Colorectal Cancer: A Comprehensive Approach. J. Altern Complement Med. 2018, 24, 890–901. [Google Scholar] [CrossRef]
- Jentzsch, V.; Davis, J.A.A.; Djamgoz, M.B.A. Pancreatic Cancer (PDAC): Introduction of Evidence-Based Complementary Measures into Integrative Clinical Management. Cancers 2020, 12, 3096. [Google Scholar] [CrossRef]
- Abe, K.; Uwagawa, T.; Haruki, K.; Takano, Y.; Onda, S.; Sakamoto, T.; Gocho, T.; Yanaga, K. Effects of ω-3 Fatty Acid Supplementation in Patients with Bile Duct or Pancreatic Cancer Undergoing Chemotherapy. Anticancer Res. 2018, 38, 2369–2375. [Google Scholar] [CrossRef]
- Yu, W.D.; Ma, Y.; Flynn, G.; Muindi, J.R.; Kong, R.X.; Trump, D.L.; Johnson, C.S. Calcitriol enhances gemcitabine anti-tumor activity in vitro and in vivo by promoting apoptosis in a human pancreatic carcinoma model system. Cell Cycle 2010, 9, 3022–3029. [Google Scholar] [CrossRef] [PubMed]
- Fountzilas, C.; Javle, M.; Tan, W.; Ma, Y.; Fetterly, G.; Iyer, R.; Johnson, C. A phase 1, open-label, dose escalation study of intravenous paricalcitol in combination with gemcitabine in patients with advanced malignancies. Cancer 2018, 124, 3890–3899. [Google Scholar] [CrossRef] [PubMed]


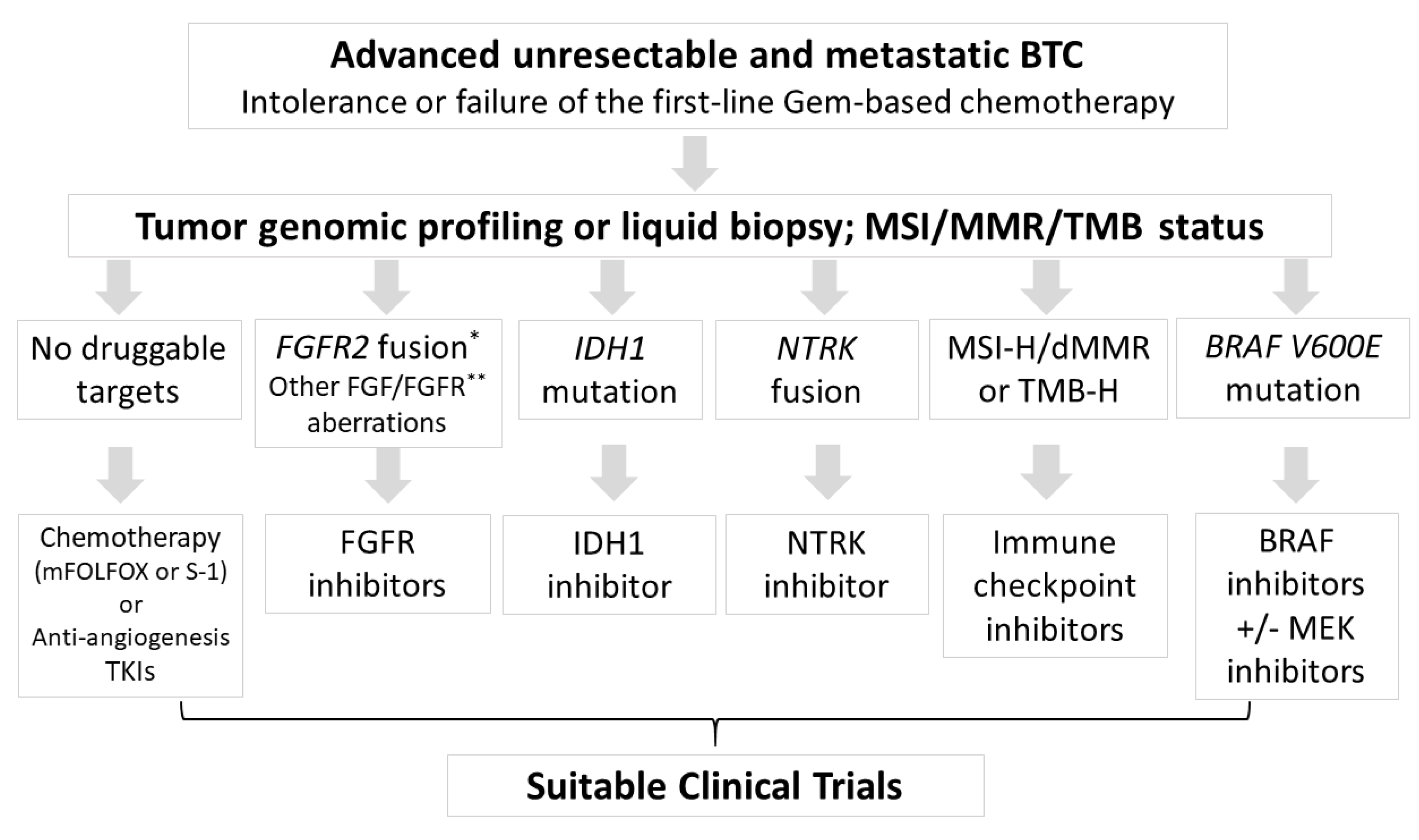{kind=link}
| Drug(s) | Study | Case (n) | Target | ORR (%) | SD (%) | PFS (mos) | OS (mos) |
|---|---|---|---|---|---|---|---|
| chemotherapy | |||||||
| mFOLFOX vs. ASC [22] | ABC06 Phase III | 162 (81 vs. 81) | - | 4% | 28% | 4 | 6.2 |
| S-1 [24] | Japan Phase II | 40 | - | 7.5% | 55% | 2.5 | 6.8 |
| FOLFIRINOX [27] | Phase II | 30 | - | 10% | 57% | 6.2 | 10.7 |
| Targeted therapy | |||||||
| Pemigatinib [34] | FIGHT202Phase II | 107 * | FGFR1-3 VEGFR2 | 35.5% | 46.5% | 6.9 | 21.1 |
| Infigratinib (BGJ398) [36] | Phase II | 71 * | FGFR1-3 | 26.9% & | 56.7% | 6.8 | 12.5 |
| Derazantinib (ARQ087) [38] | Phase I/II | 30 * | FGFR1-3 RET, PDGFR, KIT, SRC | 20.7% | 62.1% | 5.7 | - |
| Fuibatinib (TAS-120) [39] | Phase I/II | 28 * | FGFR1-4 | 25% | 54% | - | - |
| Ivosidenib vs. placebo [50] | ClarIDHy Phase III | 185 ** (124 vs. 61) | Mutant IDH1 | 2.4% | 50.8% | 2.7 | 10.8 |
| Dabrafenib + trametinib [88] | ROAR Phase II | 43 *** | BRAFV600E | 51% | - | 9 | 14 |
| Regorafenib vs. placebo [108] | REACHIN Phase II | 66(33 vs. 33) | VEGF1-3, PDGFR, FGFR | 0% | 74% | 3 | 5.3 |
| Immunotherapy | |||||||
| Pembrolizumab [127] | KN-158 Phase II | 104 | PD-1 | 5.8% | - | 2 | 7.4 |
| Nivolumab [131] | Korea Phase II | 54 | PD-1 | 22% $ 11% # | - | 3.7 | 14.2 |
| Durvalumab + Tremelimumb [134] | Phase I | 42 (D) 65 (D + T) | PD-L1 + CTLA-4 | 5% (D) 11% (D + T) | - | - | 8.1 (D) 10.1 (T + D) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, N.-J.; Chen, L.-T.; Shan, Y.-S.; Yeh, C.-N.; Chen, M.-H. Development of Possible Next Line of Systemic Therapies for Gemcitabine-Resistant Biliary Tract Cancers: A Perspective from Clinical Trials. Biomolecules 2021, 11, 97. https://doi.org/10.3390/biom11010097
Chiang N-J, Chen L-T, Shan Y-S, Yeh C-N, Chen M-H. Development of Possible Next Line of Systemic Therapies for Gemcitabine-Resistant Biliary Tract Cancers: A Perspective from Clinical Trials. Biomolecules. 2021; 11(1):97. https://doi.org/10.3390/biom11010097
Chicago/Turabian StyleChiang, Nai-Jung, Li-Tzong Chen, Yan-Shen Shan, Chun-Nan Yeh, and Ming-Huang Chen. 2021. "Development of Possible Next Line of Systemic Therapies for Gemcitabine-Resistant Biliary Tract Cancers: A Perspective from Clinical Trials" Biomolecules 11, no. 1: 97. https://doi.org/10.3390/biom11010097
APA StyleChiang, N.-J., Chen, L.-T., Shan, Y.-S., Yeh, C.-N., & Chen, M.-H. (2021). Development of Possible Next Line of Systemic Therapies for Gemcitabine-Resistant Biliary Tract Cancers: A Perspective from Clinical Trials. Biomolecules, 11(1), 97. https://doi.org/10.3390/biom11010097