RNA Metabolism Guided by RNA Modifications: The Role of SMUG1 in rRNA Quality Control

 , ,
, , 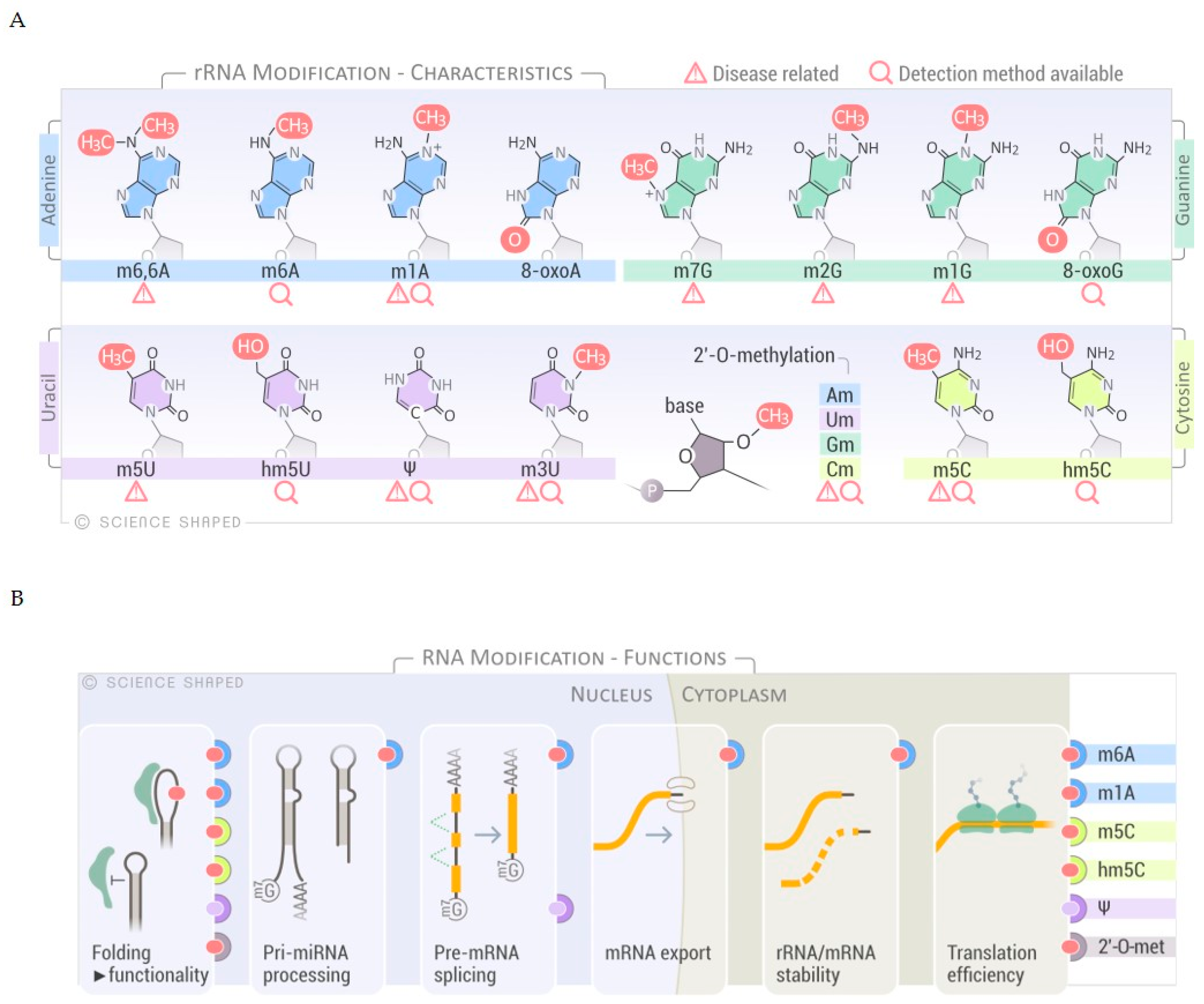{kind=link}
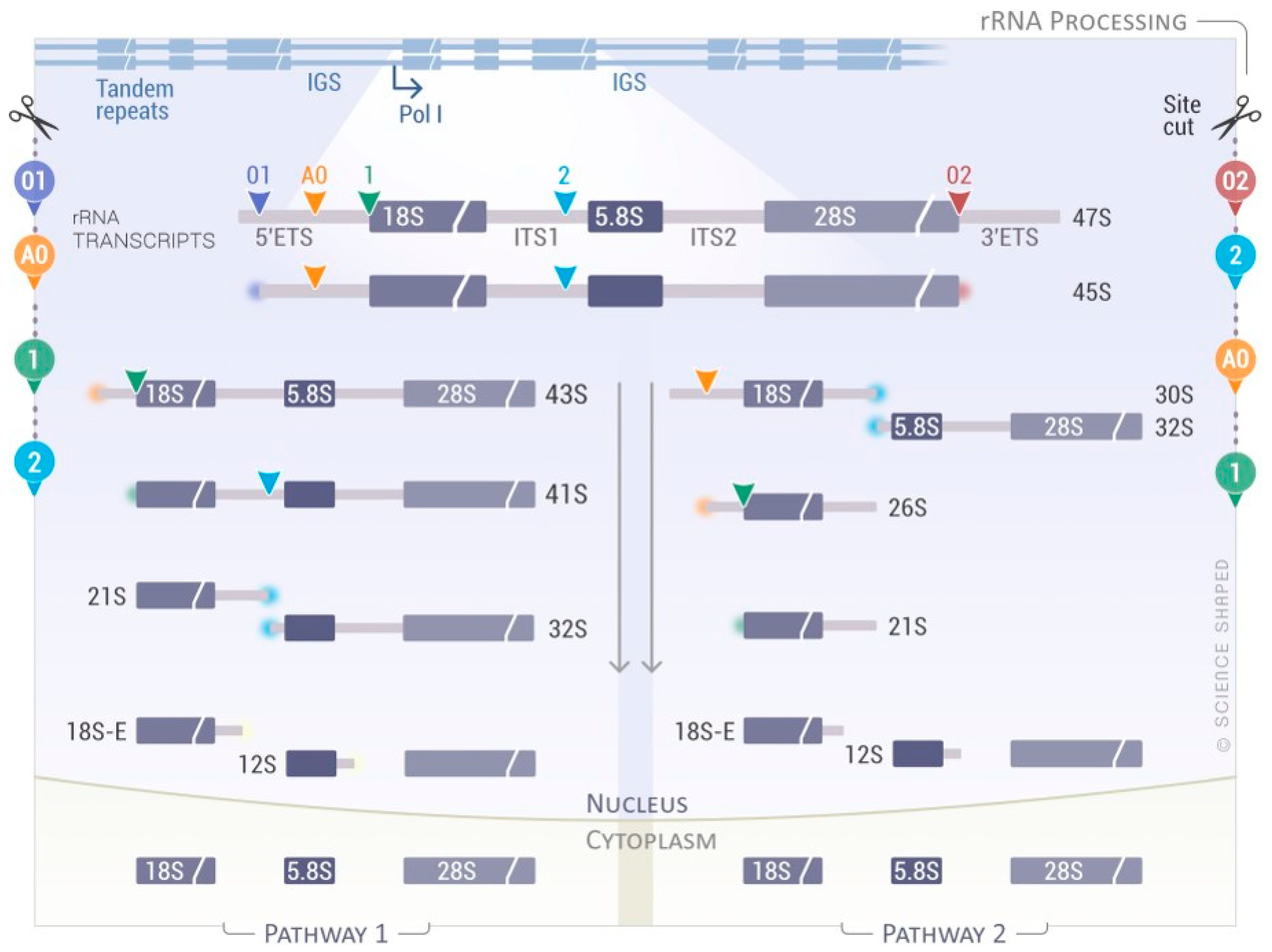{kind=link}
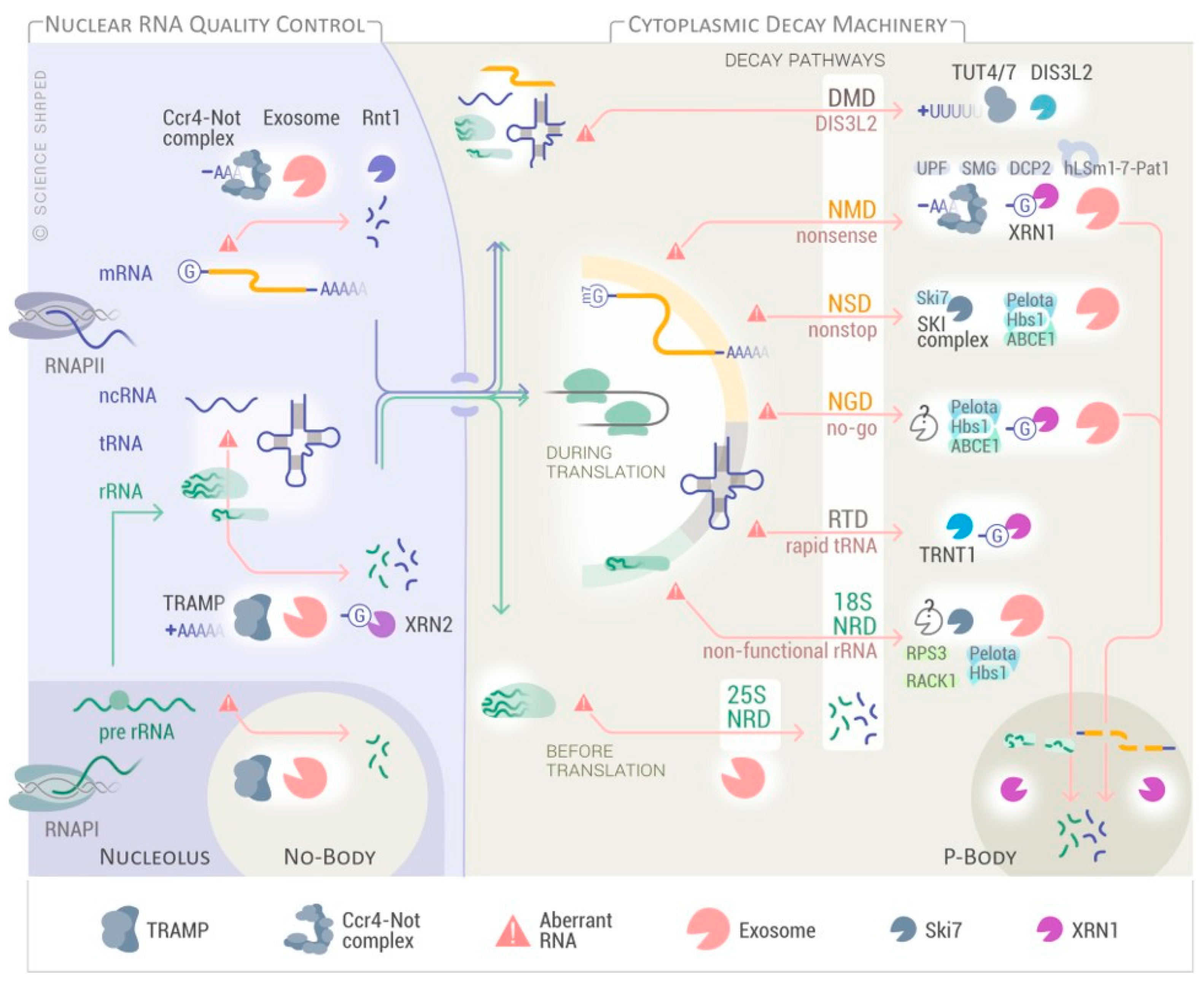{kind=link}
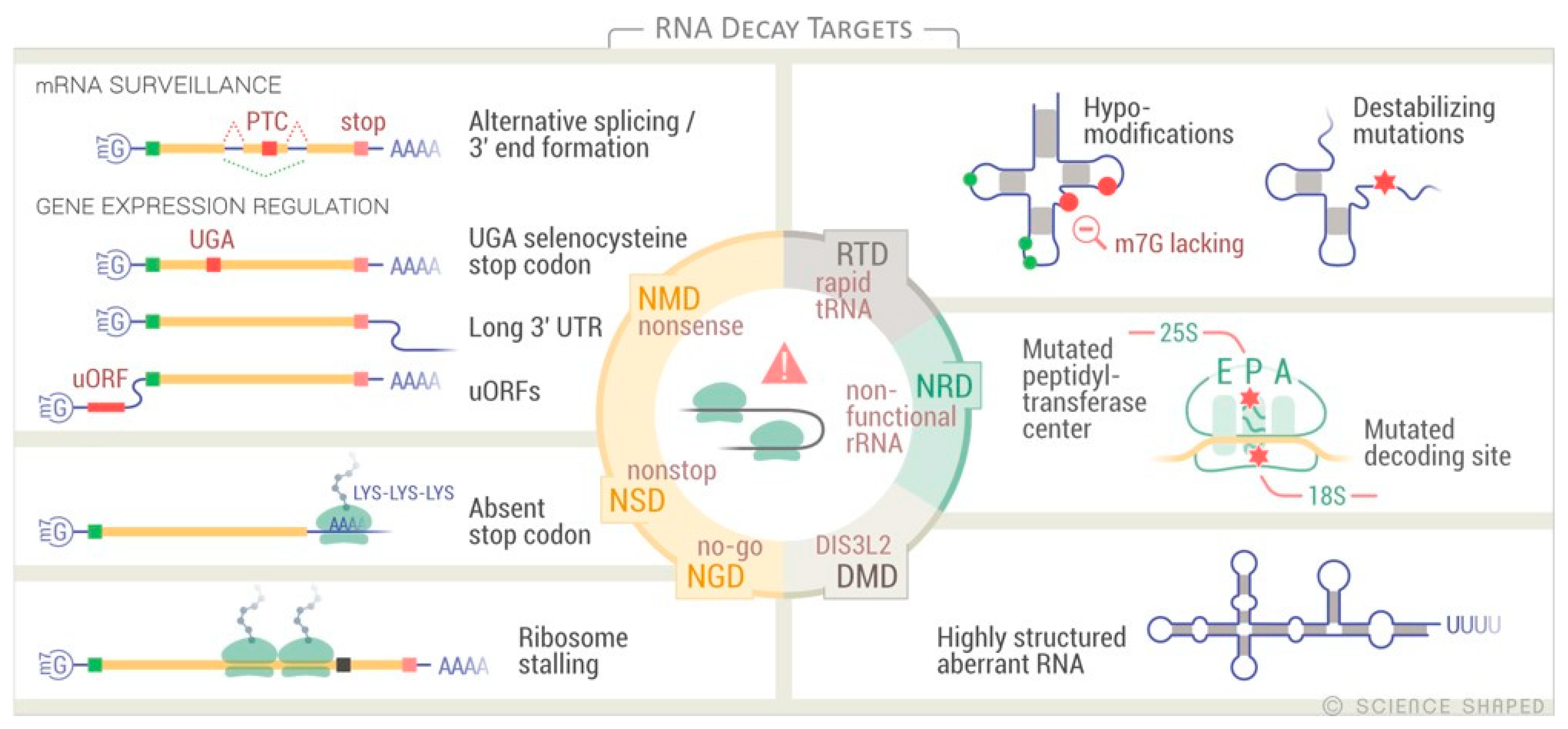{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Ribosomal RNA Modifications and Their Biological Functions
2.1. Pseudouridylation
2.2. 2′-O-Methylation
2.3. Other Base Modifications
3. rRNA Processing and Maturation
4. RNA Quality Control Mechanisms
4.1. mRNA Surveillance Pathways
4.2. rRNA Quality Control
4.3. tRNA Quality Control
4.4. ncRNA Decay Pathway
5. Processing of Damaged RNA
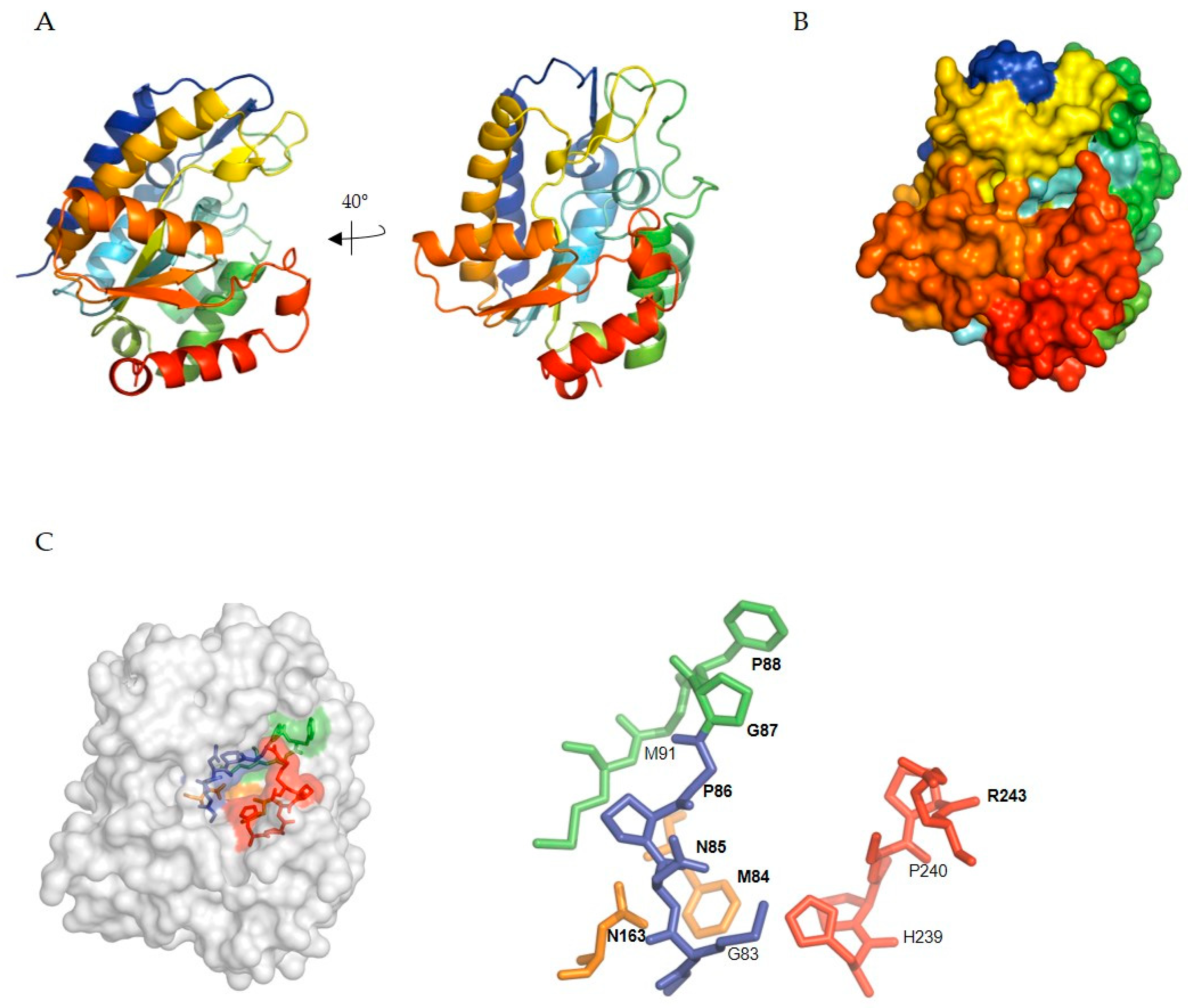
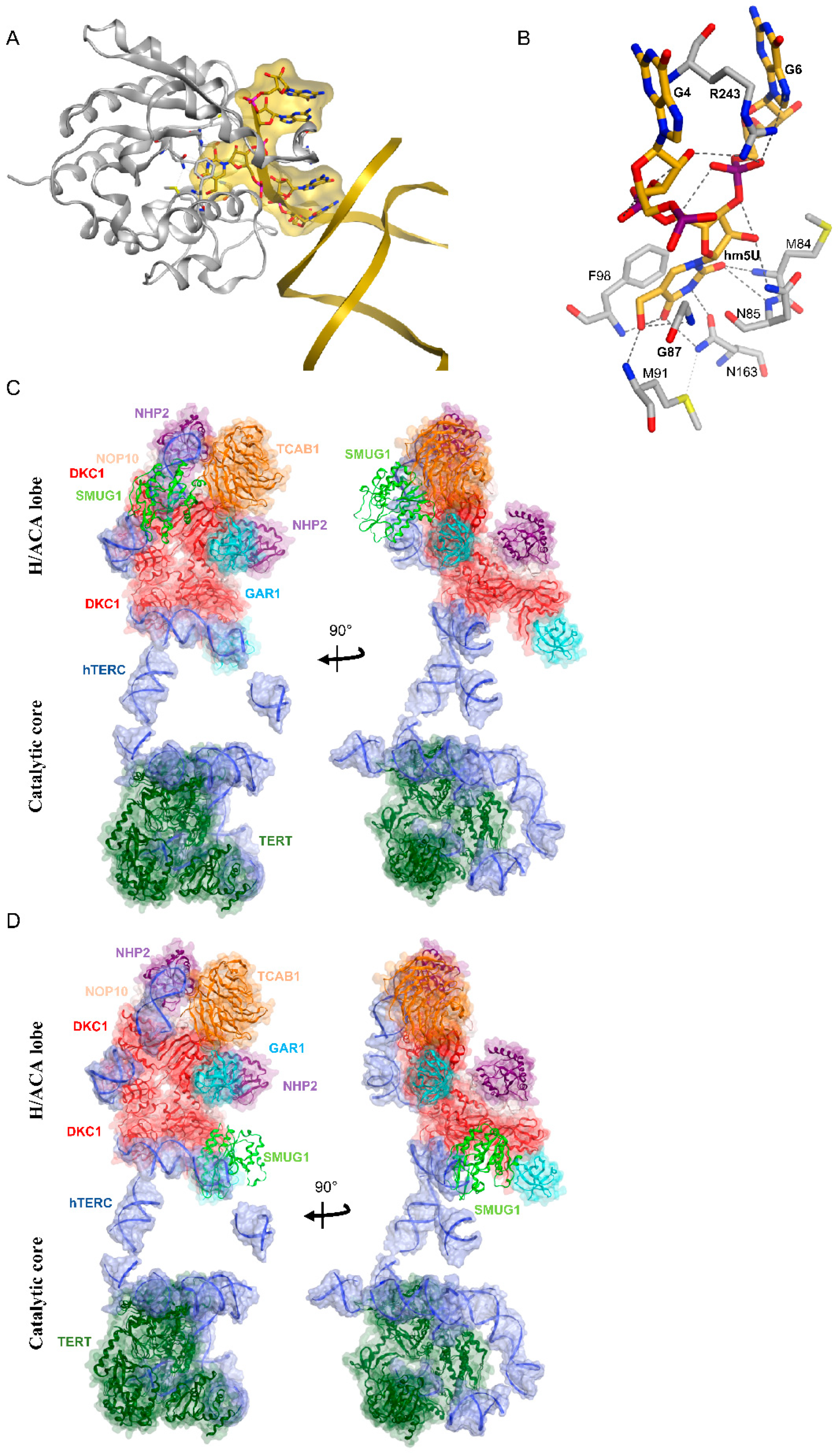
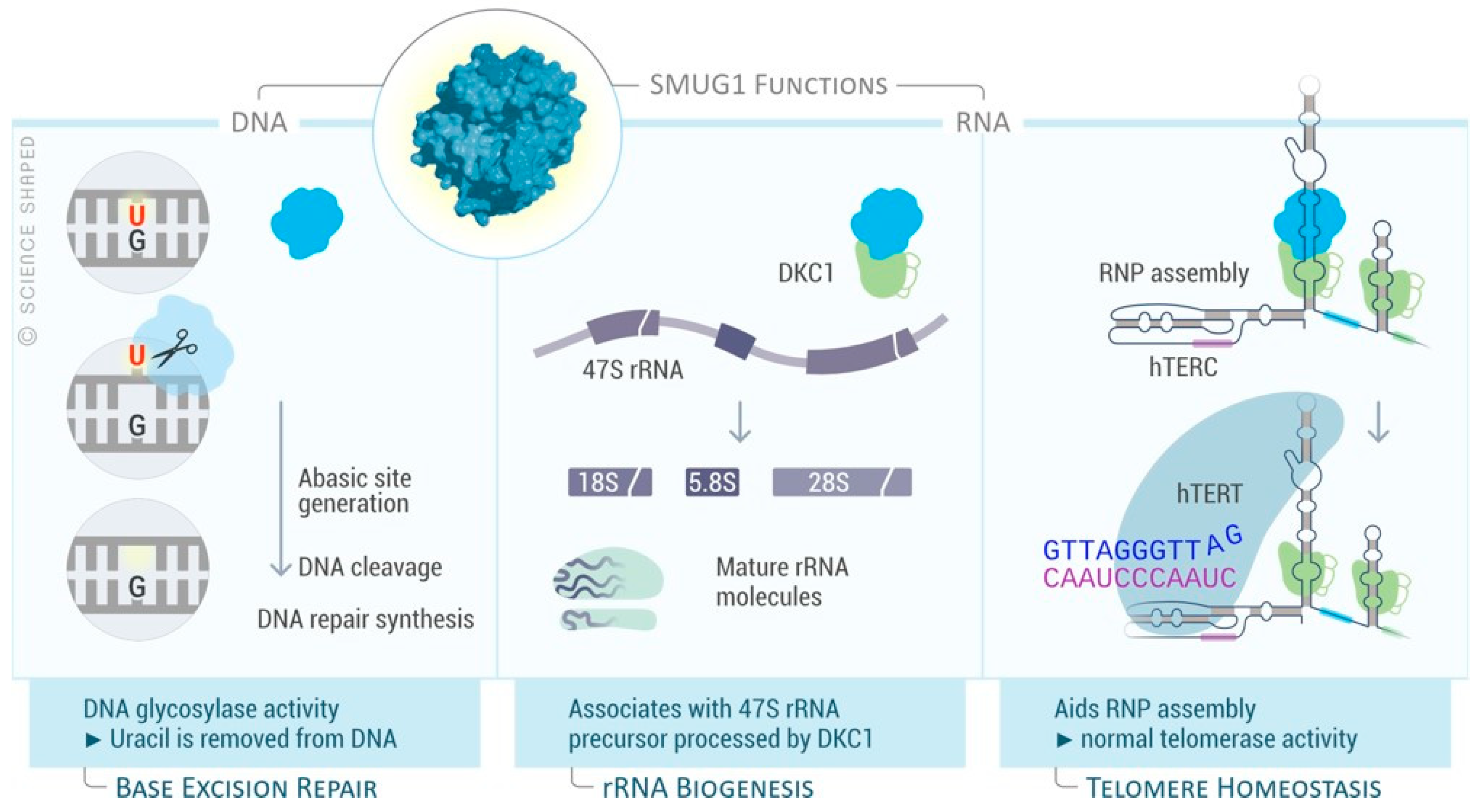
5.1. SMUG1 Structure and Function
5.2. SMUG1 in Regulating a Highly Structured RNA
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piekna-Przybylska, D.; Decatur, W.A.; Fournier, M.J. The 3D rRNA modification maps database: With interactive tools for ribosome analysis. Nucleic Acids Res. 2008, 36, D178–D183. [Google Scholar] [CrossRef] [PubMed]
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.; Fabris, D.; Agris, P.F. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011, 39, D195–D201. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, H.; Wei, Z.; Zhang, S.; Hua, G.; Zhang, S.W.; Zhang, L.; Gao, S.J.; Meng, J.; Chen, X.; et al. MeT-DB V2.0: Elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018, 46, D281–D287. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Noack, F.; Calegari, F. Epitranscriptomics: A New Regulatory Mechanism of Brain Development and Function. Front. Neurosci. 2018, 12, 85. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Warda, A.S.; Sharma, S.; Entian, K.D.; Lafontaine, D.L.J.; Bohnsack, M.T. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017, 14, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, E.M.; Kietrys, A.M.; Kool, E.T. Chemical and structural effects of base modifications in messenger RNA. Nature 2017, 541, 339–346. [Google Scholar] [CrossRef]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef]
- Khoddami, V.; Cairns, B.R. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 2013, 31, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Delatte, B.; Wang, F.; Ngoc, L.V.; Collignon, E.; Bonvin, E.; Deplus, R.; Calonne, E.; Hassabi, B.; Putmans, P.; Awe, S.; et al. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 2016, 351, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Leisegang, M.S.; Doebele, C.; Ramirez, A.S.; Simm, S.; Safferthal, C.; Kretschmer, J.; Schorge, T.; Markoutsa, S.; Haag, S.; et al. The association of late-acting snoRNPs with human pre-ribosomal complexes requires the RNA helicase DDX21. Nucleic Acids Res. 2015, 43, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Knox, A.A.; Wells, G.R.; Schneider, C.; Watkins, N.J. Interactions and activities of factors involved in the late stages of human 18S rRNA maturation. RNA Biol. 2019, 16, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.L.; Tollervey, D. Birth of the snoRNPs: The evolution of the modification-guide snoRNAs. Trends Biochem. Sci. 1998, 23, 383–388. [Google Scholar] [CrossRef]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; Leon-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef]
- Penzo, M.; Montanaro, L. Turning Uridines around: Role of rRNA Pseudouridylation in Ribosome Biogenesis and Ribosomal Function. Biomolecules 2018, 8, 38. [Google Scholar] [CrossRef]
- Taoka, M.; Nobe, Y.; Yamaki, Y.; Sato, K.; Ishikawa, H.; Izumikawa, K.; Yamauchi, Y.; Hirota, K.; Nakayama, H.; Takahashi, N.; et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018, 46, 9289–9298. [Google Scholar] [CrossRef]
- Lane, B.G.; Ofengand, J.; Gray, M.W. Pseudouridine in the large-subunit (23 S-like) ribosomal RNA. The site of peptidyl transfer in the ribosome? FEBS Lett. 1992, 302, 1–4. [Google Scholar] [CrossRef]
- Ontiveros, R.J.; Stoute, J.; Liu, K.F. The chemical diversity of RNA modifications. Biochem. J. 2019, 476, 1227–1245. [Google Scholar] [CrossRef] [PubMed]
- Bates, C.; Hubbard, S.J.; Ashe, M.P. Ribosomal flavours: An acquired taste for specific mRNAs? Biochem. Soc. Trans. 2018, 46, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Spenkuch, F.; Motorin, Y.; Helm, M. Pseudouridine: Still mysterious, but never a fake (uridine)! RNA Biol. 2014, 11, 1540–1554. [Google Scholar] [CrossRef] [PubMed]
- Ofengand, J.; Bakin, A.; Wrzesinski, J.; Nurse, K.; Lane, B.G. The pseudouridine residues of ribosomal RNA. Biochem. Cell Biol. 1995, 73, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Charette, M.; Gray, M.W. Pseudouridine in RNA: What, where, how, and why. IUBMB Life 2000, 49, 341–351. [Google Scholar] [CrossRef]
- Angrisani, A.; Vicidomini, R.; Turano, M.; Furia, M. Human dyskerin: Beyond telomeres. Biol. Chem. 2014, 395, 593–610. [Google Scholar] [CrossRef]
- Meier, U.T. RNA modification in Cajal bodies. RNA Biol. 2017, 14, 693–700. [Google Scholar] [CrossRef]
- Zaringhalam, M.; Papavasiliou, F.N. Pseudouridylation meets next-generation sequencing. Methods 2016, 107, 63–72. [Google Scholar] [CrossRef]
- Yoon, A.; Peng, G.; Brandenburger, Y.; Zollo, O.; Xu, W.; Rego, E.; Ruggero, D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 2006, 312, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Kopmar, N.; Ruggero, D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 2010, 29, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Krasnykh, O.; Haynes, N.; Theodoropoulou, M.; Peng, G.; Montanaro, L.; Ruggero, D. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 2010, 70, 6026–6035. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D.; Grisendi, S.; Piazza, F.; Rego, E.; Mari, F.; Rao, P.H.; Cordon-Cardo, C.; Pandolfi, P.P. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 2003, 299, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Thumati, N.R.; Zeng, X.L.; Au, H.H.; Jang, C.J.; Jan, E.; Wong, J.M. Severity of X-linked dyskeratosis congenita (DKCX) cellular defects is not directly related to dyskerin (DKC1) activity in ribosomal RNA biogenesis or mRNA translation. Hum. Mutat. 2013, 34, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Wood, E.; Collins, K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 1999, 402, 551–555. [Google Scholar] [CrossRef]
- Heiss, N.S.; Knight, S.W.; Vulliamy, T.J.; Klauck, S.M.; Wiemann, S.; Mason, P.J.; Poustka, A.; Dokal, I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998, 19, 32–38. [Google Scholar] [CrossRef]
- Bykhovskaya, Y.; Casas, K.; Mengesha, E.; Inbal, A.; Fischel-Ghodsian, N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am. J. Hum. Genet. 2004, 74, 1303–1308. [Google Scholar] [CrossRef]
- Shaheen, R.; Han, L.; Faqeih, E.; Ewida, N.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 2016, 135, 707–713. [Google Scholar] [CrossRef]
- Ofengand, J.; Del Campo, M.; Kaya, Y. Mapping pseudouridines in RNA molecules. Methods 2001, 25, 365–373. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Clark, W.C.; Pan, D.W.; Eckwahl, M.J.; Dai, Q.; Pan, T. Pseudouridines have context-dependent mutation and stop rates in high-throughput sequencing. RNA Biol. 2018, 15, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Marchand, V.; Pichot, F.; Neybecker, P.; Ayadi, L.; Bourguignon-Igel, V.; Wacheul, L.; Lafontaine, D.L.J.; Pinzano, A.; Helm, M.; Motorin, Y. HydraPsiSeq: A method for systematic and quantitative mapping of pseudouridines in RNA. Nucleic Acids Res. 2020, 48, e110. [Google Scholar] [CrossRef] [PubMed]
- Ayadi, L.; Galvanin, A.; Pichot, F.; Marchand, V.; Motorin, Y. RNA ribose methylation (2′-O-methylation): Occurrence, biosynthesis and biological functions. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Natchiar, S.K.; Myasnikov, A.G.; Hazemann, I.; Klaholz, B.P. Visualizing the Role of 2′-OH rRNA Methylations in the Human Ribosome Structure. Biomolecules 2018, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lin, J.; Ye, K. Box C/D guide RNAs recognize a maximum of 10 nt of substrates. Proc. Natl. Acad. Sci. USA 2016, 113, 10878–10883. [Google Scholar] [CrossRef]
- Van Nues, R.W.; Watkins, N.J. Unusual C/D motifs enable box C/D snoRNPs to modify multiple sites in the same rRNA target region. Nucleic Acids Res. 2017, 45, 2016–2028. [Google Scholar] [CrossRef]
- Birkedal, U.; Christensen-Dalsgaard, M.; Krogh, N.; Sabarinathan, R.; Gorodkin, J.; Nielsen, H. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pirnie, S.P.; Carmichael, G.G. High-throughput and site-specific identification of 2′-O-methylation sites using ribose oxidation sequencing (RibOxi-seq). RNA 2017, 23, 1303–1314. [Google Scholar] [CrossRef]
- Sharma, S.; Marchand, V.; Motorin, Y.; Lafontaine, D.L.J. Identification of sites of 2′-O-methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci. Rep. 2017, 7, 11490. [Google Scholar] [CrossRef]
- Motorin, Y.; Marchand, V. Detection and Analysis of RNA Ribose 2′-O-Methylations: Challenges and Solutions. Genes 2018, 9, 642. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Nielsen, H. Sequencing-based methods for detection and quantitation of ribose methylations in RNA. Methods 2019, 156, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Ayadi, L.; Motorin, Y.; Marchand, V. Quantification of 2′-O-Me Residues in RNA Using Next-Generation Sequencing (Illumina RiboMethSeq Protocol). Methods Mol. Biol. 2018, 1649, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Monaco, P.L.; Marcel, V.; Diaz, J.J.; Catez, F. 2′-O-Methylation of Ribosomal RNA: Towards an Epitranscriptomic Control of Translation? Biomolecules 2018, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, B.; Henning, D.; So, R.B.; Dixon, J.; Dixon, M.J.; Valdez, B.C. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum. Mol. Genet. 2005, 14, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Jansson, M.D.; Hafner, S.J.; Tehler, D.; Birkedal, U.; Christensen-Dalsgaard, M.; Lund, A.H.; Nielsen, H. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016, 44, 7884–7895. [Google Scholar] [CrossRef] [PubMed]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef]
- Sergiev, P.V.; Golovina, A.Y.; Osterman, I.A.; Nesterchuk, M.V.; Sergeeva, O.V.; Chugunova, A.A.; Evfratov, S.A.; Andreianova, E.S.; Pletnev, P.I.; Laptev, I.G.; et al. N6-Methylated Adenosine in RNA: From Bacteria to Humans. J. Mol. Biol. 2016, 428, 2134–2145. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Cai, J.; Dai, Q.; Natchiar, S.K.; Lv, R.; Chen, K.; Lu, Z.; Chen, H.; Shi, Y.G.; et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 2019, 15, 88–94. [Google Scholar] [CrossRef]
- Chen, K.; Zhao, B.S.; He, C. Nucleic Acid Modifications in Regulation of Gene Expression. Cell Chem. Biol. 2016, 23, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Leismann, J.; Spagnuolo, M.; Pradhan, M.; Wacheul, L.; Vu, M.A.; Musheev, M.; Mier, P.; Andrade-Navarro, M.A.; Graille, M.; Niehrs, C.; et al. The 18S ribosomal RNA m(6) A methyltransferase Mettl5 is required for normal walking behavior in Drosophila. EMBO Rep. 2020, 21, e49443. [Google Scholar] [CrossRef] [PubMed]
- Van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Graille, M.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; He, C. RNA epigenetics—Chemical messages for posttranscriptional gene regulation. Curr. Opin. Chem. Biol. 2016, 30, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Knuckles, P.; Buhler, M. Adenosine methylation as a molecular imprint defining the fate of RNA. FEBS Lett. 2018, 592, 2845–2859. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Grozhik, A.V.; Linder, B.; Olarerin-George, A.O.; Jaffrey, S.R. Mapping m(6)A at Individual-Nucleotide Resolution Using Crosslinking and Immunoprecipitation (miCLIP). Methods Mol. Biol. 2017, 1562, 55–78. [Google Scholar] [CrossRef]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef]
- Zorbas, C.; Nicolas, E.; Wacheul, L.; Huvelle, E.; Heurgue-Hamard, V.; Lafontaine, D.L. The human 18S rRNA base methyltransferases DIMT1L and WBSCR22-TRMT112 but not rRNA modification are required for ribosome biogenesis. Mol. Biol. Cell 2015, 26, 2080–2095. [Google Scholar] [CrossRef]
- Sergiev, P.V.; Aleksashin, N.A.; Chugunova, A.A.; Polikanov, Y.S.; Dontsova, O.A. Structural and evolutionary insights into ribosomal RNA methylation. Nat. Chem. Biol. 2018, 14, 226–235. [Google Scholar] [CrossRef]
- Peifer, C.; Sharma, S.; Watzinger, P.; Lamberth, S.; Kotter, P.; Entian, K.D. Yeast Rrp8p, a novel methyltransferase responsible for m1A 645 base modification of 25S rRNA. Nucleic Acids Res. 2013, 41, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Hartmann, J.D.; Watzinger, P.; Klepper, A.; Peifer, C.; Kotter, P.; Lafontaine, D.L.J.; Entian, K.D. A single N(1)-methyladenosine on the large ribosomal subunit rRNA impacts locally its structure and the translation of key metabolic enzymes. Sci. Rep. 2018, 8, 11904. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Peng, J.; Yi, C. Transcriptome-Wide Mapping of N (1)-Methyladenosine Methylome. Methods Mol. Biol. 2017, 1562, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jia, G. Reversible RNA Modification N(1)-methyladenosine (m(1)A) in mRNA and tRNA. Genom. Proteom. Bioinform. 2018, 16, 155–161. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D’Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol. Cell. Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef]
- Willi, J.; Kupfer, P.; Evequoz, D.; Fernandez, G.; Katz, A.; Leumann, C.; Polacek, N. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 2018, 46, 1945–1957. [Google Scholar] [CrossRef]
- Limoncelli, K.A. Identification of Factors Involved in 18S Nonfunctional Ribosomal RNA Decay and a Method for Detecting 8-oxoguanosine by RNA-Seq. Ph.D. Thesis, RNA Therapeutics Institute; UMASS Medical School, Worcester, MA, USA, 18 December 2017. Available online: https://escholarship.umassmed.edu/gsbs_diss/945 (accessed on 15 April 2020).
- Kino, K.; Hirao-Suzuki, M.; Morikawa, M.; Sakaga, A.; Miyazawa, H. Generation, repair and replication of guanine oxidation products. Genes Envrion. 2017, 39, 21. [Google Scholar] [CrossRef]
- Hayrapetyan, A.; Grosjean, H.; Helm, M. Effect of a quaternary pentamine on RNA stabilization and enzymatic methylation. Biol. Chem. 2009, 390, 851–861. [Google Scholar] [CrossRef]
- Bourgeois, G.; Ney, M.; Gaspar, I.; Aigueperse, C.; Schaefer, M.; Kellner, S.; Helm, M.; Motorin, Y. Eukaryotic rRNA Modification by Yeast 5-Methylcytosine-Methyltransferases and Human Proliferation-Associated Antigen p120. PLoS ONE 2015, 10, e0133321. [Google Scholar] [CrossRef]
- Maden, B.E. Locations of methyl groups in 28 S rRNA of Xenopus laevis and man. Clustering in the conserved core of molecule. J. Mol. Biol. 1988, 201, 289–314. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009, 37, e12. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Minois, N.; Angerer, T.B.; Amring, M.; Dellago, H.; Harreither, E.; Calle-Perez, A.; Pircher, A.; Gerstl, M.P.; Pfeifenberger, S.; et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 2015, 6, 6158. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, K.E.; Hobartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m(5)C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef]
- Huang, W.; Lan, M.D.; Qi, C.B.; Zheng, S.J.; Wei, S.Z.; Yuan, B.F.; Feng, Y.Q. Formation and determination of the oxidation products of 5-methylcytosine in RNA. Chem. Sci. 2016, 7, 5495–5502. [Google Scholar] [CrossRef]
- Jobert, L.; Skjeldam, H.K.; Dalhus, B.; Galashevskaya, A.; Vagbo, C.B.; Bjoras, M.; Nilsen, H. The human base excision repair enzyme SMUG1 directly interacts with DKC1 and contributes to RNA quality control. Mol. Cell 2013, 49, 339–345. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef]
- Hrabeta-Robinson, E.; Marcus, E.; Cozen, A.E.; Phizicky, E.M.; Lowe, T.M. High-Throughput Small RNA Sequencing Enhanced by AlkB-Facilitated RNA de-Methylation (ARM-Seq). Methods Mol. Biol. 2017, 1562, 231–243. [Google Scholar] [CrossRef]
- Khoddami, V.; Yerra, A.; Mosbruger, T.L.; Fleming, A.M.; Burrows, C.J.; Cairns, B.R. Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc. Natl. Acad. Sci. USA 2019, 116, 6784–6789. [Google Scholar] [CrossRef]
- Wescoe, Z.L.; Schreiber, J.; Akeson, M. Nanopores discriminate among five C5-cytosine variants in DNA. J. Am. Chem. Soc. 2014, 136, 16582–16587. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate detection of m(6)A RNA modifications in native RNA sequences. Nat. Commun. 2019, 10, 4079. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, R.T.; Challand, M.R.; Ganzinger, K.A.; Lewis, B.W.; Softley, C.; Schmied, W.H.; Horrocks, M.H.; Shivji, N.; Chin, J.W.; Spencer, J.; et al. Detecting RNA base methylations in single cells by in situ hybridization. Nat. Commun. 2018, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Incarnato, D.; Morandi, E.; Simon, L.M.; Oliviero, S. RNA Framework: An all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Res. 2018, 46, e97. [Google Scholar] [CrossRef] [PubMed]
- Aubert, M.; O’Donohue, M.F.; Lebaron, S.; Gleizes, P.E. Pre-Ribosomal RNA Processing in Human Cells: From Mechanisms to Congenital Diseases. Biomolecules 2018, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Moss, T.; Mars, J.C.; Tremblay, M.G.; Sabourin-Felix, M. The chromatin landscape of the ribosomal RNA genes in mouse and human. Chromosome Res. 2019, 27, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Henras, A.K.; Plisson-Chastang, C.; O’Donohue, M.F.; Chakraborty, A.; Gleizes, P.E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscipl. Rev. RNA 2015, 6, 225–242. [Google Scholar] [CrossRef]
- Mullineux, S.T.; Lafontaine, D.L. Mapping the cleavage sites on mammalian pre-rRNAs: Where do we stand? Biochimie 2012, 94, 1521–1532. [Google Scholar] [CrossRef]
- Tomecki, R.; Sikorski, P.J.; Zakrzewska-Placzek, M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells—Focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 2017, 591, 1801–1850. [Google Scholar] [CrossRef]
- Pirouz, M.; Munafo, M.; Ebrahimi, A.G.; Choe, J.; Gregory, R.I. Exonuclease requirements for mammalian ribosomal RNA biogenesis and surveillance. Nat. Struct. Mol. Biol. 2019, 26, 490–500. [Google Scholar] [CrossRef]
- Makino, D.L.; Halbach, F.; Conti, E. The RNA exosome and proteasome: Common principles of degradation control. Nat. Rev. Mol. Cell Biol. 2013, 14, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, A.; Lubas, M.; Jensen, T.H.; Dziembowski, A. RNA decay machines: The exosome. Biochim. Biophys. Acta 2013, 1829, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Tomecki, R.; Jensen, T.H.; Dziembowski, A. The eukaryotic RNA exosome: Same scaffold but variable catalytic subunits. RNA Biol. 2014, 8, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Parker, R.P. The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 1998, 17, 1497–1506. [Google Scholar] [CrossRef]
- Muhlrad, D.; Decker, C.J.; Parker, R. Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′-->3′ digestion of the transcript. Genes Dev. 1994, 8, 855–866. [Google Scholar] [CrossRef]
- Miller, J.E.; Reese, J.C. Ccr4-Not complex: The control freak of eukaryotic cells. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 315–333. [Google Scholar] [CrossRef]
- Simms, C.L.; Zaher, H.S. Quality control of chemically damaged RNA. Cell. Mol. Life Sci. CMLS 2016, 73, 3639–3653. [Google Scholar] [CrossRef]
- Manickam, N.; Nag, N.; Abbasi, A.; Patel, K.; Farabaugh, P.J. Studies of translational misreading in vivo show that the ribosome very efficiently discriminates against most potential errors. RNA 2014, 20, 9–15. [Google Scholar] [CrossRef]
- Hug, N.; Longman, D.; Caceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef]
- Lykke-Andersen, J.; Bennett, E.J. Protecting the proteome: Eukaryotic cotranslational quality control pathways. J. Cell Biol. 2014, 204, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Kervestin, S.; Jacobson, A. NMD: A multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 2012, 13, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Muhlrad, D.; Parker, R. Recognition of yeast mRNAs as “nonsense containing” leads to both inhibition of mRNA translation and mRNA degradation: Implications for the control of mRNA decapping. Mol. Biol. Cell 1999, 10, 3971–3978. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Isken, O.; Kim, Y.K.; Hosoda, N.; Mayeur, G.L.; Hershey, J.W.; Maquat, L.E. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 2008, 133, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Harigaya, Y.; Parker, R. No-go decay: A quality control mechanism for RNA in translation. Wiley Interdiscipl. Rev. RNA 2010, 1, 132–141. [Google Scholar] [CrossRef]
- Chen, L.; Muhlrad, D.; Hauryliuk, V.; Cheng, Z.; Lim, M.K.; Shyp, V.; Parker, R.; Song, H. Structure of the Dom34-Hbs1 complex and implications for no-go decay. Nat. Struct. Mol. Biol. 2010, 17, 1233–1240. [Google Scholar] [CrossRef]
- Doma, M.K.; Parker, R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 2006, 440, 561–564. [Google Scholar] [CrossRef]
- Lee, H.H.; Kim, Y.S.; Kim, K.H.; Heo, I.; Kim, S.K.; Kim, O.; Kim, H.K.; Yoon, J.Y.; Kim, H.S.; Kim, D.J.; et al. Structural and functional insights into Dom34, a key component of no-go mRNA decay. Mol. Cell 2007, 27, 938–950. [Google Scholar] [CrossRef]
- Passos, D.O.; Doma, M.K.; Shoemaker, C.J.; Muhlrad, D.; Green, R.; Weissman, J.; Hollien, J.; Parker, R. Analysis of Dom34 and its function in no-go decay. Mol. Biol. Cell 2009, 20, 3025–3032. [Google Scholar] [CrossRef]
- Van den Elzen, A.M.; Henri, J.; Lazar, N.; Gas, M.E.; Durand, D.; Lacroute, F.; Nicaise, M.; van Tilbeurgh, H.; Seraphin, B.; Graille, M. Dissection of Dom34-Hbs1 reveals independent functions in two RNA quality control pathways. Nat. Struct. Mol. Biol. 2010, 17, 1446–1452. [Google Scholar] [CrossRef]
- Becker, T.; Armache, J.P.; Jarasch, A.; Anger, A.M.; Villa, E.; Sieber, H.; Motaal, B.A.; Mielke, T.; Berninghausen, O.; Beckmann, R. Structure of the no-go mRNA decay complex Dom34-Hbs1 bound to a stalled 80S ribosome. Nat. Struct. Mol. Biol. 2011, 18, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Hosoda, N.; Hoshino, S. The Hbs1-Dom34 protein complex functions in non-stop mRNA decay in mammalian cells. J. Biol. Chem. 2013, 288, 17832–17843. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, C.J.; Eyler, D.E.; Green, R. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science 2010, 330, 369–372. [Google Scholar] [CrossRef]
- Pisareva, V.P.; Skabkin, M.A.; Hellen, C.U.; Pestova, T.V.; Pisarev, A.V. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J. 2011, 30, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Peltz, S.W.; Wilusz, C.J. Non-stop decay—A new mRNA surveillance pathway. Bioessays 2002, 24, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Klauer, A.A.; van Hoof, A. Degradation of mRNAs that lack a stop codon: A decade of nonstop progress. Wiley Interdiscipl. Rev. RNA 2012, 3, 649–660. [Google Scholar] [CrossRef]
- Danin-Kreiselman, M.; Lee, C.Y.; Chanfreau, G. RNAse III-mediated degradation of unspliced pre-mRNAs and lariat introns. Mol. Cell 2003, 11, 1279–1289. [Google Scholar] [CrossRef]
- Assenholt, J.; Mouaikel, J.; Saguez, C.; Rougemaille, M.; Libri, D.; Jensen, T.H. Implication of Ccr4-Not complex function in mRNA quality control in Saccharomyces cerevisiae. RNA 2011, 17, 1788–1794. [Google Scholar] [CrossRef]
- LaRiviere, F.J.; Cole, S.E.; Ferullo, D.J.; Moore, M.J. A late-acting quality control process for mature eukaryotic rRNAs. Mol. Cell 2006, 24, 619–626. [Google Scholar] [CrossRef]
- Cole, S.E.; LaRiviere, F.J.; Merrikh, C.N.; Moore, M.J. A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol. Cell 2009, 34, 440–450. [Google Scholar] [CrossRef]
- Parker, R. RNA degradation in Saccharomyces cerevisae. Genetics 2012, 191, 671–702. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Kitabatake, M.; Sakata, T.; Ohno, M. 40S subunit dissociation and proteasome-dependent RNA degradation in nonfunctional 25S rRNA decay. EMBO J. 2012, 31, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Limoncelli, K.A.; Merrikh, C.N.; Moore, M.J. ASC1 and RPS3: New actors in 18S nonfunctional rRNA decay. RNA 2017, 23, 1946–1960. [Google Scholar] [CrossRef] [PubMed]
- Vanacova, S.; Stefl, R. The exosome and RNA quality control in the nucleus. EMBO Rep. 2007, 8, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, N.; Panasenko, O.O.; Colau, G.; Collart, M.A. The CCR4-NOT complex physically and functionally interacts with TRAMP and the nuclear exosome. PLoS ONE 2009, 4, e6760. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, N.; Panasenko, O.O.; Deluen, C.; Hsieh, J.; Theiler, G.; Collart, M.A. Specific roles for the Ccr4-Not complex subunits in expression of the genome. RNA 2009, 15, 377–383. [Google Scholar] [CrossRef]
- Dez, C.; Houseley, J.; Tollervey, D. Surveillance of nuclear-restricted pre-ribosomes within a subnucleolar region of Saccharomyces cerevisiae. EMBO J. 2006, 25, 1534–1546. [Google Scholar] [CrossRef]
- Lafontaine, D.L.; Preiss, T.; Tollervey, D. Yeast 18S rRNA dimethylase Dim1p: A quality control mechanism in ribosome synthesis? Mol. Cell. Biol. 1998, 18, 2360–2370. [Google Scholar] [CrossRef] [PubMed]
- Hopper, A.K.; Huang, H.Y. Quality Control Pathways for Nucleus-Encoded Eukaryotic tRNA Biosynthesis and Subcellular Trafficking. Mol. Cell. Biol. 2015, 35, 2052–2058. [Google Scholar] [CrossRef]
- Megel, C.; Morelle, G.; Lalande, S.; Duchene, A.M.; Small, I.; Marechal-Drouard, L. Surveillance and cleavage of eukaryotic tRNAs. Int. J. Mol. Sci 2015, 16, 1873–1893. [Google Scholar] [CrossRef]
- Whipple, J.M.; Lane, E.A.; Chernyakov, I.; D’Silva, S.; Phizicky, E.M. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes Dev. 2011, 25, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Guy, M.P.; Young, D.L.; Payea, M.J.; Zhang, X.; Kon, Y.; Dean, K.M.; Grayhack, E.J.; Mathews, D.H.; Fields, S.; Phizicky, E.M. Identification of the determinants of tRNA function and susceptibility to rapid tRNA decay by high-throughput in vivo analysis. Genes Dev. 2014, 28, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Payea, M.J.; Sloma, M.F.; Kon, Y.; Young, D.L.; Guy, M.P.; Zhang, X.; De Zoysa, T.; Fields, S.; Mathews, D.H.; Phizicky, E.M. Widespread temperature sensitivity and tRNA decay due to mutations in a yeast tRNA. RNA 2018, 24, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Payea, M.J.; Hauke, A.C.; De Zoysa, T.; Phizicky, E.M. Mutations in the anticodon stem of tRNA cause accumulation and Met22-dependent decay of pre-tRNA in yeast. RNA 2020, 26, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.D.; Wilusz, J.E.; Zheng, Y.; Beal, P.A.; Joshua-Tor, L. On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme. Cell 2015, 160, 644–658. [Google Scholar] [CrossRef]
- Wilusz, J.E.; Whipple, J.M.; Phizicky, E.M.; Sharp, P.A. tRNAs marked with CCACCA are targeted for degradation. Science 2011, 334, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Ijiri, K.; Ohtsuki, T. mTOR regulates the nucleoplasmic diffusion of Xrn2 under conditions of heat stress. FEBS Lett. 2014, 588, 3454–3460. [Google Scholar] [CrossRef]
- Dewe, J.M.; Whipple, J.M.; Chernyakov, I.; Jaramillo, L.N.; Phizicky, E.M. The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA 2012, 18, 1886–1896. [Google Scholar] [CrossRef]
- Pirouz, M.; Ebrahimi, A.G.; Gregory, R.I. Unraveling 3′-end RNA uridylation at nucleotide resolution. Methods 2019, 155, 10–19. [Google Scholar] [CrossRef]
- Ustianenko, D.; Pasulka, J.; Feketova, Z.; Bednarik, L.; Zigackova, D.; Fortova, A.; Zavolan, M.; Vanacova, S. TUT-DIS3L2 is a mammalian surveillance pathway for aberrant structured non-coding RNAs. EMBO J. 2016, 35, 2179–2191. [Google Scholar] [CrossRef]
- Ustianenko, D.; Hrossova, D.; Potesil, D.; Chalupnikova, K.; Hrazdilova, K.; Pachernik, J.; Cetkovska, K.; Uldrijan, S.; Zdrahal, Z.; Vanacova, S. Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. RNA 2013, 19, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Pirouz, M.; Du, P.; Munafo, M.; Gregory, R.I. Dis3l2-Mediated Decay Is a Quality Control Pathway for Noncoding RNAs. Cell Rep. 2016, 16, 1861–1873. [Google Scholar] [CrossRef] [PubMed]
- Malecki, M.; Viegas, S.C.; Carneiro, T.; Golik, P.; Dressaire, C.; Ferreira, M.G.; Arraiano, C.M. The exoribonuclease Dis3L2 defines a novel eukaryotic RNA degradation pathway. EMBO J. 2013, 32, 1842–1854. [Google Scholar] [CrossRef]
- Lubas, M.; Damgaard, C.K.; Tomecki, R.; Cysewski, D.; Jensen, T.H.; Dziembowski, A. Exonuclease hDIS3L2 specifies an exosome-independent 3′-5′ degradation pathway of human cytoplasmic mRNA. EMBO J. 2013, 32, 1855–1868. [Google Scholar] [CrossRef]
- Labno, A.; Warkocki, Z.; Kulinski, T.; Krawczyk, P.S.; Bijata, K.; Tomecki, R.; Dziembowski, A. Perlman syndrome nuclease DIS3L2 controls cytoplasmic non-coding RNAs and provides surveillance pathway for maturing snRNAs. Nucleic Acids Res. 2016, 44, 10437–10453. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Morris, M.R.; Cooper, W.N.; Staals, R.H.; Wake, N.C.; Fews, G.A.; Gill, H.; Gentle, D.; Shuib, S.; Ricketts, C.J.; et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat. Genet. 2012, 44, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Pirouz, M.; Gregory, R.I. A Single Let-7 MicroRNA Bypasses LIN28-Mediated Repression. Cell Rep. 2015, 13, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, H.S.; Visnes, T.; Vagbo, C.B.; Svaasand, E.K.; Doseth, B.; Slupphaug, G.; Kavli, B.; Krokan, H.E. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011, 39, 8430–8444. [Google Scholar] [CrossRef] [PubMed]
- Jobert, L.; Nilsen, H. Regulatory mechanisms of RNA function: Emerging roles of DNA repair enzymes. Cell. Mol. Life Sci. CMLS 2014, 71, 2451–2465. [Google Scholar] [CrossRef]
- Wood, R.D.; Mitchell, M.; Lindahl, T. Human DNA repair genes, 2005. Mutat. Res. 2005, 577, 275–283. [Google Scholar] [CrossRef]
- Seeberg, E.; Eide, L.; Bjoras, M. The base excision repair pathway. Trends Biochem. Sci. 1995, 20, 391–397. [Google Scholar] [CrossRef]
- Lindahl, T. Keynote: Past, present, and future aspects of base excision repair. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 68, xvii–xxx. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Nilsen, H.; Skorpen, F.; Otterlei, M.; Slupphaug, G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000, 476, 73–77. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Tye, B.K.; Chien, J.; Lehman, I.R.; Duncan, B.K.; Warner, H.R. Uracil incorporation: A source of pulse-labeled DNA fragments in the replication of the Escherichia coli chromosome. Proc. Natl. Acad. Sci. USA 1978, 75, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H. Structure and function in the uracil-DNA glycosylase superfamily. Mutat. Res. 2000, 460, 165–181. [Google Scholar] [CrossRef]
- Kim, Y.J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharm. 2012, 5, 3–13. [Google Scholar] [CrossRef]
- Masaoka, A.; Matsubara, M.; Hasegawa, R.; Tanaka, T.; Kurisu, S.; Terato, H.; Ohyama, Y.; Karino, N.; Matsuda, A.; Ide, H. Mammalian 5-formyluracil-DNA glycosylase. 2. Role of SMUG1 uracil-DNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry 2003, 42, 5003–5012. [Google Scholar] [CrossRef]
- Alexeeva, M.; Moen, M.N.; Grosvik, K.; Tesfahun, A.N.; Xu, X.M.; Muruzabal-Lecumberri, I.; Olsen, K.M.; Rasmussen, A.; Ruoff, P.; Kirpekar, F.; et al. Excision of uracil from DNA by hSMUG1 includes strand incision and processing. Nucleic Acids Res. 2019, 47, 779–793. [Google Scholar] [CrossRef]
- Schormann, N.; Ricciardi, R.; Chattopadhyay, D. Uracil-DNA glycosylases-structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. A Publ. Protein Soc. 2014, 23, 1667–1685. [Google Scholar] [CrossRef]
- Haushalter, K.A.; Todd Stukenberg, M.W.; Kirschner, M.W.; Verdine, G.L. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol. 1999, 9, 174–185. [Google Scholar] [CrossRef]
- Kroustallaki, P.; Lirussi, L.; Carracedo, S.; You, P.; Esbensen, Q.Y.; Gotz, A.; Jobert, L.; Alsoe, L.; Saetrom, P.; Gagos, S.; et al. SMUG1 Promotes Telomere Maintenance through Telomerase RNA Processing. Cell Rep. 2019, 28, 1690–1702.e10. [Google Scholar] [CrossRef] [PubMed]
- Egan, E.D.; Collins, K. An enhanced H/ACA RNP assembly mechanism for human telomerase RNA. Mol. Cell. Biol. 2012, 32, 2428–2439. [Google Scholar] [CrossRef]
- Schmidt, J.C.; Cech, T.R. Human telomerase: Biogenesis, trafficking, recruitment, and activation. Genes Dev. 2015, 29, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.D.; Tam, J.; Wu, R.A.; Greber, B.J.; Toso, D.; Nogales, E.; Collins, K. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 2018, 557, 190–195. [Google Scholar] [CrossRef]
- Dominguez, P.M.; Shaknovich, R. Epigenetic function of activation-induced cytidine deaminase and its link to lymphomagenesis. Front. Immunol. 2014, 5, 642. [Google Scholar] [CrossRef]
- Lirussi, L.; Nilsen, H. Telomere maintenance: Regulating hTERC fatethrough RNA modifications. Mol. & Cellular Oncology 2019, 6, e1670489. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lirussi, L.; Demir, Ö.; You, P.; Sarno, A.; Amaro, R.E.; Nilsen, H. RNA Metabolism Guided by RNA Modifications: The Role of SMUG1 in rRNA Quality Control. Biomolecules 2021, 11, 76. https://doi.org/10.3390/biom11010076
Lirussi L, Demir Ö, You P, Sarno A, Amaro RE, Nilsen H. RNA Metabolism Guided by RNA Modifications: The Role of SMUG1 in rRNA Quality Control. Biomolecules. 2021; 11(1):76. https://doi.org/10.3390/biom11010076
Chicago/Turabian StyleLirussi, Lisa, Özlem Demir, Panpan You, Antonio Sarno, Rommie E. Amaro, and Hilde Nilsen. 2021. "RNA Metabolism Guided by RNA Modifications: The Role of SMUG1 in rRNA Quality Control" Biomolecules 11, no. 1: 76. https://doi.org/10.3390/biom11010076
APA StyleLirussi, L., Demir, Ö., You, P., Sarno, A., Amaro, R. E., & Nilsen, H. (2021). RNA Metabolism Guided by RNA Modifications: The Role of SMUG1 in rRNA Quality Control. Biomolecules, 11(1), 76. https://doi.org/10.3390/biom11010076