Bispecific Targeting of EGFR and Urokinase Receptor (uPAR) Using Ligand-Targeted Toxins in Solid Tumors



Abstract
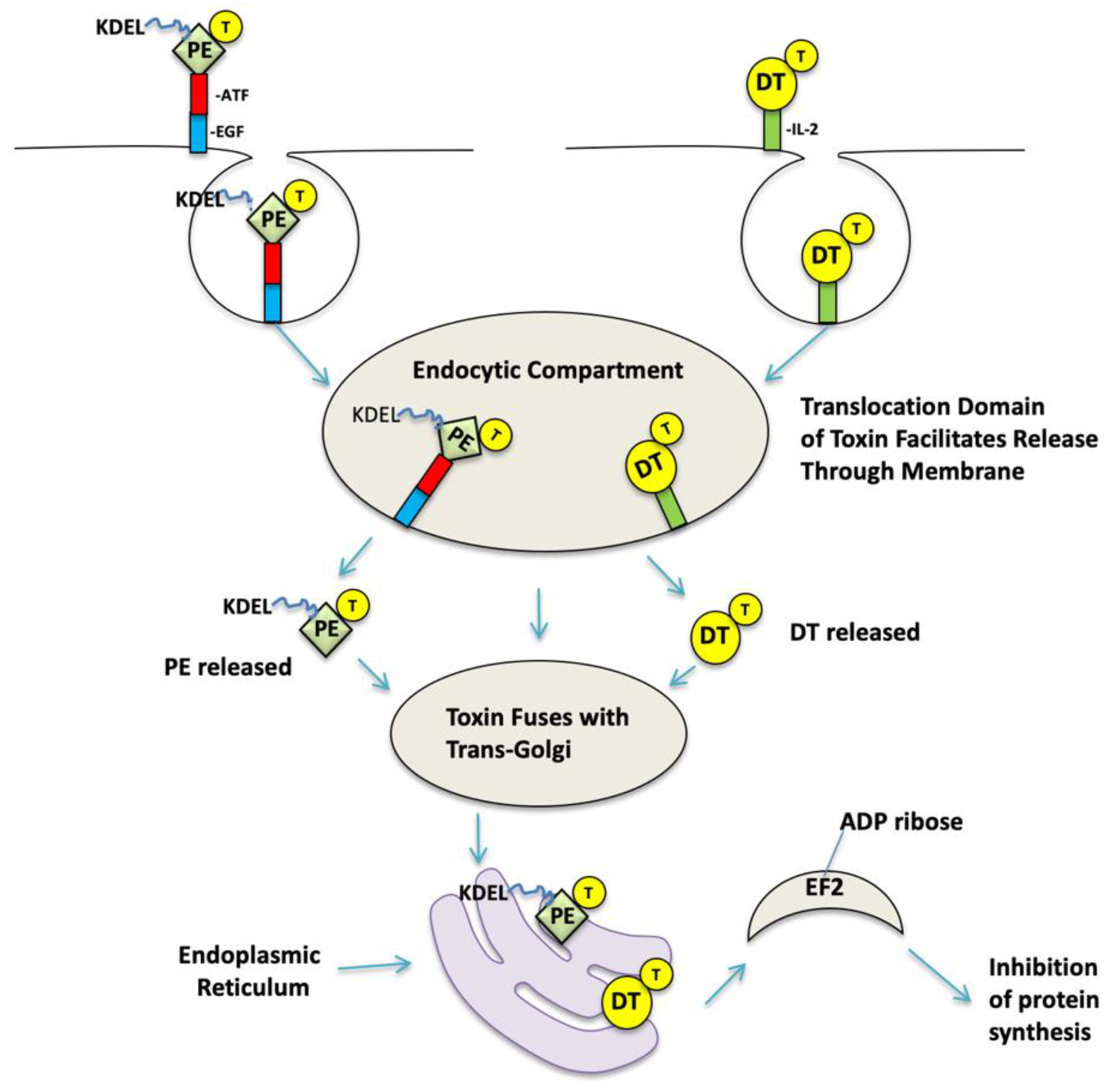
1. Targeted Toxins Have Made a Mark on Cancer Therapy
2. Bispecificity Can Be Advantageous over Monospecific Therapy
3. EGFR, A Prominent Target in Cancer Therapy
4. In Addition to Targeting Cancer Cells, Targeted Toxins Can Target the Tumor Microenvironment
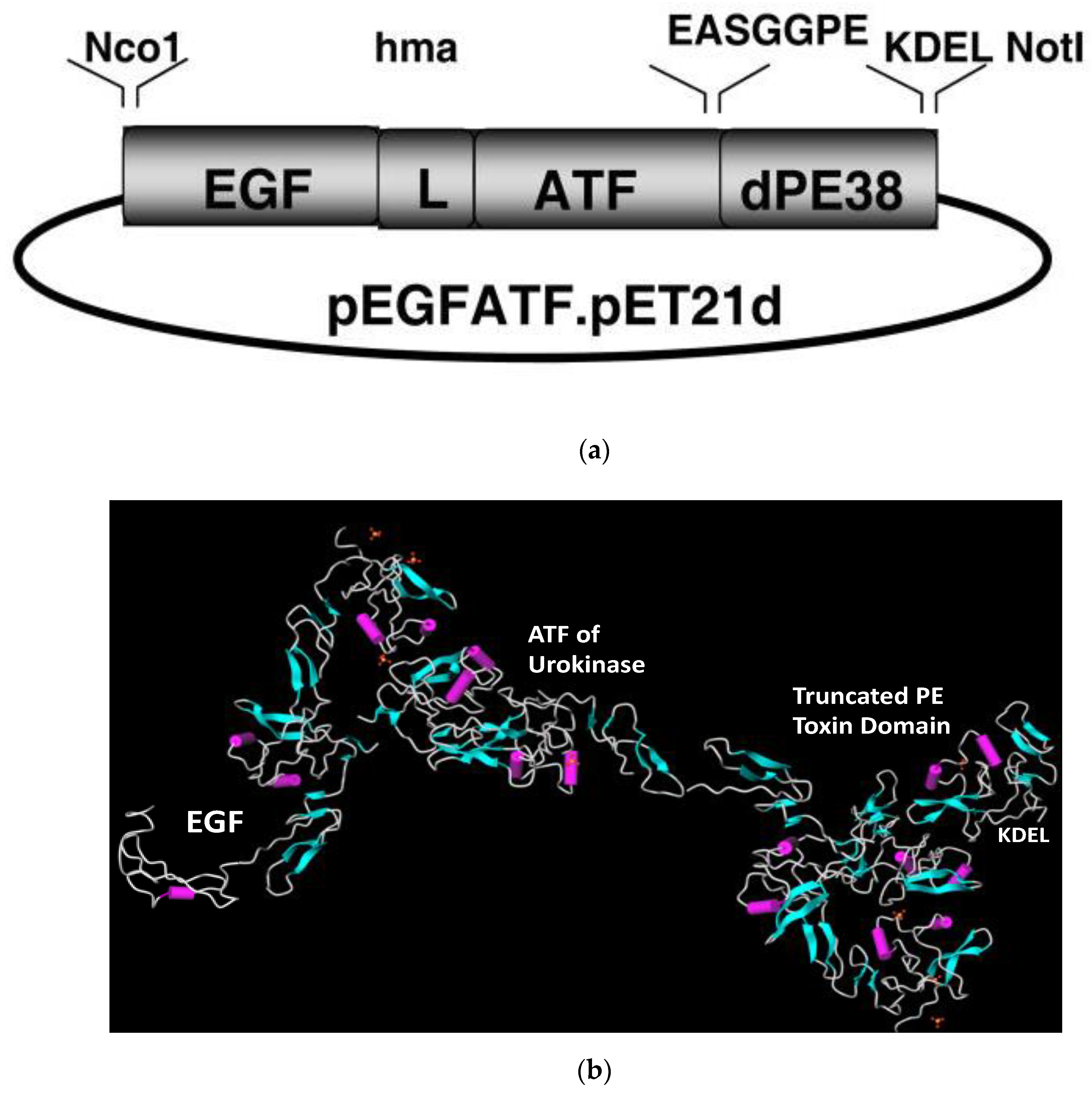
5. eBAT, a Bispecific LTT
6. eBAT Studies Show Promise for Glioma and Carcinoma Therapy
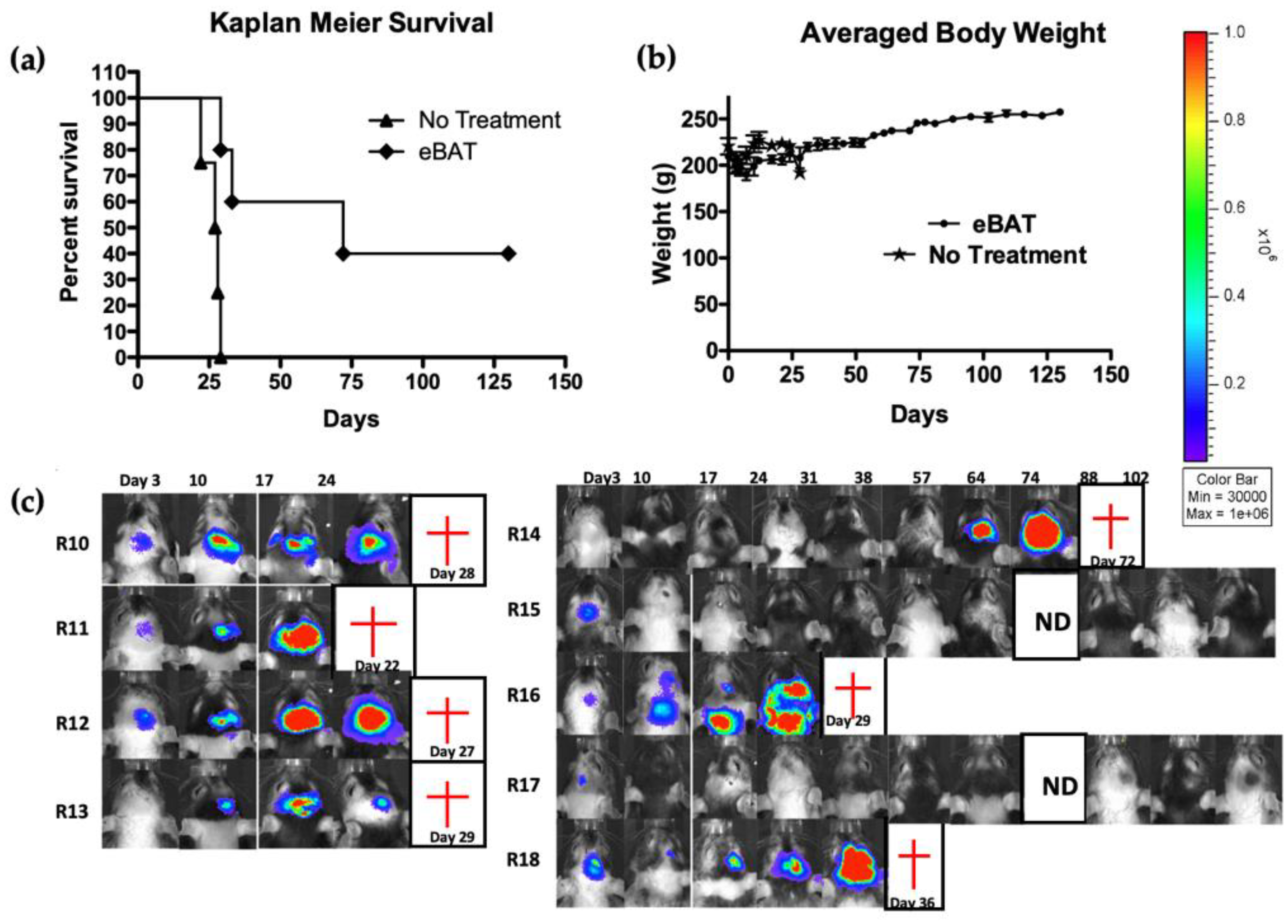
7. eBAT Shows Efficacy in Sarcoma Xenograft Models and Cancer Stem Cells
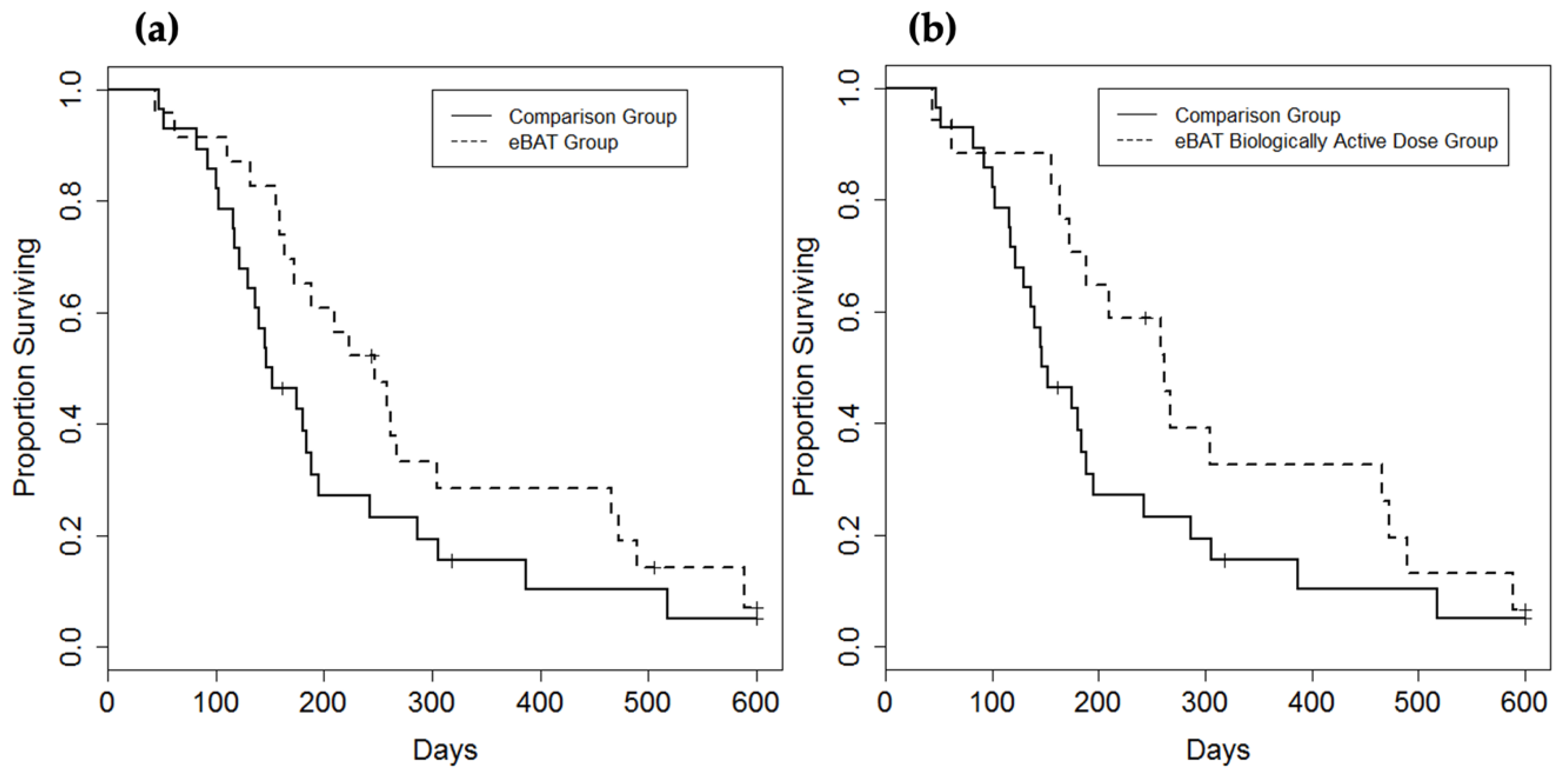
8. eBAT Is Effective in Canine Companion Clinical Trials
9. Bispecific Targeting Appears to Reduce eBAT Toxicity, a Major Drug Advantage
10. Discussion
11. The Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015. [Google Scholar] [CrossRef]
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Accumulation of a recombinant immunotoxin in a tumor in vivo: Fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998, 58, 968–975. [Google Scholar]
- Oh, S.; Stish, B.J.; Sachdev, D.; Chen, H.; Dudek, A.Z.; Vallera, D.A. A novel reduced immunogenicity bispecific targeted toxin simultaneously recognizing human epidermal growth factor and interleukin-4 receptors in a mouse model of metastatic breast carcinoma. Clin. Cancer Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Wayne, A.S.; FitzGerald, D.J.; Kreitman, R.J.; Pastan, I. Immunotoxins for leukemia. Blood 2014, 123, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Nagata, S.; FitzGerald, D.J.; Beers, R.; Fisher, R.J.; Vincent, J.J.; Lee, B.; Nakamura, M.; Hwang, J.; Kreitman, R.J.; et al. Characterization of the B Cell Epitopes Associated with a Truncated Form of Pseudomonas Exotoxin (PE38) Used to Make Immunotoxins for the Treatment of Cancer Patients. J. Immunol. 2006, 177, 8822–8834. [Google Scholar] [CrossRef] [PubMed]
- Turturro, F. Denileukin diftitox: A biotherapeutic paradigm shift in the treatment of lymphoid-derived disorders. Expert Rev. Anticancer Ther. 2007, 7, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Alkharabsheh, O.; Frankel, A.E. Clinical activity and tolerability of SL-401 (Tagraxofusp): Recombinant diphtheria toxin and interleukin-3 in hematologic malignancies. Biomedicines 2019, 7, E6. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Tallman, M.S.; Robak, T.; Coutre, S.; Wilson, W.H.; Stetler-Stevenson, M.; FitzGerald, D.J.; Lechleider, R.; Pastan, I. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Karlin, L.; Robak, T.; Gladstone, D.E.; le Coutre, P.; Dietrich, S.; Gotic, M.; et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 2018, 32, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; Song, J.; Stiller, T.; Nguyen, T.; Palmer, J.; O’Donnell, M.; Stein, A.S.; Marcucci, G.; Forman, S.; Pullarkat, V. Correlates of resistance and relapse during blinatumomab therapy for relapsed/refractory acute lymphoblastic leukemia. Am. J. Hematol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Todhunter, D.A.; Kuroki, D.W.; Shu, Y.; Sicheneder, A.; Chen, H. A bispecific recombinant immunotoxin, DT2219, targeting human CD19 and CD22 receptors in a mouse xenograft model of B-cell leukemia/lymphoma. Clin. Cancer Res. 2005, 11, 3879–3888. [Google Scholar] [CrossRef]
- Bachanova, V.; Cao, Q.; Weisdorf, D.J.; Curtsinger, J.M.; Cooley, S.A.; Miller, J.; Vallera, D. Bispecific ligand-directed toxin targeting CD22 and CD19 (DT2219) for refractory B-cell malignancies: Results of phase I-II trial. J. Clin. Oncol. 2019, 37, e19066. [Google Scholar] [CrossRef]
- Bachanova, V.; Frankel, A.E.; Cao, Q.; Lewis, D.; Grzywacz, B.; Verneris, M.R.; Ustun, C.; Lazaryan, A.; McClune, B.; Warlick, E.D.; et al. Phase i study of a bispecific ligand-directed toxin targeting CD22 and CD19 (DT2219) for refractory B-cell malignancies. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef]
- Vallera, D.A.; Chen, H.; Sicheneder, A.R.; Panoskaltsis-Mortari, A.; Taras, E.P. Genetic alteration of a bispecific ligand-directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic B cell malignancy. Leuk. Res. 2009, 33, 1233–1242. [Google Scholar] [CrossRef]
- Schmidt, M.; Hynes, N.E.; Groner, B.; Wels, W. A bivalent single-chain antibody-toxin specific for ErbB-2 and the EGF receptor. Int. J. Cancer 1996. [Google Scholar] [CrossRef]
- Loew, S.; Schmidt, U.; Unterberg, A.; Halatsch, M.-E. The Epidermal Growth Factor Receptor as a Therapeutic Target in Glioblastoma Multiforme and other Malignant Neoplasms. Anticancer Agents Med. Chem. 2012. [Google Scholar] [CrossRef]
- Sunada, H.; Magun, B.E.; Mendelsohn, J.; MacLeod, C.L. Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation. Proc. Natl. Acad. Sci. USA 1986. [Google Scholar] [CrossRef]
- Galizia, G.; Lieto, E.; De Vita, F.; Orditura, M.; Castellano, P.; Troiani, T.; Imperatore, V.; Ciardiello, F. Cetuximab, a chimeric human mouse anti-epidermal growth factor receptor monoclonal antibody, in the treatment of human colorectal cancer. Oncogene 2007, 26, 3654–3660. [Google Scholar] [CrossRef]
- Yang, X.D.; Jia, X.C.; Corvalan, J.R.F.; Wang, P.; Davis, C.G.; Jakobovits, A. Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Res. 1999, 59, 1236–1243. [Google Scholar] [PubMed]
- Simon, N.; FitzGerald, D. Immunotoxin therapies for the treatment of epidermal growth factor receptor-dependent cancers. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Wels, W.; Beerli, R.; Hellmann, P.; Schmidt, M.; Marte, B.M.; Kornilova, E.S.; Hekele, A.; Mendelsohn, J.; Groner, B.; Hynes, N.E. EGF receptor and p185erbB-2-specific single-chain antibody toxins differ in their cell-killing activity on tumor cells expressing both receptor proteins. Int. J. Cancer 1995. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Harandi, A.; Zaidi, A.S.; Stocker, A.M.; Laber, D.A. Clinical efficacy and toxicity of anti-EGFR therapy in common cancers. J. Oncol. 2009, 2009. [Google Scholar] [CrossRef]
- Zhong, H.; Bowen, J.P. Molecular design and clinical development of VEGFR kinase inhibitors. Curr. Top. Med. Chem. 2007, 2, 161–168. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Olson, T.A.; Bautch, V.L.; Mohanraj, D. Vascular endothelial growth factor-toxin conjugate specifically inhibits KDR/flk-1-positive endothelial cell proliferation in vitro and angiogenesis in vivo. Cancer Res. 1996, 56, 1324–1330. [Google Scholar]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005. [Google Scholar] [CrossRef]
- Haigh, J.J. Role of VEGF in organogenesis. Organogenesis 2008, 4, 247–256. [Google Scholar] [CrossRef]
- Rajagopal, V.; Kreitman, R.J. Recombinant toxins that bind to the urokinase receptor are cytotoxic without requiring binding to the α2-macroglobulin receptor. J. Biol. Chem. 2000. [Google Scholar] [CrossRef]
- Di Mauro, C.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A. Targeting Urokinase-Type Plasminogen Activator Receptor on Human Glioblastoma Tumors With Diphtheria Toxin Fusion Protein DTAT. CancerSpectrum Knowl. Environ. 2002, 94, 597–606. [Google Scholar] [CrossRef]
- Waldron, N.N.; Oh, S.; Vallera, D.A. Bispecific targeting of EGFR and uPAR in a mouse model of head and neck squamous cell carcinoma. Oral Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Hall, W.A.; Vallera, D.A. Efficacy of antiangiogenic targeted toxins against glioblastoma multiforme. Neurosurg. Focus 2006. [Google Scholar] [CrossRef]
- Borgatti, A.; Koopmeiners, J.S.; Sarver, A.L.; Winter, A.L.; Stuebner, K.; Todhunter, D.; Rizzardi, A.E.; Henriksen, J.C.; Schmechel, S.; Forster, C.L.; et al. Safe and effective sarcoma therapy through bispecific targeting of EGFR and uPAR. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Chaudhary, V.K.; Jinno, Y.; FitzGerald, D.; Pastan, I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc. Natl. Acad. Sci. USA 1990. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem. J. 1995. [Google Scholar] [CrossRef]
- Tsai, A.K.; Oh, S.; Chen, H.; Shu, Y.; Ohlfest, J.R.; Vallera, D.A. A novel bispecific ligand-directed toxin designed to simultaneously target EGFR on human glioblastoma cells and uPAR on tumor neovasculature. J. Neurooncol. 2011. [Google Scholar] [CrossRef]
- Oh, S.; Ohlfest, J.R.; Todhunter, D.A.; Vallera, V.D.; Hall, W.A.; Chen, H.; Vallera, D.A. Intracranial elimination of human glioblastoma brain tumors in nude rats using the bispecific ligand-directed toxin, DTEGF13 and convection enhanced delivery. J. Neurooncol. 2009. [Google Scholar] [CrossRef]
- Oh, S.; Tsai, A.K.; Ohlfest, J.R.; Panoskaltsis-Mortari, A.; Vallera, D.A. Evaluation of a bispecific biological drug designed to simultaneously target glioblastoma and its neovasculature in the brain: Laboratory investigation. J. Neurosurg. 2011. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ayodele, O.; Abdul Razak, A.R. Immunotherapy in soft-tissue sarcoma. Curr. Oncol. 2019, 26, 17–23. [Google Scholar] [CrossRef]
- Pilbeam, K.; Wang, H.; Taras, E.; Bergerson, R.J.; Ettestad, B.; DeFor, T.; Borgatti, A.; Vallera, D.A.; Verneris, M.R. Targeting pediatric sarcoma with a bispecific ligand immunotoxin targeting urokinase and epidermal growth factor receptors. Oncotarget 2018, 9, 11938–11947. [Google Scholar] [CrossRef] [PubMed]
- Oh, F.; Todhunter, D.; Taras, E.; Vallera, D.A.; Borgatti, A. Targeting EGFR and uPAR on human rhabdomyosarcoma, osteosarcoma, and ovarian adenocarcinoma with a bispecific ligand-directed toxin. Clin. Pharmacol. Adv. Appl. 2018. [Google Scholar] [CrossRef]
- Sell, S. On the stem cell origin of cancer. Am. J. Pathol. 2010, 176, 2584–2594. [Google Scholar] [CrossRef]
- Schappa, J.T.; Frantz, A.M.; Gorden, B.H.; Dickerson, E.B.; Vallera, D.A.; Modiano, J.F. Hemangiosarcoma and its cancer stem cell subpopulation are effectively killed by a toxin targeted through epidermal growth factor and urokinase receptors. Int. J. Cancer 2013. [Google Scholar] [CrossRef]
- Khanna, C.; Lindblad-Toh, K.; Vail, D.; London, C.; Bergman, P.; Barber, L.; Breen, M.; Kitchell, B.; McNeil, E.; Modiano, J.F.; et al. The dog as a cancer model. Nat. Biotechnol. 2006, 24, 1065–1066. [Google Scholar] [CrossRef]
- Koopmeiners, J.S.; Modiano, J. A Bayesian adaptive Phase I-II clinical trial for evaluating efficacy and toxicity with delayed outcomes. Clin. Trials 2014. [Google Scholar] [CrossRef]
- Borgatti, A.; Fieberg, A.; Winter, A.L.; Stuebner, K.; Taras, E.; Todhunter, D.; Masyr, A.; Rendhal, A.; Vallera, D.A.; Koopmeiners, J.S.; et al. Impact of Repeated Cycles of EGF Bispecific Angiotoxin (eBAT) Administered at a Reduced Interval from Doxorubicin Chemotherapy in Dogs with Splenic Hemangiosarcoma. Vet. Comp. Oncol. 2020. [Google Scholar] [CrossRef]
- Smallshaw, J.E.; Ghetie, V.; Rizo, J.; Fulmer, J.R.; Trahan, L.L.; Ghetie, M.A.; Vitetta, E.S. Genetic engineering of an immunotoxin to eliminate pulmonary vascular leak in mice. Nat. Biotechnol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Pastan, I. Contextualizing the Use of Moxetumomab Pasudotox in the Treatment of Relapsed or Refractory Hairy Cell Leukemia. Oncologist 2020. [Google Scholar] [CrossRef]
- Patnaik, A.; Gordon, M.; Tsai, F.; Papadopoulous, K.; Rasco, D.; Beeram, S.M.; Fu, S.; Janku, F.; Hynes, S.M.; Gundala, S.R.; et al. A phase I study of LY3164530, a bispecific antibody targeting MET and EGFR, in patients with advanced or metastatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 407–418. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, C.F.; Huhalov, A.; Harms, B.D.; Adams, S.; Paragas, V.; Oyama, S.; Zhang, B.; Luus, L.; Overland, R.; Nguyen, S.; et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol. Cancer Ther. 2012, 11, 582–593. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advantages | -New molecule (first in class dual targeting tumor and its microenvironment) |
| -Targets tumor cells at picomolar concentrations | |
| -Applicable against a wide variety of solid tumor types | |
| -Exceptionally safe in vivo | |
| -Targets cancer stem cells (CSCs) and transit amplifying tumor cells | |
| -Potential targeting of neoangiogenic vasculature and immunosuppressive TAMs | |
| -Biologically effective dose does not need to reach maximum tolerated dose | |
| Disadvantages | -Modest liver toxicity from dog data-appears to be self-limiting and reversible |
| -Likely will need to be used in combination with other therapies | |
| -Still may generate some antibody response |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, F.; Modiano, J.F.; Bachanova, V.; Vallera, D.A. Bispecific Targeting of EGFR and Urokinase Receptor (uPAR) Using Ligand-Targeted Toxins in Solid Tumors. Biomolecules 2020, 10, 956. https://doi.org/10.3390/biom10060956
Oh F, Modiano JF, Bachanova V, Vallera DA. Bispecific Targeting of EGFR and Urokinase Receptor (uPAR) Using Ligand-Targeted Toxins in Solid Tumors. Biomolecules. 2020; 10(6):956. https://doi.org/10.3390/biom10060956
Chicago/Turabian StyleOh, Felix, Jaime F. Modiano, Veronika Bachanova, and Daniel A. Vallera. 2020. "Bispecific Targeting of EGFR and Urokinase Receptor (uPAR) Using Ligand-Targeted Toxins in Solid Tumors" Biomolecules 10, no. 6: 956. https://doi.org/10.3390/biom10060956
APA StyleOh, F., Modiano, J. F., Bachanova, V., & Vallera, D. A. (2020). Bispecific Targeting of EGFR and Urokinase Receptor (uPAR) Using Ligand-Targeted Toxins in Solid Tumors. Biomolecules, 10(6), 956. https://doi.org/10.3390/biom10060956