Why COVID-19 Transmission Is More Efficient and Aggressive Than Viral Transmission in Previous Coronavirus Epidemics?



{kind=link}
{kind=link}
Abstract
1. Introduction
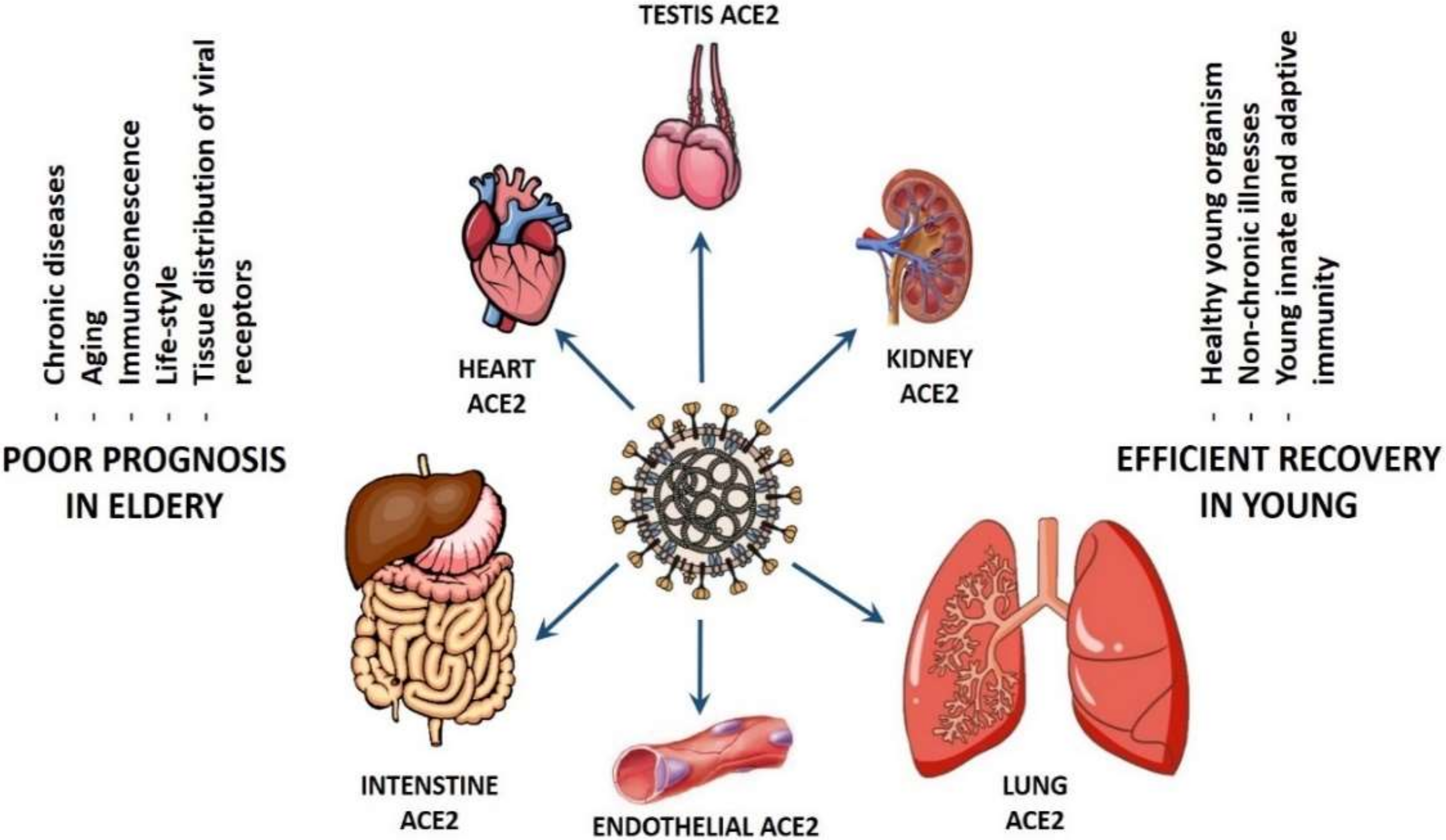
2. Intrinsic Viral Factors
3. Human (Host) Factors
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Bruckova, M.; McIntosh, K.; Kapikian, A.Z.; Chanock, R.M. The adaptation of two human coronavirus strains (OC38 and OC43) to growth in cell monolayers. Proc. Soc. Exp. Biol. Med. 1970, 135, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, C.; Chen, L.; Xu, B.; Zhou, Y.; Cao, L.; Shang, Y.; Fu, Z.; Chen, A.; Deng, L.; et al. A novel human coronavirus OC43 genotype detected in mainland China. Emerg. Microbes Infect. 2018, 7, 173. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Hierholzer, J.C. Purification and biophysical properties of human coronavirus 229E. Virology 1976, 75, 155–165. [Google Scholar] [CrossRef]
- Kaye, H.S.; Ong, S.B.; Dowdle, W.R. Detection of coronavirus 229E antibody by indirect hemagglutination. Appl. Microbiol. 1972, 24, 703–707. [Google Scholar] [CrossRef]
- Fouchier, R.A.; Hartwig, N.G.; Bestebroer, T.M.; Niemeyer, B.; de Jong, J.C.; Simon, J.H.; Osterhaus, A.D. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 6212–6216. [Google Scholar] [CrossRef]
- Van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Larson, H.E.; Reed, S.E.; Tyrrell, D.A. Isolation of rhinoviruses and coronaviruses from 38 colds in adults. J. Med. Virol. 1980, 5, 221–229. [Google Scholar] [CrossRef]
- Bradburne, A.F.; Bynoe, M.L.; Tyrrell, D.A. Effects of a “new” human respiratory virus in volunteers. Br. Med. J. 1967, 3, 767–769. [Google Scholar] [CrossRef]
- McIntosh, K.; Dees, J.H.; Becker, W.B.; Kapikian, A.Z.; Chanock, R.M. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc. Natl. Acad. Sci. USA 1967, 57, 933–940. [Google Scholar] [CrossRef]
- Almeida, J.D.; Tyrrell, D.A. The morphology of three previously uncharacterized human respiratory viruses that grow in organ culture. J. Gen. Virol. 1967, 1, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Hamre, D.; Procknow, J.J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 1966, 121, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Esper, F.; Weibel, C.; Ferguson, D.; Landry, M.L.; Kahn, J.S. Evidence of a novel human coronavirus that is associated with respiratory tract disease in infants and young children. J. Infect. Dis. 2005, 191, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Gerna, G.; Percivalle, E.; Sarasini, A.; Campanini, G.; Piralla, A.; Rovida, F.; Genini, E.; Marchi, A.; Baldanti, F. Human respiratory coronavirus HKU1 versus other coronavirus infections in Italian hospitalised patients. J. Clin. Virol. 2007, 38, 244–250. [Google Scholar] [CrossRef]
- Gerna, G.; Campanini, G.; Rovida, F.; Percivalle, E.; Sarasini, A.; Marchi, A.; Baldanti, F. Genetic variability of human coronavirus OC43-, 229E-, and NL63-like strains and their association with lower respiratory tract infections of hospitalized infants and immunocompromised patients. J. Med. Virol. 2006, 78, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Geller, C.; Varbanov, M.; Duval, R.E. Human coronaviruses: Insights into environmental resistance and its influence on the development of new antiseptic strategies. Viruses 2012, 4, 3044–3068. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family Coronaviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A., Adams, M., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Amsterdam, The Netherlands; Academic Press: Boston, MA, USA, 2012; pp. 806–820. [Google Scholar]
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res. 2014, 101, 45–56. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Benvenuto, D.; Giovanetti, M.; Ciccozzi, A.; Spoto, S.; Angeletti, S.; Ciccozzi, M. The 2019-new coronavirus epidemic: Evidence for virus evolution. J. Med. Virol. 2020, 92, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S. Another Decade, Another Coronavirus. N. Engl. J. Med. 2020, 382, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Haagmans, B.L.; Al Dhahiry, S.H.; Reusken, C.B.; Raj, V.S.; Galiano, M.; Myers, R.; Godeke, G.J.; Jonges, M.; Farag, E.; Diab, A.; et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet. Infect. Dis. 2014, 14, 140–145. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.M.; Van Ranst, M. Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef]
- Lau, S.K.; Lee, P.; Tsang, A.K.; Yip, C.C.; Tse, H.; Lee, R.A.; So, L.Y.; Lau, Y.L.; Chan, K.H.; Woo, P.C.; et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- Huynh, J.; Li, S.; Yount, B.; Smith, A.; Sturges, L.; Olsen, J.C.; Nagel, J.; Johnson, J.B.; Agnihothram, S.; Gates, J.E.; et al. Evidence supporting a zoonotic origin of human coronavirus strain NL63. J. Virol. 2012, 86, 12816–12825. [Google Scholar] [CrossRef]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an ancestral association of human coronavirus 229E with bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Oostra, M.; de Haan, C.A.; Rottier, P.J. The 29-nucleotide deletion present in human but not in animal severe acute respiratory syndrome coronaviruses disrupts the functional expression of open reading frame 8. J. Virol. 2007, 81, 13876–13888. [Google Scholar] [CrossRef] [PubMed]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 100682. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Shell disorder analysis predicts greater resilience of the SARS-CoV-2 (COVID-19) outside the body and in body fluids. Microb. Pathog. 2020, 144, 104177. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Rigidity of the outer shell predicted by a protein intrinsic disorder model sheds light on the COVID-19 (Wuhan-2019-nCoV) infectivity. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- De Groot, R.J. Structure, function and evolution of the hemagglutinin-esterase proteins of corona- and toroviruses. Glycoconj. J. 2006, 23, 59–72. [Google Scholar] [CrossRef]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a novel influenza virus in cattle and Swine: Proposal for a new genus in the Orthomyxoviridae family. mBio 2014, 5, e00014–e00031. [Google Scholar] [CrossRef]
- Matrosovich, M.; Herrler, G.; Klenk, H.D. Sialic acid receptors of viruses. Top. Curr. Chem. 2015, 367, 1–28. [Google Scholar] [CrossRef]
- Vlasak, R.; Luytjes, W.; Spaan, W.; Palese, P. Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc. Natl. Acad. Sci. USA 1988, 85, 4526–4529. [Google Scholar] [CrossRef]
- Wan, H.; Perez, D.R. Quail carry sialic acid receptors compatible with binding of avian and human influenza viruses. Virology 2006, 346, 278–286. [Google Scholar] [CrossRef]
- Traving, C.; Schauer, R. Structure, function and metabolism of sialic acids. Cell Mol. Life Sci. 1998, 54, 1330–1349. [Google Scholar] [CrossRef]
- Desforges, M.; Desjardins, J.; Zhang, C.; Talbot, P.J. The acetyl-esterase activity of the hemagglutinin-esterase protein of human coronavirus OC43 strongly enhances the production of infectious virus. J. Virol. 2013, 87, 3097–3107. [Google Scholar] [CrossRef]
- Bakkers, M.J.; Lang, Y.; Feitsma, L.J.; Hulswit, R.J.; de Poot, S.A.; van Vliet, A.L.; Margine, I.; de Groot-Mijnes, J.D.; van Kuppeveld, F.J.; Langereis, M.A.; et al. Betacoronavirus adaptation to humans involved progressive loss of hemagglutinin-esterase lectin activity. Cell Host Microbe 2017, 21, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Mubarak, A.; Alturaiki, W.; Hemida, M.G. Middle east respiratory syndrome coronavirus (MERS-CoV): Infection, immunological response, and vaccine development. J. Immunol. Res. 2019, 2019, 6491738. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Fantini, J.; Di Scala, C.; Chahinian, H.; Yahi, N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int. J. Antimicrob. Agents 2020, 105960. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Receptor recognition and cross-species infections of SARS coronavirus. Antivir. Res. 2013, 100, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Receptor recognition mechanisms of coronaviruses: A decade of structural studies. J. Virol. 2015, 89, 1954–1964. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 2020, 19, 410–417. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
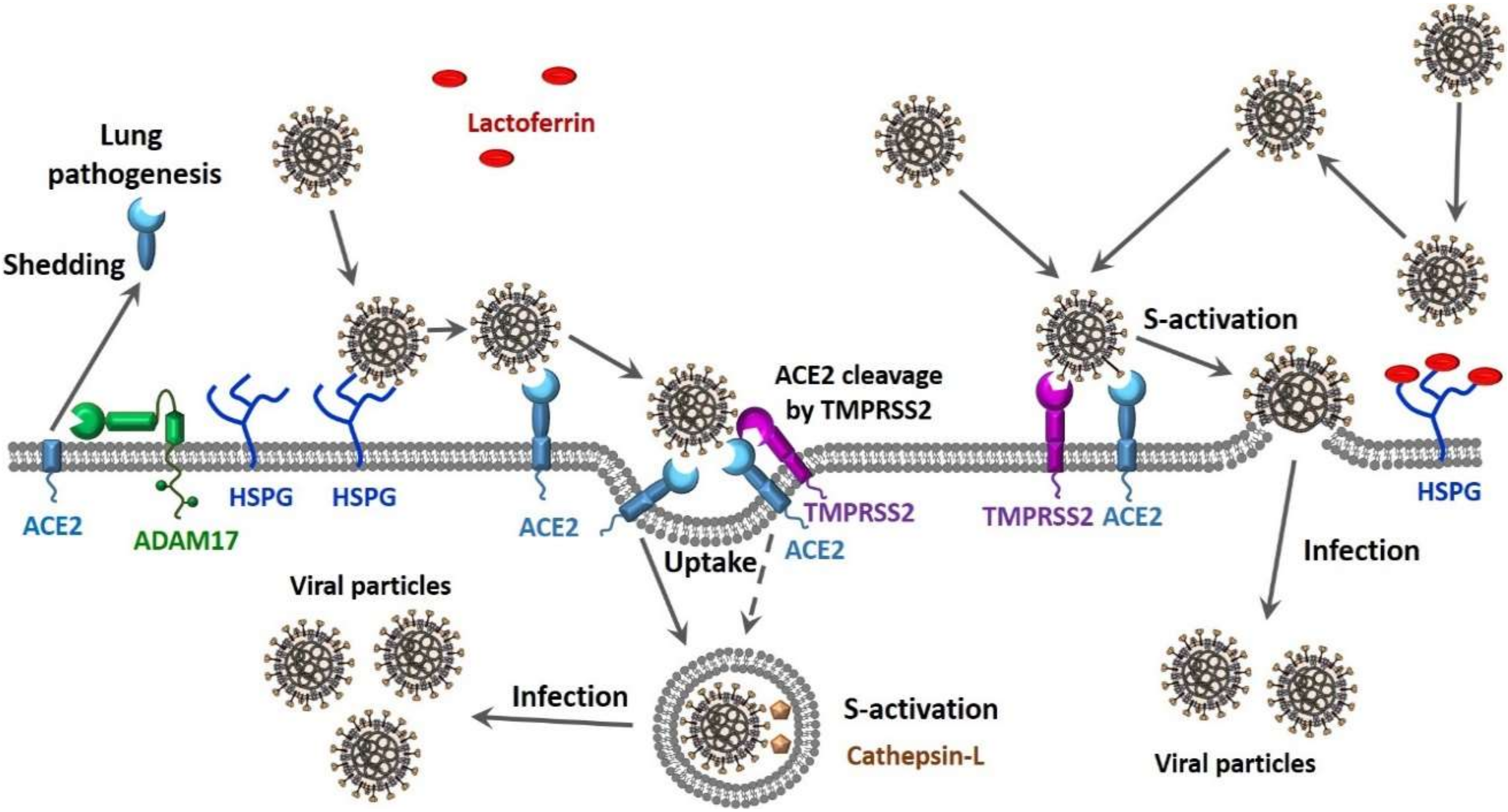
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, X.; Qiu, Y.; Feng, F.; Feng, J.; Jia, Y.; Zhu, H.; Hu, K.; Liu, J.; Liu, Z.; et al. Clinical characteristics of 82 death cases with COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Nao, N.; Yamagishi, J.; Miyamoto, H.; Igarashi, M.; Manzoor, R.; Ohnuma, A.; Tsuda, Y.; Furuyama, W.; Shigeno, A.; Kajihara, M.; et al. Genetic predisposition to acquire a polybasic cleavage site for highly pathogenic avian influenza virus hemagglutinin. mBio 2017, 8. [Google Scholar] [CrossRef]
- Kam, Y.W.; Okumura, Y.; Kido, H.; Ng, L.F.; Bruzzone, R.; Altmeyer, R. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS ONE 2009, 4, e7870. [Google Scholar] [CrossRef]
- Milewska, A.; Falkowski, K.; Kalinska, M.; Bielecka, E.; Naskalska, A.; Mak, P.; Lesner, A.; Ochman, M.; Urlik, M.; Potempa, J.; et al. Kallikrein 13: A new player in coronaviral infections. bioRxiv 2020. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Ji, H.L.; Zhao, R.; Matalon, S.; Matthay, M.A. Elevated plasmin(ogen) as a common risk factor for COVID-19 susceptibility. Physiol. Rev. 2020, 100, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Linkins, L.A.; Takach Lapner, S. Review of D-dimer testing: Good, bad, and ugly. Int. J. Lab. Hematol. 2017, 39 (Suppl. S1), 98–103. [Google Scholar] [CrossRef]
- Zhao, R.; Ali, G.; Nie, H.G.; Chang, Y.; Bhattarai, D.; Su, X.; Zhao, X.; Matthay, M.A.; Ji, H.L. Plasmin improves oedematous blood-gas barrier by cleaving epithelial sodium channels. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, T.S.; Staruschenko, A. Involvement of ENaC in the development of salt-sensitive hypertension. Am. J. Physiol. Ren. Physiol. 2017, 313, F135–F140. [Google Scholar] [CrossRef]
- Matalon, S.; Bartoszewski, R.; Collawn, J.F. Role of epithelial sodium channels in the regulation of lung fluid homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1229–L1238. [Google Scholar] [CrossRef]
- Kleyman, T.R.; Kashlan, O.B.; Hughey, R.P. Epithelial Na(+) channel regulation by extracellular and intracellular factors. Annu. Rev. Physiol. 2018, 80, 263–281. [Google Scholar] [CrossRef]
- Kone, B.C. Epigenetics and the control of the collecting duct epithelial sodium channel. Semin. Nephrol. 2013, 33, 383–391. [Google Scholar] [CrossRef]
- Hanukoglu, I.; Hanukoglu, A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 2016, 579, 95–132. [Google Scholar] [CrossRef]
- Eaton, D.C.; Helms, M.N.; Koval, M.; Bao, H.F.; Jain, L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu. Rev. Physiol. 2009, 71, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhao, R.; Zhao, X.; Matthay, M.A.; Nie, H.G.; Ji, H.L. ENaCs as both effectors and regulators of MiRNAs in lung epithelial development and regeneration. Cell. Physiol. Biochem. 2017, 44, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Boscardin, E.; Alijevic, O.; Hummler, E.; Frateschi, S.; Kellenberger, S. The function and regulation of acid-sensing ion channels (ASICs) and the epithelial Na(+) channel (ENaC): IUPHAR review 19. Br. J. Pharmacol. 2016, 173, 2671–2701. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, J.W.; Wu, M.S.; Chen, K.C.; Peng, C.C.; Kang, Y.N. Effects of calcium channel blockers comparing to angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in patients with hypertension and chronic kidney disease stage 3 to 5 and dialysis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0188975. [Google Scholar] [CrossRef] [PubMed]
- Berri, F.; Rimmelzwaan, G.F.; Hanss, M.; Albina, E.; Foucault-Grunenwald, M.L.; Le, V.B.; Vogelzang-van Trierum, S.E.; Gil, P.; Camerer, E.; Martinez, D.; et al. Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog. 2013, 9, e1003229. [Google Scholar] [CrossRef]
- Goto, H.; Wells, K.; Takada, A.; Kawaoka, Y. Plasminogen-binding activity of neuraminidase determines the pathogenicity of influenza A virus. J. Virol. 2001, 75, 9297–9301. [Google Scholar] [CrossRef]
- LeBouder, F.; Lina, B.; Rimmelzwaan, G.F.; Riteau, B. Plasminogen promotes influenza A virus replication through an annexin 2-dependent pathway in the absence of neuraminidase. J. Gen. Virol. 2010, 91, 2753–2761. [Google Scholar] [CrossRef]
- Murakami, M.; Towatari, T.; Ohuchi, M.; Shiota, M.; Akao, M.; Okumura, Y.; Parry, M.A.; Kido, H. Mini-plasmin found in the epithelial cells of bronchioles triggers infection by broad-spectrum influenza A viruses and Sendai virus. Eur. J. Biochem. 2001, 268, 2847–2855. [Google Scholar] [CrossRef]
- Su, H.; Yang, X.; Wang, S.; Shi, H.; Liu, X. Effect of annexin II-mediated conversion of plasmin from plasminogen on airborne transmission of H9N2 avian influenza virus. Vet. Microbiol. 2018, 223, 100–106. [Google Scholar] [CrossRef]
- Tse, L.V.; Marcano, V.C.; Huang, W.; Pocwierz, M.S.; Whittaker, G.R. Plasmin-mediated activation of pandemic H1N1 influenza virus hemagglutinin is independent of the viral neuraminidase. J. Virol. 2013, 87, 5161–5169. [Google Scholar] [CrossRef]
- Sun, X.; Tse, L.V.; Ferguson, A.D.; Whittaker, G.R. Modifications to the hemagglutinin cleavage site control the virulence of a neurotropic H1N1 influenza virus. J. Virol. 2010, 84, 8683–8690. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Klenk, H.D.; Rott, R. Proteolytic cleavage of the viral glycoproteins and its significance for the virulence of Newcastle disease virus. Virology 1976, 72, 494–508. [Google Scholar] [CrossRef]
- Hamilton, B.S.; Whittaker, G.R. Cleavage activation of human-adapted influenza virus subtypes by kallikrein-related peptidases 5 and 12. J. Biol. Chem. 2013, 288, 17399–17407. [Google Scholar] [CrossRef] [PubMed]
- Dubovi, E.J.; Geratz, J.D.; Tidwell, R.R. Enhancement of respiratory syncytial virus-induced cytopathology by trypsin, thrombin, and plasmin. Infect. Immun. 1983, 40, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.H.; Hirsh, A.; Li, D.C.; Holloway, G.; Chao, J.; Boucher, R.C.; Gabriel, S.E. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002, 277, 8338–8345. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. Jama 2020. [Google Scholar] [CrossRef]
- Antalis, T.M.; Bugge, T.H.; Wu, Q. Membrane-anchored serine proteases in health and disease. Prog. Mol. Biol. Transl. Sci. 2011, 99, 1–50. [Google Scholar] [CrossRef]
- Zmora, P.; Hoffmann, M.; Kollmus, H.; Moldenhauer, A.S.; Danov, O.; Braun, A.; Winkler, M.; Schughart, K.; Pohlmann, S. TMPRSS11A activates the influenza A virus hemagglutinin and the MERS coronavirus spike protein and is insensitive against blockade by HAI-1. J. Biol. Chem. 2018, 293, 13863–13873. [Google Scholar] [CrossRef]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pohlmann, S.; Soilleux, E.J. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef]
- Simmons, G.; Zmora, P.; Gierer, S.; Heurich, A.; Pohlmann, S. Proteolytic activation of the SARS-coronavirus spike protein: Cutting enzymes at the cutting edge of antiviral research. Antivir. Res. 2013, 100, 605–614. [Google Scholar] [CrossRef]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef] [PubMed]
- Bottcher-Friebertshauser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar] [CrossRef]
- Chaipan, C.; Kobasa, D.; Bertram, S.; Glowacka, I.; Steffen, I.; Solomon Tsegaye, T.; Takeda, M.; Bugge, T.H.; Kim, S.; Park, Y.; et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 2009, 83, 3200. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Palau, V.; Riera, M.; Soler, M.J. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol. Dial. Transplant. 2020. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef]
- Haga, S.; Nagata, N.; Okamura, T.; Yamamoto, N.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antivir. Res. 2010, 85, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Peck, K.M.; Scobey, T.; Swanstrom, J.; Jensen, K.L.; Burch, C.L.; Baric, R.S.; Heise, M.T. Permissivity of dipeptidyl peptidase 4 orthologs to Middle East respiratory syndrome coronavirus is governed by glycosylation and other complex determinants. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Gramberg, T.; Simmons, G.; Moller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef]
- Wang, S.; Guo, F.; Liu, K.; Wang, H.; Rao, S.; Yang, P.; Jiang, C. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008, 136, 8–15. [Google Scholar] [CrossRef]
- Sigrist, C.J.; Bridge, A.; Le Mercier, P. A potential role for integrins in host cell entry by SARS-CoV-2. Antivir. Res. 2020, 177, 104759. [Google Scholar] [CrossRef]
- Hussein, H.A.; Walker, L.R.; Abdel-Raouf, U.M.; Desouky, S.A.; Montasser, A.K.; Akula, S.M. Beyond RGD: Virus interactions with integrins. Arch. Virol. 2015, 160, 2669–2681. [Google Scholar] [CrossRef]
- Wickham, T.J.; Filardo, E.J.; Cheresh, D.A.; Nemerow, G.R. Integrin alpha v beta 5 selectively promotes adenovirus mediated cell membrane permeabilization. J. Cell Biol. 1994, 127, 257–264. [Google Scholar] [CrossRef]
- Williams, C.H.; Kajander, T.; Hyypia, T.; Jackson, T.; Sheppard, D.; Stanway, G. Integrin alpha v beta 6 is an RGD-dependent receptor for coxsackievirus A9. J. Virol. 2004, 78, 6967–6973. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, Y.; Cai, H.; Mirza, A.M.; Iorio, R.M.; Peeples, M.E.; Niewiesk, S.; Li, J. Roles of the putative integrin-binding motif of the human metapneumovirus fusion (f) protein in cell-cell fusion, viral infectivity, and pathogenesis. J. Virol. 2014, 88, 4338–4352. [Google Scholar] [CrossRef]
- Chang, A.; Masante, C.; Buchholz, U.J.; Dutch, R.E. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulfate. J. Virol. 2012, 86, 3230–3243. [Google Scholar] [CrossRef]
- Xiao, J.; Palefsky, J.M.; Herrera, R.; Berline, J.; Tugizov, S.M. The Epstein-Barr virus BMRF-2 protein facilitates virus attachment to oral epithelial cells. Virology 2008, 370, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Feire, A.L.; Koss, H.; Compton, T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. USA 2004, 101, 15470–15475. [Google Scholar] [CrossRef] [PubMed]
- Zarate, S.; Romero, P.; Espinosa, R.; Arias, C.F.; Lopez, S. VP7 mediates the interaction of rotaviruses with integrin alphavbeta3 through a novel integrin-binding site. J. Virol. 2004, 78, 10839–10847. [Google Scholar] [CrossRef]
- Altan-Bonnet, N. Extracellular vesicles are the Trojan horses of viral infection. Curr. Opin. Microbiol. 2016, 32, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, N.; Perales, C.; Domingo, E. Extracellular vesicles: Vehicles of en bloc viral transmission. Virus Res. 2019, 265, 143–149. [Google Scholar] [CrossRef]
- Gunasekaran, M.; Bansal, S.; Ravichandran, R.; Sharma, M.; Perincheri, S.; Rodriguez, F.; Hachem, R.; Fisher, C.E.; Limaye, A.P.; Omar, A.; et al. Respiratory viral infection in lung transplantation induces exosomes that trigger chronic rejection. J. Heart Lung Transplant. 2020, 39, 379–388. [Google Scholar] [CrossRef]
- Elrashdy, F.; Aljaddawi, A.A.; Redwan, E.M.; Uversky, V.N. On the potential role of exosomes in the COVID-19 reinfection/reactivation opportunity. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Naskalska, A.; Dabrowska, A.; Szczepanski, A.; Milewska, A.; Jasik, K.P.; Pyrc, K. Membrane protein of human coronavirus NL63 is responsible for interaction with the adhesion receptor. J. Virol. 2019, 93, e00319–e00355. [Google Scholar] [CrossRef]
- Milewska, A.; Zarebski, M.; Nowak, P.; Stozek, K.; Potempa, J.; Pyrc, K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 2014, 88, 13221–13230. [Google Scholar] [CrossRef]
- Belting, M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem. Sci. 2003, 28, 145–151. [Google Scholar] [CrossRef]
- Lang, J.; Yang, N.; Deng, J.; Liu, K.; Yang, P.; Zhang, G.; Jiang, C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE 2011, 6, e23710. [Google Scholar] [CrossRef] [PubMed]
- Redwan, E.M.; Uversky, V.N.; El-Fakharany, E.M.; Al-Mehdar, H. Potential lactoferrin activity against pathogenic viruses. Comptes Rendus Biol. 2014, 337, 581–595. [Google Scholar] [CrossRef]
- Albar, A.H.; Almehdar, H.A.; Uversky, V.N.; Redwan, E.M. Structural heterogeneity and multifunctionality of lactoferrin. Curr. Protein Pept. Sci. 2014, 15, 778–797. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; El-Fakkarany, E.; Lonnerdal, B.; Redwan, E.M. Inhibitory effects of native and recombinant full-length camel lactoferrin and its N and C lobes on hepatitis C virus infection of Huh7.5 cells. J. Med. Microbiol. 2012, 61, 375–383. [Google Scholar] [CrossRef]
- Reghunathan, R.; Jayapal, M.; Hsu, L.Y.; Chng, H.H.; Tai, D.; Leung, B.P.; Melendez, A.J. Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 2005, 6, 2. [Google Scholar] [CrossRef]
- Jenssen, H.; Hancock, R.E. Antimicrobial properties of lactoferrin. Biochimie 2009, 91, 19–29. [Google Scholar] [CrossRef]
- Qinfen, Z.; Jinming, C.; Xiaojun, H.; Huanying, Z.; Jicheng, H.; Ling, F.; Kunpeng, L.; Jingqiang, Z. The life cycle of SARS coronavirus in vero E6 cells. J. Med. Virol. 2004, 73, 332–337. [Google Scholar] [CrossRef]
- Simmons, G.; Reeves, J.D.; Rennekamp, A.J.; Amberg, S.M.; Piefer, A.J.; Bates, P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA 2004, 101, 4240–4245. [Google Scholar] [CrossRef]
- Ng, M.L.; Tan, S.H.; See, E.E.; Ooi, E.E.; Ling, A.E. Early events of SARS coronavirus infection in vero cells. J. Med. Virol. 2003, 71, 323–331. [Google Scholar] [CrossRef]
- Pelkmans, L.; Helenius, A. Insider information: What viruses tell us about endocytosis. Curr. Opin. Cell Biol. 2003, 15, 414–422. [Google Scholar] [CrossRef]
- Sieczkarski, S.B.; Whittaker, G.R. Dissecting virus entry via endocytosis. J. Gen. Virol. 2002, 83, 1535–1545. [Google Scholar] [CrossRef]
- Nunes-Correia, I.; Eulalio, A.; Nir, S.; Pedroso de Lima, M.C. Caveolae as an additional route for influenza virus endocytosis in MDCK cells. Cell. Mol. Biol. Lett. 2004, 9, 47–60. [Google Scholar] [PubMed]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Peterlin, B.M. Endocytic entry of HIV-1. Curr. Biol. 2000, 10, 1005–1008. [Google Scholar] [CrossRef]
- Matlin, K.S.; Reggio, H.; Helenius, A.; Simons, K. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell Biol. 1981, 91, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 12543. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, e105114. [Google Scholar] [CrossRef]
- Fung, S.Y.; Yuen, K.S.; Ye, Z.W.; Chan, C.P.; Jin, D.Y. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: Lessons from other pathogenic viruses. Emerg. Microbes Infect. 2020, 9, 558–570. [Google Scholar] [CrossRef]
- Gandon, S.; Michalakis, Y. Evolution of parasite virulence against qualitative or quantitative host resistance. Proc. Biol. Sci. R. Soc. 2000, 267, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Thrall, P.H.; Burdon, J.J. Evolution of virulence in a plant host-pathogen metapopulation. Science 2003, 299, 1735–1737. [Google Scholar] [CrossRef] [PubMed]
- Thrall, P.H.; Burdon, J.J.; Bever, J.D. Local adaptation in the Linum marginale—Melampsora lini host-pathogen interaction. Evolution. 2002, 56, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Gandon, S.; van Baalen, M.; Jansen, V.A. The evolution of parasite virulence, superinfection, and host resistance. Am. Nat. 2002, 159, 658–669. [Google Scholar] [CrossRef]
- Kubinak, J.L.; Potts, W.K. Host resistance influences patterns of experimental viral adaptation and virulence evolution. Virulence 2013, 4, 410–418. [Google Scholar] [CrossRef]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef]
- Welsh, R.M.; Selin, L.K. No one is naive: The significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2002, 2, 417–426. [Google Scholar] [CrossRef]
- St. John, A.L.; Rathore, A.P.S. Adaptive immune responses to primary and secondary dengue virus infections. Nat. Rev. Immunol. 2019, 19, 218–230. [Google Scholar] [CrossRef]
- Kim, C.W.; Yoo, H.J.; Park, J.H.; Oh, J.E.; Lee, H.K. Exogenous interleukin-33 contributes to protective immunity via cytotoxic T-cell priming against mucosal influenza viral infection. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Mullbacher, A.; Lobigs, M.; Alsharifi, M.; Regner, M. Cytotoxic T-cell immunity as a target for influenza vaccines. Lancet. Infect. Dis. 2006, 6, 255–256. [Google Scholar] [CrossRef]
- McMichael, A.J.; Gotch, F.M.; Noble, G.R.; Beare, P.A. Cytotoxic T-cell immunity to influenza. N. Engl. J. Med. 1983, 309, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Schmitz, K.S.; Raadsen, M.P.; Grifoni, A.; Okba, N.M.A.; Endeman, H.; van den Akker, J.P.C.; Molenkamp, R.; Koopmans, M.P.G.; van Gorp, E.C.M.; et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4 (+) and CD8 (+) T cells induced by SARS-CoV-2 in UK convalescent COVID-19 patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, W.; Yang, L.; You, R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharm. Res. 2020, 157, 104833. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020. [Google Scholar] [CrossRef]
- Barkauskas, C.; Cronce, M.; Rackley, C.; Bowie, E.; Keene, D.; Stripp, B.; Randell, S.; Noble, P.; Hogan, B. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Q.; Sun, X.; Shen, J.; Chen, H. Organoids as a powerful model for respiratory diseases. Stem Cells Int. 2020, 2020, 5847876. [Google Scholar] [CrossRef]
- Rivellese, F.; Prediletto, E. ACE2 at the centre of COVID-19 from paucisymptomatic infections to severe pneumonia. Autoimmun. Rev. 2020, 102536. [Google Scholar] [CrossRef]
- Kass, D.A.; Duggal, P.; Cingolani, O. Obesity could shift severe COVID-19 disease to younger ages. Lancet 2020, 395, 1544–1545. [Google Scholar] [CrossRef]
- Heialy, S.A.; Hachim, M.; Senok, A.; Tayoun, A.A.; Hamoudi, R.; Alsheikh-Ali, A.; Hamid, Q. Regulation of angiotensin converting enzyme 2 (ACE2) in obesity: Implications for COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, M.; Shen, W.; Rowan, N.R.; Kulaga, H.; Hillel, A.; Ramanathan, M., Jr.; Lane, A.P. Elevated ACE2 expression in the olfactory neuroepithelium: Implications for anosmia and upper respiratory SARS-CoV-2 entry and replication. Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- Lechien, J.R.; Chiesa-Estomba, C.M.; De Siati, D.R.; Horoi, M.; Le Bon, S.D.; Rodriguez, A.; Dequanter, D.; Blecic, S.; El Afia, F.; Distinguin, L.; et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): A multicenter European study. Eur. Arch. Otorhinolaryngol. 2020, 277, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Bunyavanich, S.; Do, A.; Vicencio, A. Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. Jama 2020. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Barbry, P.; Brazma, A.; Desai, T.; Duong, T.E.; Eickelberg, O.; Haniffa, M.; et al. SARS-CoV-2 entry genes are most highly expressed in nasal goblet and ciliated cells within human airways. arXiv 2020, arXiv:2003.06122. [Google Scholar]
- Hui, D.S.C.; Zumla, A. Severe acute respiratory syndrome: Historical, epidemiologic, and clinical features. Infect. Dis. Clin. North. Am. 2019, 33, 869–889. [Google Scholar] [CrossRef]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Pan, Y.; Zhao, Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharm. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol. Metab. 2004, 15, 166–169. [Google Scholar] [CrossRef]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China. Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Delanghe, J.R.; Speeckaert, M.M.; De Buyzere, M.L. The host’s angiotensin-converting enzyme polymorphism may explain epidemiological findings in COVID-19 infections. Clin. Chim. Acta 2020, 505, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Penninger, J.M. Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 2006, 6, 271–276. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.; Amanullah, A.; Aziz, B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Elrashdy, F.; Redwan, E.M.; Uversky, V.N. Intrinsic disorder perspective of an interplay between the renin-angiotensin-aldosterone system and SARS-CoV-2. Infect. Genet. Evol. 2020, 85, 104510. [Google Scholar] [CrossRef]
- Shen, M.; Liu, C.; Xu, R.; Ruan, Z.; Zhao, S.; Zhang, H.; Wang, W.; Huang, X.; Yang, L.; Tang, Y.; et al. SARS-CoV-2 infection of cats and dogs? Preprints. 2020. 2020040116.
- Zhai, X.; Sun, J.; Yan, Z.; Zhang, J.; Zhao, J.; Zhao, Z.; Gao, Q.; He, W.T.; Veit, M.; Su, S. Comparison of SARS-CoV-2 spike protein binding to ACE2 receptors from human, pets, farm animals, and putative intermediate hosts. J. Virol. 2020. [Google Scholar] [CrossRef]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 2020. [Google Scholar] [CrossRef]
- Patterson, E.I.; Elia, G.; Grassi, A.; Giordano, A.; Desario, C.; Medardo, M.; Smith, S.L.; Anderson, E.R.; Prince, T.; Patterson, G.T.; et al. Evidence of exposure to SARS-CoV-2 in cats and dogs from households in Italy. bioRxiv 2020. [Google Scholar] [CrossRef]
- Abdel-Moneim, A.S.; Abdelwhab, E.M. Evidence for SARS-CoV-2 infection of animal hosts. Pathogens 2020, 9. [Google Scholar] [CrossRef]
- Hossain, M.G.; Javed, A.; Akter, S.; Saha, S. SARS-CoV-2 host diversity: An update of natural infections and experimental evidence. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.; Abad, D.; Eiros, J.M.; Rodriguez-Lazaro, D. Are animals a neglected transmission route of SARS-CoV-2? Pathogens 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Sit, T.H.C.; Brackman, C.J.; Ip, S.M.; Tam, K.W.S.; Law, P.Y.T.; To, E.M.W.; Yu, V.Y.T.; Sims, L.D.; Tsang, D.N.C.; Chu, D.K.W.; et al. Infection of dogs with SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Dhama, K.; Sharun, K.; Iqbal Yatoo, M.; Malik, Y.S.; Singh, R.; Michalak, I.; Sah, R.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. COVID-19: Animals, veterinary and zoonotic links. Vet. Q. 2020, 40, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Gollakner, R.; Capua, I. Is COVID-19 the first pandemic that evolves into a panzootic? Vet. Ital. 2020, 56, 7–8. [Google Scholar] [CrossRef]
- Mao, L.J.; Xu, J.; Xu, Z.H.; Xia, X.P.; Li, B.; He, J.G.; Zhao, P.; Pan, J.W.; Zhang, D.; Su, Y.; et al. A child with household transmitted COVID-19. BMC Infect. Dis. 2020, 20, 329. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Z.; Xia, S.; Shi, B.; Zhou, X.N.; Shi, Y.; Liu, J. What are the underlying transmission patterns of COVID-19 outbreak?—An age-specific social contact characterization. EClinicalMedicine 2020, 100354. [Google Scholar] [CrossRef]
- Uversky, V.N.; Elrashdy, F.; Aljadawi, A.; Redwan, E.M. Household pets and SARS-CoV2 transmissibility in the light of the ACE2 intrinsic disorder status. J. Biomol. Struct. Dyn. 2020, in press. [Google Scholar]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.; Castoe, T.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. bioRxiv 2020. [Google Scholar] [CrossRef]
- Karamitros, T.; Papadopoulou, G.; Bousali, M.; Mexias, A.; Tsiodras, S.; Mentis, A. SARS-CoV-2 exhibits intra-host genomic plasticity and low-frequency polymorphic quasispecies. bioRxiv 2020. 2020.2003.2027.009480. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Rueca, M.; Messina, F.; Giombini, E.; Carletti, F.; Colavita, F.; Castilletti, C.; Lalle, E.; Bordi, L.; Vairo, F.; et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020. [Google Scholar] [CrossRef]
- Altan-Bonnet, N.; Chen, Y.H. Intercellular transmission of viral populations with vesicles. J. Virol. 2015, 89, 12242–12244. [Google Scholar] [CrossRef]
- Borderia, A.V.; Isakov, O.; Moratorio, G.; Henningsson, R.; Aguera-Gonzalez, S.; Organtini, L.; Gnadig, N.F.; Blanc, H.; Alcover, A.; Hafenstein, S.; et al. Group selection and contribution of minority variants during virus adaptation determines virus fitness and phenotype. PLoS Pathog. 2015, 11, e1004838. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Baranowski, E.; Ruiz-Jarabo, C.M.; Martin-Hernandez, A.M.; Saiz, J.C.; Escarmis, C. Quasispecies structure and persistence of RNA viruses. Emerg. Infect. Dis. 1998, 4, 521–527. [Google Scholar] [CrossRef]
- Park, D.; Huh, H.J.; Kim, Y.; Son, D.-S.; Jeon, H.-J.; Im, E.-H.; Kim, J.-W.; Lee, N.; Kang, E.-S.; Kang, C.; et al. Analysis of intra-patient heterogeneity uncovers the microevolution of Middle East respiratory syndrome coronavirus. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001214. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Z.; Wang, F.S. SARS-associated coronavirus quasispecies in individual patients. N. Engl. J. Med. 2004, 350, 1366–1367. [Google Scholar] [CrossRef] [PubMed]
- Mahy, B.W.J. The evolution and emergence of RNA viruses. Emerg. Infect. Dis. 2010, 16, 899. [Google Scholar] [CrossRef]
- Kostaki, E.G.; Karamitros, T.; Bobkova, M.; Oikonomopoulou, M.; Magiorkinis, G.; Garcia, F.; Hatzakis, A.; Paraskevis, D. Spatiotemporal characteristics of the HIV-1 CRF02_AG/CRF63_02A1 epidemic in Russia and Central Asia. AIDS Res. Hum. Retrovir. 2018, 34, 415–420. [Google Scholar] [CrossRef]
- Baric, R.S.; Fu, K.; Schaad, M.C.; Stohlman, S.A. Establishing a genetic recombination map for murine coronavirus strain A59 complementation groups. Virology 1990, 177, 646–656. [Google Scholar] [CrossRef]
- Lai, M.M. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef]
- Hon, C.C.; Lam, T.Y.; Shi, Z.L.; Drummond, A.J.; Yip, C.W.; Zeng, F.; Lam, P.Y.; Leung, F.C. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 2008, 82, 1819–1826. [Google Scholar] [CrossRef]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Bisht, H.; Roberts, A.; Vogel, L.; Bukreyev, A.; Collins, P.L.; Murphy, B.R.; Subbarao, K.; Moss, B. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc. Natl. Acad. Sci. USA 2004, 101, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- Enjuanes, L.; Dediego, M.L.; Alvarez, E.; Deming, D.; Sheahan, T.; Baric, R. Vaccines to prevent severe acute respiratory syndrome coronavirus-induced disease. Virus Res. 2008, 133, 45–62. [Google Scholar] [CrossRef]
- Jiaming, L.; Yanfeng, Y.; Yao, D.; Yawei, H.; Linlin, B.; Baoying, H.; Jinghua, Y.; Gao, G.F.; Chuan, Q.; Wenjie, T. The recombinant N-terminal domain of spike proteins is a potential vaccine against Middle East respiratory syndrome coronavirus (MERS-CoV) infection. Vaccine 2017, 35, 10–18. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Escarmís, C.; Menéndez-Arias, L.; Perales, C.; Herrera, M.; Novella, I.S.; Holland, J.J. CHAPTER 4—Viral quasispecies: Dynamics, interactions, and pathogenesis ** dedicated to Manfred Eigen on the occasion of his 80th birthday, for the insights that his pioneer studies have represented for virology. In Origin and Evolution of Viruses, 2nd ed.; Domingo, E., Parrish, C.R., Holland, J.J., Eds.; Academic Press: London, UK, 2008; pp. 87–118. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Rezaei, N. Immune-epidemiological parameters of the novel coronavirus—A perspective. Expert Rev. Clin. Immunol. 2020, 1–6. [Google Scholar] [CrossRef]
- Koff, W.C.; Williams, M.A. Covid-19 and immunity in aging populations—A new research agenda. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington state. JAMA 2020. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, D.; Yang, P.; Poon, L.L.M.; Wang, Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 2020, 20, 411–412. [Google Scholar] [CrossRef]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, L.M.; Wan, L.; Xiang, T.X.; Le, A.; Liu, J.M.; Peiris, M.; Poon, L.L.M.; Zhang, W. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Pawelec, G.; Weng, N.P. Can an effective SARS-CoV-2 vaccine be developed for the older population? Immun. Ageing 2020, 17, 8. [Google Scholar] [CrossRef]
- Weng, N.P. Aging of the immune system: How much can the adaptive immune system adapt? Immunity 2006, 24, 495–499. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, J.K.; Shin, O.S. Aging and the immune system: The impact of immunosenescence on viral infection, immunity and vaccine immunogenicity. Immune Netw. 2019, 19, e37. [Google Scholar] [CrossRef]
- Miyashita, N.; Kawai, Y.; Akaike, H.; Ouchi, K.; Hayashi, T.; Kurihara, T.; Okimoto, N. Influence of age on the clinical differentiation of atypical pneumonia in adults. Respirology 2012, 17, 1073–1079. [Google Scholar] [CrossRef]
- Pawelec, G.; Akbar, A.; Caruso, C.; Solana, R.; Grubeck-Loebenstein, B.; Wikby, A. Human immunosenescence: Is it infectious? Immunol. Rev. 2005, 205, 257–268. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, S.R.; Choi, I.; Jung, H. Causes and mechanisms of hematopoietic stem cell aging. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Sauce, D.; Larsen, M.; Fastenackels, S.; Duperrier, A.; Keller, M.; Grubeck-Loebenstein, B.; Ferrand, C.; Debre, P.; Sidi, D.; Appay, V. Evidence of premature immune aging in patients thymectomized during early childhood. J. Clin. Invest. 2009, 119, 3070–3078. [Google Scholar] [CrossRef] [PubMed]
- Zlamy, M.; Almanzar, G.; Parson, W.; Schmidt, C.; Leierer, J.; Weinberger, B.; Jeller, V.; Unsinn, K.; Eyrich, M.; Wurzner, R.; et al. Efforts of the human immune system to maintain the peripheral CD8+ T cell compartment after childhood thymectomy. Immun. Ageing 2016, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 2014, 192, 2689–2698. [Google Scholar] [CrossRef] [PubMed]
- Sansoni, P.; Vescovini, R.; Fagnoni, F.; Biasini, C.; Zanni, F.; Zanlari, L.; Telera, A.; Lucchini, G.; Passeri, G.; Monti, D.; et al. The immune system in extreme longevity. Exp. Gerontol. 2008, 43, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Weinberger, B.; Grubeck-Loebenstein, B. The aging of the immune system. Transpl. Int. 2009, 22, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- LeMaoult, J.; Messaoudi, I.; Manavalan, J.S.; Potvin, H.; Nikolich-Zugich, D.; Dyall, R.; Szabo, P.; Weksler, M.E.; Nikolich-Zugich, J. Age-related dysregulation in CD8 T cell homeostasis: Kinetics of a diversity loss. J. Immunol. 2000, 165, 2367–2373. [Google Scholar] [CrossRef]
- Posnett, D.N.; Sinha, R.; Kabak, S.; Russo, C. Clonal populations of T cells in normal elderly humans: The T cell equivalent to “benign monoclonal gammapathy”. J. Exp. Med. 1994, 179, 609–618. [Google Scholar] [CrossRef]
- Callahan, J.E.; Kappler, J.W.; Marrack, P. Unexpected expansions of CD8-bearing cells in old mice. J. Immunol. 1993, 151, 6657–6669. [Google Scholar]
- Blackman, M.A.; Woodland, D.L. The narrowing of the CD8 T cell repertoire in old age. Curr. Opin. Immunol. 2011, 23, 537–542. [Google Scholar] [CrossRef]
- Yager, E.J.; Ahmed, M.; Lanzer, K.; Randall, T.D.; Woodland, D.L.; Blackman, M.A. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med. 2008, 205, 711–723. [Google Scholar] [CrossRef]
- Messaoudi, I.; Lemaoult, J.; Guevara-Patino, J.A.; Metzner, B.M.; Nikolich-Zugich, J. Age-related CD8 T cell clonal expansions constrict CD8 T cell repertoire and have the potential to impair immune defense. J. Exp. Med. 2004, 200, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Cambier, J.C. Ageing, autoimmunity and arthritis: Senescence of the B cell compartment—Implications for humoral immunity. Arthritis Res. Ther. 2004, 6, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Allman, D.; Miller, J.P. B cell development and receptor diversity during aging. Curr. Opin. Immunol. 2005, 17, 463–467. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp. Gerontol. 2017, 87, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Diaz, A.; Romero, M.; Phillips, M.; Mendez, N.V.; Landin, A.M.; Blomberg, B.B. Unique biomarkers for B-cell function predict the serum response to pandemic H1N1 influenza vaccine. Int. Immunol. 2012, 24, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamazaki, T.; Okubo, Y.; Uehara, Y.; Sugane, K.; Agematsu, K. Regulation of aged humoral immune defense against pneumococcal bacteria by IgM memory B cell. J. Immunol. 2005, 175, 3262–3267. [Google Scholar] [CrossRef]
- Khurana, S.; Frasca, D.; Blomberg, B.; Golding, H. AID activity in B cells strongly correlates with polyclonal antibody affinity maturation in-vivo following pandemic 2009-H1N1 vaccination in humans. PLoS Pathog. 2012, 8, e1002920. [Google Scholar] [CrossRef]
- Frasca, D.; Landin, A.M.; Alvarez, J.P.; Blackshear, P.J.; Riley, R.L.; Blomberg, B.B. Tristetraprolin, a negative regulator of mRNA stability, is increased in old B cells and is involved in the degradation of E47 mRNA. J. Immunol. 2007, 179, 918–927. [Google Scholar] [CrossRef]
- Muramatsu, M.; Nagaoka, H.; Shinkura, R.; Begum, N.A.; Honjo, T. Discovery of activation-induced cytidine deaminase, the engraver of antibody memory. Adv. Immunol. 2007, 94, 1–36. [Google Scholar] [CrossRef]
- Nikolich-Zugich, J.; Li, G.; Uhrlaub, J.L.; Renkema, K.R.; Smithey, M.J. Age-related changes in CD8 T cell homeostasis and immunity to infection. Semin. Immunol. 2012, 24, 356–364. [Google Scholar] [CrossRef]
- Gorina, Y.; Kelly, T.; Lubitz, J.; Hines, Z. Trends in influenza and pneumonia among older persons in the United States. Aging Trends 2008, 8, 1–11. [Google Scholar]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Greene, M.A.; Loeser, R.F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1966–1971. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.M.; Marsland, B.J. Lung homeostasis: Influence of age, microbes, and the immune system. Immunity 2017, 46, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Boe, D.M.; Boule, L.A.; Kovacs, E.J. Innate immune responses in the ageing lung. Clin. Exp. Immunol. 2017, 187, 16–25. [Google Scholar] [CrossRef]
- Bahadoran, A.; Lee, S.H.; Wang, S.M.; Manikam, R.; Rajarajeswaran, J.; Raju, C.S.; Sekaran, S.D. Immune responses to influenza virus and its correlation to age and inherited factors. Front. Microbiol. 2016, 7, 1841. [Google Scholar] [CrossRef]
- Krone, C.L.; van de Groep, K.; Trzcinski, K.; Sanders, E.A.; Bogaert, D. Immunosenescence and pneumococcal disease: An imbalance in host-pathogen interactions. Lancet Respir. Med. 2014, 2, 141–153. [Google Scholar] [CrossRef]
- Leung, G.M.; Hedley, A.J.; Ho, L.M.; Chau, P.; Wong, I.O.; Thach, T.Q.; Ghani, A.C.; Donnelly, C.A.; Fraser, C.; Riley, S.; et al. The epidemiology of severe acute respiratory syndrome in the 2003 Hong Kong epidemic: An analysis of all 1755 patients. Ann. Intern. Med. 2004, 141, 662–673. [Google Scholar] [CrossRef]
- Hakim, F.T.; Gress, R.E. Immunosenescence: Deficits in adaptive immunity in the elderly. Tissue Antigens 2007, 70, 179–189. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Monie, T.P. The canonical inflammasome: A macromolecular complex driving inflammation. Subcell Biochem. 2017, 83, 43–73. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Cho, S.J.; Plataki, M.; Mitzel, D.; Lowry, G.; Rooney, K.; Stout-Delgado, H. Decreased NLRP3 inflammasome expression in aged lung may contribute to increased susceptibility to secondary Streptococcus pneumoniae infection. Exp. Gerontol. 2018, 105, 40–46. [Google Scholar] [CrossRef]
- Hoegen, T.; Tremel, N.; Klein, M.; Angele, B.; Wagner, H.; Kirschning, C.; Pfister, H.W.; Fontana, A.; Hammerschmidt, S.; Koedel, U. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J. Immunol. 2011, 187, 5440–5451. [Google Scholar] [CrossRef]
- Deftereos, S.G.; Siasos, G.; Giannopoulos, G.; Vrachatis, D.A.; Angelidis, C.; Giotaki, S.G.; Gargalianos, P.; Giamarellou, H.; Gogos, C.; Daikos, G.; et al. The Greek study in the effects of colchicine in COvid-19 complications prevention (GRECCO-19 study): Rationale and study design. Hell. J. Cardiol. 2020. [Google Scholar] [CrossRef]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, N.; Cho, Y.; Lockey, R.F.; Kolliputi, N. The role of the NLRP3 inflammasome in pulmonary diseases. Ther. Adv. Respir. Dis. 2015, 9, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Farag, N.S.; Breitinger, U.; Breitinger, H.G.; El Azizi, M.A. Viroporins and inflammasomes: A key to understand virus-induced inflammation. Int. J. Biochem. Cell Biol. 2020, 122, 105738. [Google Scholar] [CrossRef]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Danthi, P. Viruses and the diversity of cell death. Annu. Rev. Virol. 2016, 3, 533–553. [Google Scholar] [CrossRef]
- Chen, C.Y.; Ping, Y.H.; Lee, H.C.; Chen, K.H.; Lee, Y.M.; Chan, Y.J.; Lien, T.C.; Jap, T.S.; Lin, C.H.; Kao, L.S.; et al. Open reading frame 8a of the human severe acute respiratory syndrome coronavirus not only promotes viral replication but also induces apoptosis. J. Infect. Dis. 2007, 196, 405–415. [Google Scholar] [CrossRef]
- Yue, Y.; Nabar, N.R.; Shi, C.S.; Kamenyeva, O.; Xiao, X.; Hwang, I.Y.; Wang, M.; Kehrl, J.H. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018, 9, 904. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome—A key player in antiviral responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef]
- Wei, L.; Sun, S.; Xu, C.H.; Zhang, J.; Xu, Y.; Zhu, H.; Peh, S.C.; Korteweg, C.; McNutt, M.A.; Gu, J. Pathology of the thyroid in severe acute respiratory syndrome. Hum. Pathol. 2007, 38, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.L.; Ding, Y.Q.; He, L.; Wang, W.; Zhang, J.H.; Wang, H.J.; Cai, J.J.; Geng, J.; Lu, Y.D.; Luo, Y.L. Detection of cell apoptosis in the pathological tissues of patients with SARS and its significance. Di Yi Jun Yi Da Xue Xue Bao 2003, 23, 770–773. [Google Scholar] [PubMed]
- Bordi, L.; Castilletti, C.; Falasca, L.; Ciccosanti, F.; Calcaterra, S.; Rozera, G.; Di Caro, A.; Zaniratti, S.; Rinaldi, A.; Ippolito, G.; et al. Bcl-2 inhibits the caspase-dependent apoptosis induced by SARS-CoV without affecting virus replication kinetics. Arch. Virol. 2006, 151, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Yang, R.; Guo, L.; Qu, J.; Wang, J.; Hung, T. Apoptosis induced by the SARS-associated coronavirus in vero cells is replication-dependent and involves caspase. DNA Cell Biol. 2005, 24, 496–502. [Google Scholar] [CrossRef]
- Krahling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Muhlberger, E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J. Virol. 2009, 83, 2298–2309. [Google Scholar] [CrossRef]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human coronaviruses: A review of virus-host interactions. Diseases 2016, 4. [Google Scholar] [CrossRef]
- Chu, H.; Zhou, J.; Wong, B.H.; Li, C.; Chan, J.F.; Cheng, Z.S.; Yang, D.; Wang, D.; Lee, A.C.; Li, C.; et al. Middle East respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J. Infect. Dis. 2016, 213, 904–914. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, J.; Teng, Y.; Sun, H.; Tian, G.; He, L.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; et al. Complement receptor C5aR1 inhibition reduces pyroptosis in hDPP4-transgenic mice infected with MERS-CoV. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Vaz Fragoso, C.A. Epidemiology of lung disease in older persons. Clin. Geriatr. Med. 2017, 33, 491–501. [Google Scholar] [CrossRef]
- Dyer, C. The interaction of ageing and lung disease. Chron. Respir. Dis. 2012, 9, 63–67. [Google Scholar] [CrossRef]
- Akgun, K.M.; Crothers, K.; Pisani, M. Epidemiology and management of common pulmonary diseases in older persons. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2012, 67, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Olloquequi, J. COVID-19 Susceptibility in chronic obstructive pulmonary disease. Eur. J. Clin. Invest. 2020. [Google Scholar] [CrossRef] [PubMed]
- Notter, R.H. Lung Surfactants: Basic Science and Clinical Applications; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Reynolds, H.Y. Lung inflammation: Normal host defense or a complication of some diseases? Annu. Rev. Med. 1987, 38, 295–323. [Google Scholar] [CrossRef]
- Fagiolo, U.; Cossarizza, A.; Santacaterina, S.; Ortolani, C.; Monti, D.; Paganelli, R.; Franceschi, C. Increased cytokine production by peripheral blood mononuclear cells from healthy elderly people. Ann. N. Y. Acad. Sci. 1992, 663, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Moliva, J.I.; Rajaram, M.V.; Sidiki, S.; Sasindran, S.J.; Guirado, E.; Pan, X.J.; Wang, S.H.; Ross, P., Jr.; Lafuse, W.P.; Schlesinger, L.S.; et al. Molecular composition of the alveolar lining fluid in the aging lung. Age 2014, 36, 9633. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.S.; Siddiqi, T.J.; Khan, M.S.; Patel, U.K.; Shahid, I.; Ahmed, J.; Kalra, A.; Michos, E.D. Is there a smoker’s paradox in COVID-19? BMJ Evid. Based Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Chen, Q.; Xie, M. Smoking increases the risk of infectious diseases: A narrative review. Tob. Induc. Dis. 2020, 18, 60. [Google Scholar] [CrossRef]
- Kaur, G.; Lungarella, G.; Rahman, I. SARS-CoV-2 COVID-19 susceptibility and lung inflammatory storm by smoking and vaping. J. Inflamm. 2020, 17, 21. [Google Scholar] [CrossRef]
- Liu, W.; Tao, Z.W.; Wang, L.; Yuan, M.L.; Liu, K.; Zhou, L.; Wei, S.; Deng, Y.; Liu, J.; Liu, H.G.; et al. Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chin. Med. J. 2020, 133, 1032–1038. [Google Scholar] [CrossRef]
- Lippi, G.; Henry, B.M. Active smoking is not associated with severity of coronavirus disease 2019 (COVID-19). Eur. J. Intern. Med. 2020, 75, 107–108. [Google Scholar] [CrossRef]
- Chakladar, J.; Shende, N.; Li, W.T.; Rajasekaran, M.; Chang, E.Y.; Ongkeko, W.M. Smoking-mediated upregulation of the androgen pathway leads to increased SARS-CoV-2 susceptibility. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Wambier, C.G.; Goren, A. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is likely to be androgen mediated. J. Am. Acad. Derm. 2020, 83, 308–309. [Google Scholar] [CrossRef]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef]
- Penna, C.; Mercurio, V.; Tocchetti, C.G.; Pagliaro, P. Sex-related differences in COVID-19 lethality. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Li, L.; Samplaski, M. Why does COVID-19 kill more elderly men than women? Is there a role for testosterone? Andrology 2020. [Google Scholar] [CrossRef]
- Li, Y.; Jerkic, M.; Slutsky, A.S.; Zhang, H. Molecular mechanisms of sex bias differences in COVID-19 mortality. Crit. Care 2020, 24, 405. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.G.; Zhong, X.; Liaw, B.; Tremblay, D.; Tsao, C.K.; Galsky, M.D.; Oh, W.K. Does androgen deprivation therapy protect against severe complications from COVID-19? Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Caffo, O.; Zagonel, V.; Baldessari, C.; Berruti, A.; Bortolus, R.; Buti, S.; Ceresoli, G.L.; Donini, M.; Ermacora, P.; Fornarini, G.; et al. On the relationship between androgen-deprivation therapy for prostate cancer and risk of infection by SARS-CoV-2. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Montopoli, M.; Zumerle, S.; Vettor, R.; Rugge, M.; Zorzi, M.; Catapano, C.V.; Carbone, G.M.; Cavalli, A.; Pagano, F.; Ragazzi, E.; et al. Androgen-deprivation therapies for prostate cancer and risk of infection by SARS-CoV-2: A population-based study (N = 4532). Ann. Oncol. 2020, 31, 1040–1045. [Google Scholar] [CrossRef]
- Schafer, A.; Baric, R.S. Epigenetic landscape during coronavirus infection. Pathogens 2017, 6. [Google Scholar] [CrossRef]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.J.; Knipe, D.M. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Jeffrey, K.L. Beyond receptors and signaling: Epigenetic factors in the regulation of innate immunity. Immunol. Cell Biol. 2015, 93, 233–244. [Google Scholar] [CrossRef]
- Gomez-Diaz, E.; Jorda, M.; Peinado, M.A.; Rivero, A. Epigenetics of host-pathogen interactions: The road ahead and the road behind. PLoS Pathog. 2012, 8, e1003007. [Google Scholar] [CrossRef] [PubMed]
- Marazzi, I.; Ho, J.S.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Menachery, V.D.; Eisfeld, A.J.; Schafer, A.; Josset, L.; Sims, A.C.; Proll, S.; Fan, S.; Li, C.; Neumann, G.; Tilton, S.C.; et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 2014, 5, e01114–e01174. [Google Scholar] [CrossRef]
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 2020, 215, 108410. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elrashdy, F.; Redwan, E.M.; Uversky, V.N. Why COVID-19 Transmission Is More Efficient and Aggressive Than Viral Transmission in Previous Coronavirus Epidemics? Biomolecules 2020, 10, 1312. https://doi.org/10.3390/biom10091312
Elrashdy F, Redwan EM, Uversky VN. Why COVID-19 Transmission Is More Efficient and Aggressive Than Viral Transmission in Previous Coronavirus Epidemics? Biomolecules. 2020; 10(9):1312. https://doi.org/10.3390/biom10091312
Chicago/Turabian StyleElrashdy, Fatma, Elrashdy M. Redwan, and Vladimir N. Uversky. 2020. "Why COVID-19 Transmission Is More Efficient and Aggressive Than Viral Transmission in Previous Coronavirus Epidemics?" Biomolecules 10, no. 9: 1312. https://doi.org/10.3390/biom10091312
APA StyleElrashdy, F., Redwan, E. M., & Uversky, V. N. (2020). Why COVID-19 Transmission Is More Efficient and Aggressive Than Viral Transmission in Previous Coronavirus Epidemics? Biomolecules, 10(9), 1312. https://doi.org/10.3390/biom10091312