Brain NMDA Receptors in Schizophrenia and Depression


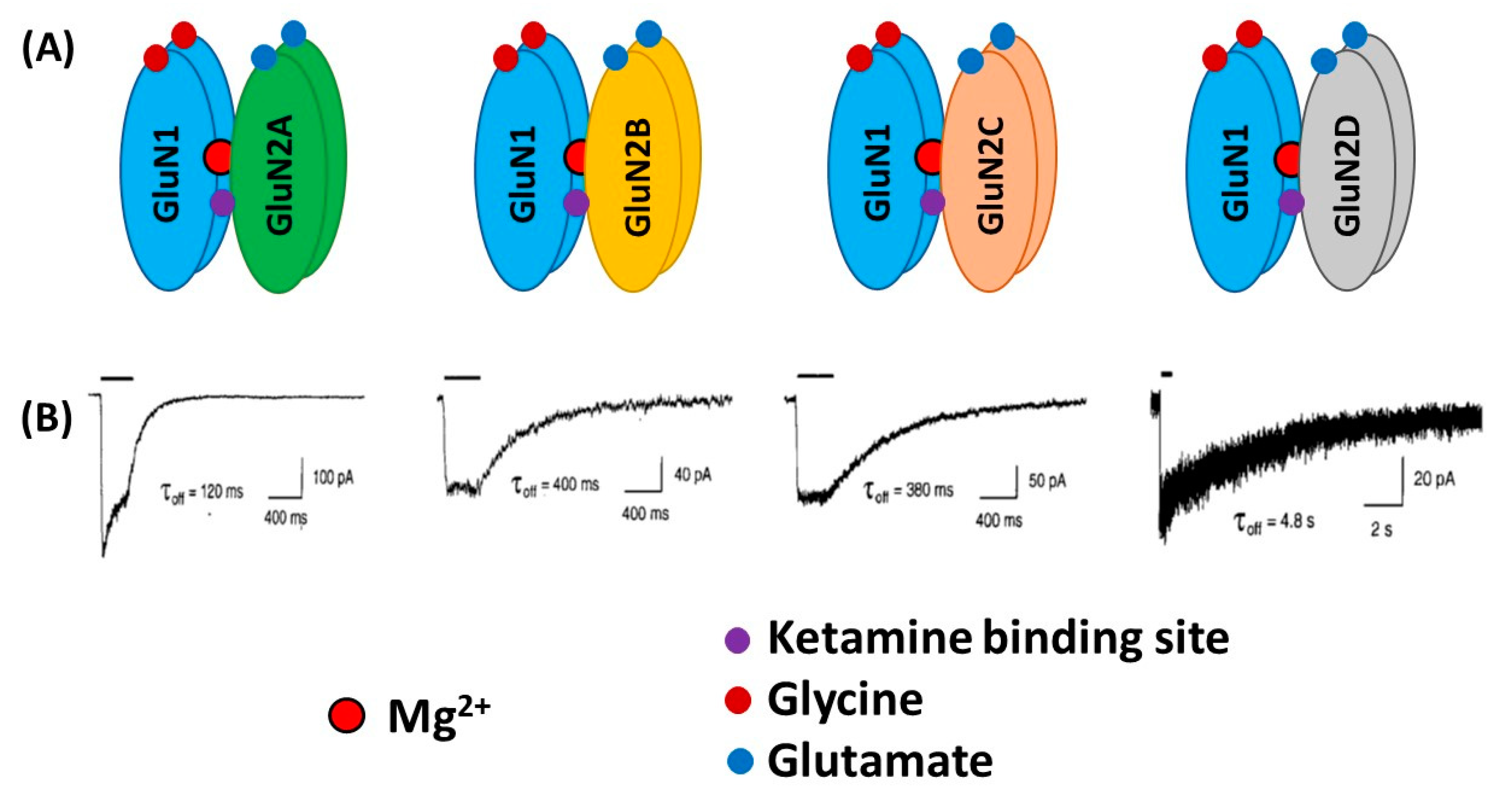{kind=link}
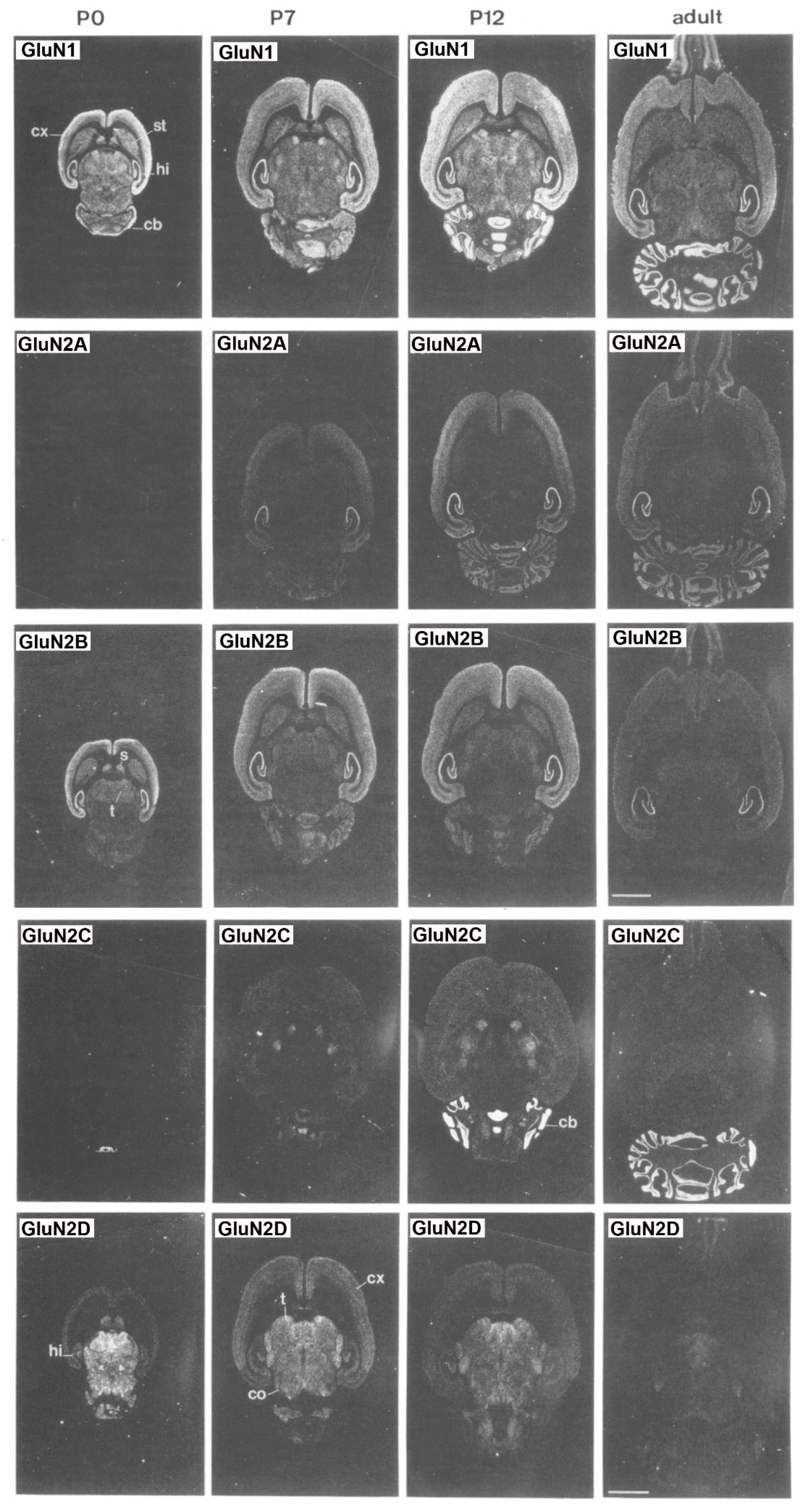{kind=link}
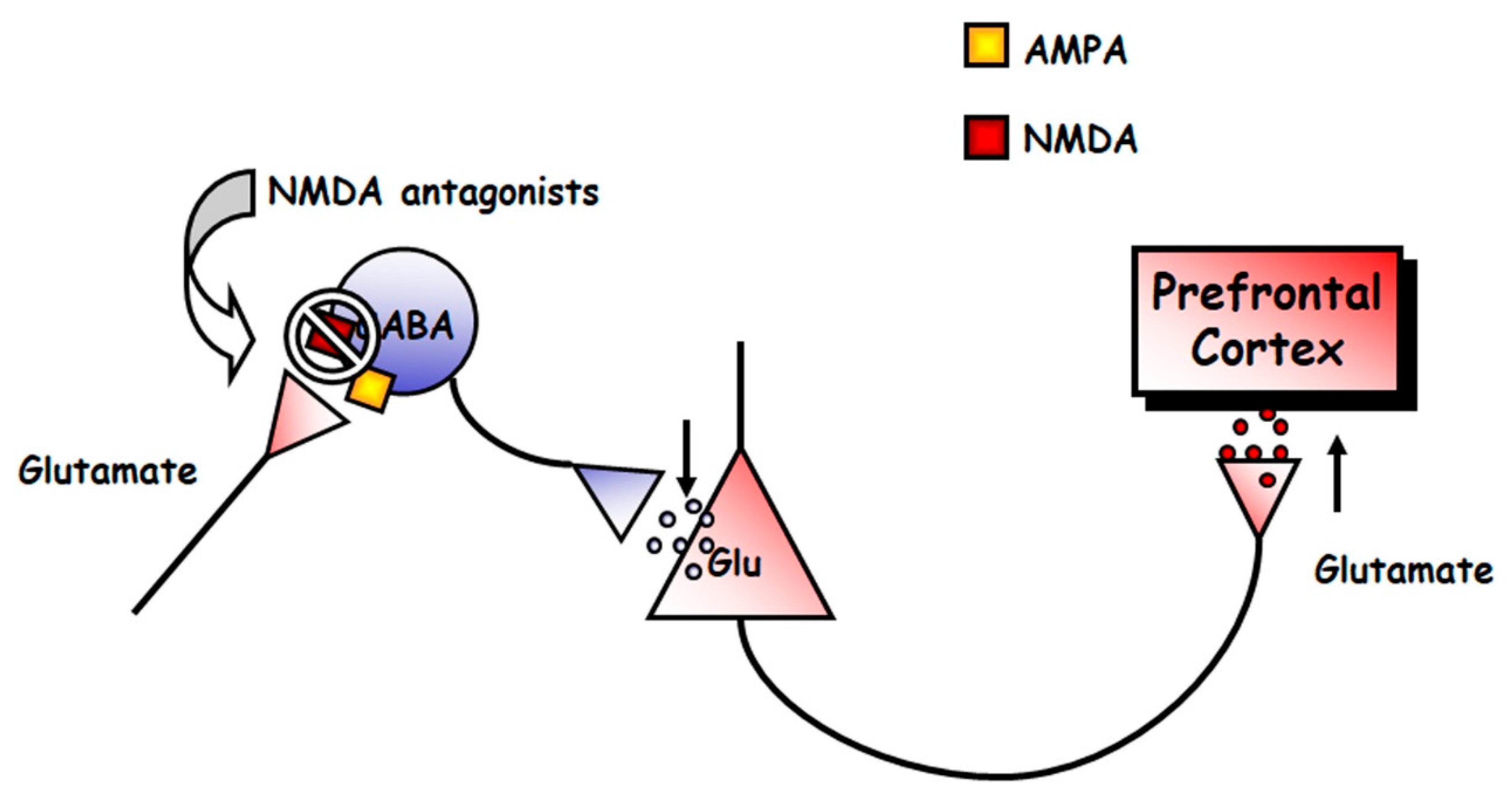{kind=link}
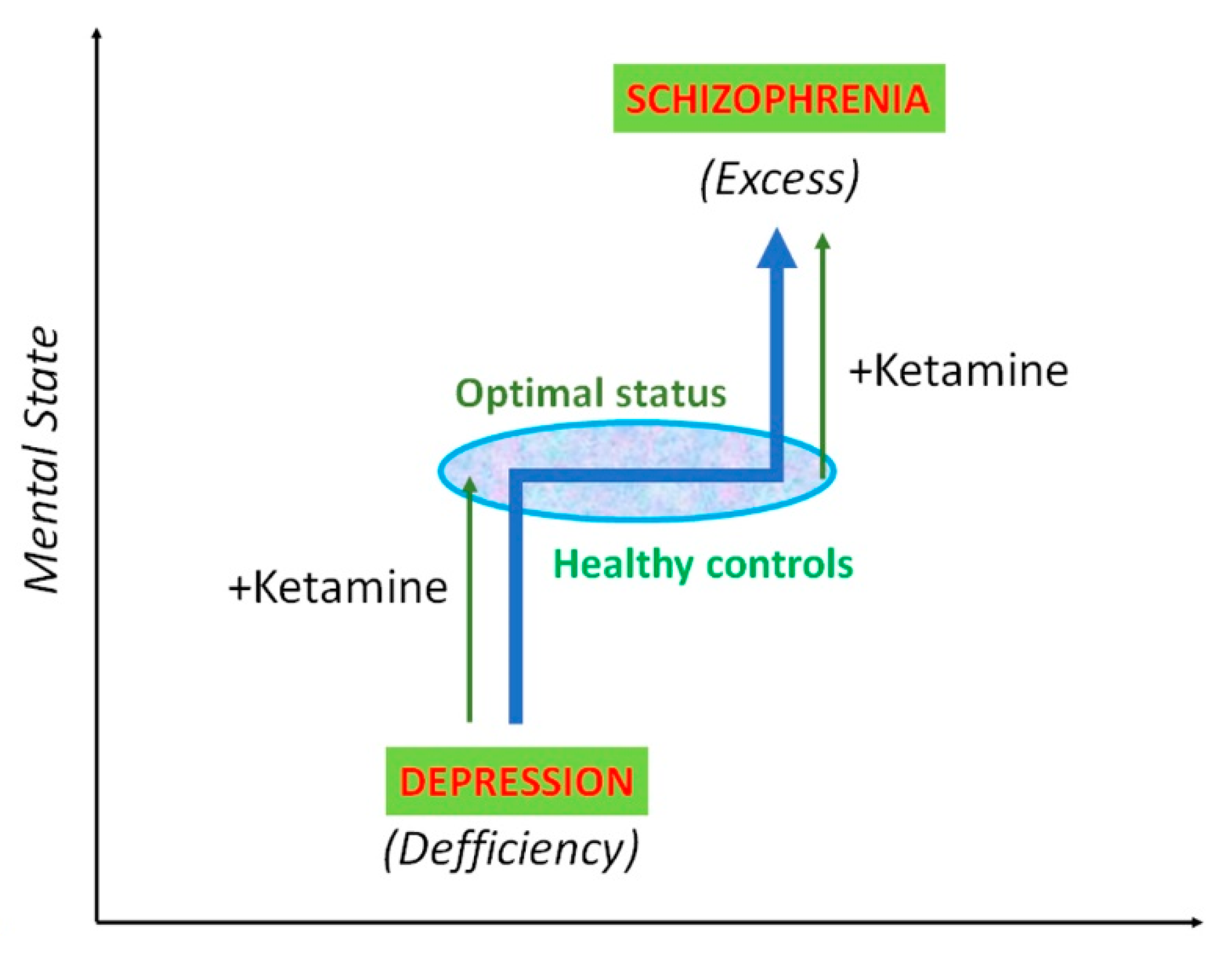{kind=link}
{kind=link}
Abstract
1. Introduction
2. NMDA Receptors in Schizophrenia
2.1. Clinical Evidence
2.1.1. Neurophysiology
2.1.2. Post-Mortem Studies
2.2. Preclinical Evidence
2.2.1. Neurophysiology
2.2.2. Animal Models
3. NMDA Receptors as Target for Treatment in Schizophrenia
4. NMDA Receptors in Depression
4.1. Clinical Evidence
Post-Mortem Studies
4.2. Preclinical Evidence
5. NMDA Receptors as Target for Treatment in Depression
6. Conclusions
Funding
Conflicts of Interest
References
- Pittenger, C.; Sanacora, G.; Krystal, J.H. The NMDA receptor as a therapeutic target in major depressive disorder. CNS Neurol. Disord. Drug Targets 2007, 6, 101–115. [Google Scholar] [CrossRef]
- Hall, J.; Trent, S.; Thomas, K.L.; O’Donovan, M.C.; Owen, M.J. Genetic risk for schizophrenia: Convergence on synaptic pathways involved in plasticity. Biol. Psychiatry 2015, 77, 52–58. [Google Scholar] [CrossRef]
- Pittenger, C.; Duman, R.S. Stress, depression, and neuroplasticity: A convergence of mechanisms. Neuropsychopharmacology 2008, 33, 88–109. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Kehl, S.J.; McLennan, H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 1983, 334, 33–46. [Google Scholar] [CrossRef]
- Malenka, R.C. Synaptic plasticity in the hippocampus: LTP and LTD. Cell 1994, 78, 535–538. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Ulbrich, M.H.; Isacoff, E.Y. Rules of engagement for NMDA receptor subunits. Proc. Natl. Acad. Sci. USA 2008, 105, 14163–14168. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Chergui, K. Dopamine induces a GluN2A-dependent form of long-term depression of NMDA synaptic responses in the nucleus accumbens. Neuropharmacology 2011, 60, 975–981. [Google Scholar] [CrossRef]
- Mullasseril, P.; Hansen, K.B.; Vance, K.M.; Ogden, K.K.; Yuan, H.; Kurtkaya, N.L.; Santangelo, R.; Orr, A.G.; Le, P.; Vellano, K.M.; et al. A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat. Commun. 2010, 1, 90. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, K.; Galante, M.; Angulo, M.C.; Audinat, E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J. Physiol. 2007, 580, 373–383. [Google Scholar] [CrossRef]
- Lozovaya, N.A.; Grebenyuk, S.E.; Tsintsadze, T.; Feng, B.; Monaghan, D.T.; Krishtal, O.A. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape ‘superslow’ afterburst EPSC in rat hippocampus. J. Physiol. 2004, 558, 451–463. [Google Scholar] [CrossRef]
- Thomas, C.G.; Miller, A.J.; Westbrook, G.L. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J. Neurophysiol. 2006, 95, 1727–1734. [Google Scholar] [CrossRef]
- Janssen, W.G.; Vissavajjhala, P.; Andrews, G.; Moran, T.; Hof, P.R.; Morrison, J.H. Cellular and synaptic distribution of NR2A and NR2B in macaque monkey and rat hippocampus as visualized with subunit-specific monoclonal antibodies. Exp. Neurol. 2005, 191, S28–S44. [Google Scholar] [CrossRef]
- Villmann, C.; Becker, C.M. On the hypes and falls in neuroprotection: Targeting the NMDA receptor. Neuroscientist 2007, 13, 594–615. [Google Scholar] [CrossRef]
- Kalia, L.V.; Kalia, S.K.; Salter, M.W. NMDA receptors in clinical neurology: Excitatory times ahead. Lancet Neurol. 2008, 7, 742–755. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.J.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.K.; Pinals, D.A.; Weingartner, H.; Sirocco, K.; Missar, C.D.; Pickar, D.; Breier, A. NMDA receptor function and human cognition: The effects of ketamine in healthy volunteers. Neuropsychopharmacology 1996, 14, 301–307. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Jevtovic-Todorovic, V.; Selke, G.; Melson, A.K.; Hershey, T.; Craft, S.; Olney, J.W. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology 1999, 20, 106–118. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Adler, C.M.; Kennison, S.D.; Elman, I.; Pickar, D.; Breier, A. Clozapine blunts N-methyl-D-aspartate antagonist-induced psychosis: A study with ketamine. Biol. Psychiatry 1997, 42, 664–668. [Google Scholar] [CrossRef]
- Lahti, A.C.; Weiler, M.A.; Michaelidis, T.; Parwani, A.; Tamminga, C.A. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 2001, 25, 455–467. [Google Scholar] [CrossRef]
- Stone, J.M.; Erlandsson, K.; Arstad, E.; Squassante, L.; Teneggi, V.; Bressan, R.A.; Krystal, J.H.; Ell, P.J.; Pilowsky, L.S. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: A [123I] CNS-1261 SPET study. Psychopharmacology (Berl) 2008, 197, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Pilowsky, L.S.; Bressan, R.A.; Stone, J.M.; Erlandsson, K.; Mulligan, R.S.; Krystal, J.H.; Ell, P.J. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry 2006, 11, 118–119. [Google Scholar] [CrossRef]
- Kumari, V.; Soni, W.; Mathew, V.M.; Sharma, T. Prepulse inhibition of the startle response in men with schizophrenia: Effects of age of onset of illness, symptoms, and medication. Arch. Gen. Psychiatry 2000, 57, 609–614. [Google Scholar] [CrossRef]
- Parwani, A.; Duncan, E.J.; Bartlett, E.; Madonick, S.H.; Efferen, T.R.; Rajan, R.; Sanfilipo, M.; Chappell, P.B.; Chakravorty, S.; Gonzenbach, S.; et al. Impaired prepulse inhibition of acoustic startle in schizophrenia. Biol. Psychiatry 2000, 47, 662–669. [Google Scholar] [CrossRef]
- Braff, D.L.; Geyer, M.A.; Swerdlow, N.R. Human studies of prepulse inhibition of startle: Normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001, 156, 234–258. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.J.; Madonick, S.H.; Parwani, A.; Angrist, B.; Rajan, R.; Chakravorty, S.; Efferen, T.R.; Szilagyi, S.; Stephanides, M.; Chappell, P.B.; et al. Clinical and sensorimotor gating effects of ketamine in normals. Neuropsychopharmacology 2001, 25, 72–83. [Google Scholar] [CrossRef]
- Shelley, A.M.; Silipo, G.; Javitt, D.C. Diminished responsiveness of ERPs in schizophrenic subjects to changes in auditory stimulation parameters: Implications for theories of cortical dysfunction. Schizophr. Res. 1999, 37, 65–79. [Google Scholar] [CrossRef]
- Jessen, F.; Fries, T.; Kucharski, C.; Nishimura, T.; Hoenig, K.; Maier, W.; Falkai, P.; Heun, R. Amplitude reduction of the mismatch negativity in first-degree relatives of patients with schizophrenia. Neurosci. Lett. 2001, 309, 185–188. [Google Scholar] [CrossRef]
- Michie, P.T.; Innes-Brown, H.; Todd, J.; Jablensky, A.V. Duration mismatch negativity in biological relatives of patients with schizophrenia spectrum disorders. Biol. Psychiatry 2002, 52, 749–758. [Google Scholar] [CrossRef]
- Umbricht, D.; Koller, R.; Vollenweider, F.X.; Schmid, L. Mismatch negativity predicts psychotic experiences induced by NMDA receptor antagonist in healthy volunteers. Biol. Psychiatry 2002, 51, 400–406. [Google Scholar] [CrossRef]
- Matsuura, M.; Yoshino, M.; Ohta, K.; Onda, H.; Nakajima, K.; Kojima, T. Clinical significance of diffuse delta EEG activity in chronic schizophrenia. Clin. Electroencephalogr. 1994, 25, 115–121. [Google Scholar] [CrossRef]
- Winterer, G.; Coppola, R.; Goldberg, T.E.; Egan, M.F.; Jones, D.W.; Sanchez, C.E.; Weinberger, D.R. Prefrontal broadband noise, working memory, and genetic risk for schizophrenia. Am. J. Psychiatry 2004, 161, 490–500. [Google Scholar] [CrossRef]
- Waberski, T.D.; Norra, C.; Kawohl, W.; Thyerlei, D.; Hock, D.; Klostermann, F.; Curio, G.; Buchner, H.; Hoff, P.; Gobbelé, R. Electrophysiological evidence for altered early cerebral somatosensory signal processing in schizophrenia. Psychophysiology 2004, 41, 361–366. [Google Scholar] [CrossRef]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 2010, 11, 100–113. [Google Scholar] [CrossRef]
- Sun, Y.; Farzan, F.; Barr, M.S.; Kirihara, K.; Fitzgerald, P.B.; Light, G.A.; Daskalakis, Z.J. Gamma oscillations in schizophrenia: Mechanisms and clinical significance. Brain Res. 2011, 1413, 98–114. [Google Scholar] [CrossRef]
- Buzsáki, G.; Wang, X.-J. Mechanisms of gamma oscillations. Annu. Rev. Neurosci. 2012, 35, 203–225. [Google Scholar] [CrossRef]
- Tada, M.; Nagai, T.; Kirihara, K.; Koike, S.; Suga Maraki, T.; Kobayashi, T.; Kasai, K. Differential alterations of auditory gamma oscillatory responses between pre-onset high-risk individuals and first-episode schizophrenia. Cereb. Cortex 2016, 26, 1027–1035. [Google Scholar] [CrossRef]
- Hirano, Y.; Oribe, N.; Kanba, S.; Onitsuka, T.; Nestor, P.G.; Spencer, K.M. Spontaneous gamma activity in schizophrenia. JAMA Psychiatry 2015, 72, 813–821. [Google Scholar] [CrossRef]
- Cardin, J.A.; Carlen, M.; Meletis, K.; Knoblich, U.; Zhang, F.; Deisseroth, K.; Tsai, L.H.; Moore, C.I. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 2009, 459, 663–667. [Google Scholar] [CrossRef]
- Sohal, V.S.; Zhang, F.; Yizhar, O.; Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009, 459, 698–702. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Lewis, D.A. GABA neurons and the mechanisms of network oscillations: Implications for understanding cortical dysfunction in schizophrenia. Schizophr. Bull. 2008, 34, 944–961. [Google Scholar] [CrossRef]
- Hashimoto, T.; Arion, D.; Unger, T.; Maldonado-Aviles, J.G.; Morris, H.M.; Volk, D.W.; Mimics, K.; Lewis, D.A. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2008, 13, 147–161. [Google Scholar] [CrossRef]
- Moyer, C.E.; Delevich, K.M.; Fish, K.N.; Asafu-Adjei, J.K.; Sampson, A.R.; Dorph-Petersen, K.A.; Lewis, D.A.; Sweet, R.A. Reduced glutamate decarboxylase 65 protein within primary auditory cortex inhibitory boutons in schizophrenia. Biol. Psychiatry 2012, 72, 734–743. [Google Scholar] [CrossRef]
- Homayoun, H.; Moghaddam, B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef]
- Pinault, D. N-methyl D-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol. Psychiatry 2008, 63, 730–735. [Google Scholar] [CrossRef]
- Hakami, T.; Jones, N.C.; Tolmacheva, E.A.; Gaudias, J.; Chaumont, J.; Salzberg, M.; O’Brien, T.J.; Pinault, D. NMDA receptor hypofunction leads to generalized and persistent aberrant gamma oscillations independent of hyperlocomotion and the state of consciousness. PLoS ONE 2009, 4, e6755. [Google Scholar] [CrossRef]
- Kocsis, B. Differential role of NR2A and NR2B subunits in N-methyl-D-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biol. Psychiatry 2012, 71, 987–995. [Google Scholar] [CrossRef]
- Sullivan, E.M.; Timi, P.; Hong, L.E.; O’Donnell, P. Reverse translation of clinical electrophysiological biomarkers in behaving rodents under acute and chronic NMDA receptor antagonism. Neuropsychopharmacology 2015, 40, 719–727. [Google Scholar] [CrossRef]
- Baldeweg, T.; Spence, S.; Hirsch, S.R.; Gruzelier, J. Gamma-band electroencephalographic oscillations in a patient with somatic hallucinations. Lancet 1998, 352, 620–621. [Google Scholar] [CrossRef]
- Lee, K.H.; Williams, L.M.; Haig, A.; Gordon, E. “Gamma (40 Hz) phase synchronicity” and symptom dimensions in schizophrenia. Cogn. Neuropsychiatry 2003, 8, 57–71. [Google Scholar] [CrossRef]
- Spencer, K.M.; Nestor, P.G.; Perlmutter, R.; Niznikiewicz, M.A.; Klump, M.C.; Frumin, M.; Martha, E.; Shenton McCarley, R.W. Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc. Natl. Acad. Sci. USA 2004, 101, 17288–17293. [Google Scholar] [CrossRef]
- Brenner, C.A.; Krishnan, G.P.; Vohs, J.L.; Ahn, W.Y.; Hetrick, W.P.; Morzorati, S.L.; O’Donnell, B.F. Steady state responses: Electrophysiological assessment of sensory function in schizophrenia. Schizophr. Bull. 2009, 35, 1065–1077. [Google Scholar] [CrossRef]
- Woo, T.U.; Spencer, K.; McCarley, R.W. Gamma oscillation deficits and the onset and early progression of schizophrenia. Harv. Rev. Psychiatry 2010, 18, 173–189. [Google Scholar] [CrossRef]
- Plourde, G.; Baribeau, J.; Bonhomme, V. Ketamine increases the amplitude of the 40-Hz auditory steady-state response in humans. Br. J. Anaesth. 1997, 78, 524–529. [Google Scholar] [CrossRef]
- Hong, L.E.; Summerfelt, A.; Buchanan, R.W.; O’Donnell, P.; Thaker, G.K.; Weiler, M.A.; Lahti, A.C. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology 2010, 35, 632–640. [Google Scholar] [CrossRef]
- Kocsis, B.; Brown, R.E.; McCarley, R.W.; Hajos, M. Impact of ketamine on neuronal network dynamics: Translational modeling of schizophrenia-relevant deficits. CNS Neurosci. Ther. 2013, 19, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Callicott, J.H.; Bertolino, A.; Mattay, V.S.; Langheim, F.J.; Duyn, J.; Coppola, R.; Goldberg, T.E.; Weinberger, D.R. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb. Cortex 2000, 10, 1078–1092. [Google Scholar] [CrossRef] [PubMed]
- Manoach, D.S.; Gollub, R.L.; Benson, E.S.; Searl, M.M.; Goff, D.C.; Halpern, E.; Saper, C.B.; Rauch, S.L. Schizophrenic subjects show aberrant fMRI activation of dorsolateral prefrontal cortex and basal ganglia during working memory performance. Biol. Psychiatry 2000, 48, 99–109. [Google Scholar] [CrossRef]
- Dienel, S.J.; Enwright, J.F., III; Hoftman, G.D.; Lewis, D.A. Markers of glutamate and GABA neurotransmission in the prefrontal cortex of schizophrenia subjects: Disease effects differ across anatomical levels of resolution. Schizophr. Res. 2020, 217, 86–94. [Google Scholar] [CrossRef]
- Lahti, A.C.; Holcomb, H.H.; Medoff, D.R.; Tamminga, C.A. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 1995, 6, 869–872. [Google Scholar] [CrossRef]
- Abdallah, C.G.; De Feyter, H.M.; Averill, L.A.; Jiang, L.; Averill, C.L.; Chowdhury, G.M.I.; Purohit, P.; de Graaf, R.A.; Esterlis, I.; Juchem, C.; et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology 2018, 43, 2154–2160. [Google Scholar] [CrossRef]
- Basar, E.; Schurmann, M.; Basar-Eroglu, C.; Karakas, S. Alpha oscillations in brain functioning: An integrative theory. Int. J. Psychophysiol. 1997, 26, 5–29. [Google Scholar] [CrossRef]
- Basar-Eroglu, C.; Basar, E.; Demiralp, T.; Schurmann, M. P300-response: Possible psychophysiological correlates in delta and theta frequency channels. A review. Int. J. Psychophysiol. 1992, 13, 161–179. [Google Scholar] [CrossRef]
- Miller, R. Cortico-Hippocampal Interplay and the Representation of Contexts in the Brain; Springer: Berlin, Germany, 1991. [Google Scholar]
- Klimesch, W.; Schimke, H.; Schwaiger, J. Episodic and semantic memory: An analysis in the EEG theta and alpha band. Electroencephalogr. Clin. Neurophysiol. 1994, 91, 428–441. [Google Scholar] [CrossRef]
- Basar, E.; Basar-Eroglu, C.; Karakas, S.; Schurmann, M. Are cognitive processes manifested in eventrelated gamma, alpha, theta and delta oscillations in the EEG? Neurosci. Lett. 1999, 259, 165–168. [Google Scholar] [CrossRef]
- Catts, V.S.; Lai, Y.L.; Weickert, C.S.; Weickert, T.W.; Catts, S.V. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biol. Psychology 2016, 116, 57–67. [Google Scholar] [CrossRef]
- Sokolov, B.P. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: Evidence on reversible up-regulation by typical neuroleptics. J. Neurochem. 1998, 71, 2454–2464. [Google Scholar] [CrossRef] [PubMed]
- Catts, V.S.; Derminio, D.S.; Hahn, C.G.; Weickert, C.S. Postsynaptic density levels of the NMDA receptor NR1 subunit and PSD-95 protein in prefrontal cortex from people with schizophrenia. NPJ Schizophr. 2015, 1, 15037. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.M.; Sakai, K.; Roberts, R.C.; Conley, R.R.; Dean, B.; Tamminga, C.A. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: Effects of schizophrenia. Am. J. Psychiatry 2000, 157, 1141–1149. [Google Scholar] [CrossRef]
- Law, A.J.; Deakin, J.F. Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport 2001, 12, 2971–2974. [Google Scholar] [CrossRef]
- Akbarian, S.; Sucher, N.J.; Bradley, D.; Tafazzoli, A.; Trinh, D.; Hetrick, W.P.; Potkin, S.G.; Sandman, C.A.; Bunney, W.E., Jr.; Jones, E.G. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996, 16, 19–30. [Google Scholar] [CrossRef]
- Weickert, C.S.; Fung, S.J.; Catts, V.S.; Schofield, P.R.; Allen, K.M.; Moore, L.T.; Newell, K.A.; Pellen, D.; Huang, X.F.; Catts, S.V.; et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry 2013, 18, 1185–1192. [Google Scholar] [CrossRef]
- Banerjee, A.; Wang, H.Y.; Borgmann-Winter, K.E.; MacDonald, M.L.; Kaprielian, H.; Stucky, A.; Kvasic, J.; Egbujo, C.; Ray, R.; Talbot, K.; et al. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol. Psychiatry 2015, 20, 1091–1100. [Google Scholar] [CrossRef]
- Seeman, P.; Lee, T. Antipsychotic drugs: Direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 1975, 188, 1217–1219. [Google Scholar] [CrossRef]
- Creese, I.; Burt, D.R.; Snyder, S.H. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 1976, 192, 481–483. [Google Scholar] [CrossRef]
- Abi-Dargham, A.; Rodenhiser, J.; Printz, D.; Zea-Ponce, Y.; Gil, R.; Kegeles, L.S.; Weiss, R.; Cooper, T.B.; Mann, J.J.; Van Heertum, R.L.; et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc. Natl. Acad. Sci. USA 2000, 97, 8104–8109. [Google Scholar] [CrossRef]
- Vollenweider, F.X.; Vontobel, P.; Oye, I.; Hell, D.; Leenders, K.L. Effects of (S)-ketamine on striatal dopamine: A [11C] raclopride PET study of a model psychosis in humans. J. Psychiatr. Res. 2000, 34, 35–43. [Google Scholar] [CrossRef]
- Kristiansen, L.V.; Huerta, I.; Beneyto, M.; Meador-Woodruff, J.H. NMDA receptors and schizophrenia. Curr. Opin. Pharmacol. 2007, 7, 48–55. [Google Scholar] [CrossRef]
- Moghaddam, B.; Javitt, D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef]
- Okabe, S.; Miwa, A.; Okado, H. Alternative splicing of the C-terminal domain regulates cell surface expression of the NMDA receptor NR1 subunit. J. Neurosci. 1999, 19, 7781–7792. [Google Scholar] [CrossRef]
- Pauly, T.; Schlicksupp, A.; Neugebauer, R.; Kuhse, J. Synaptic targeting of N-methyl-D-aspartate receptor splice variants is regulated differentially by receptor activity. Neuroscience 2005, 131, 99–111. [Google Scholar] [CrossRef]
- Bauer, D.; Gupta, D.; Harotunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Abnormal expression of glutamate transporter and transporter interacting molecules in prefrontal cortex in elderly patients with schizophrenia. Schizophr. Res. 2008, 104, 108–120. [Google Scholar] [CrossRef]
- Kalandadze, A.; Wu, Y.; Fournier, K.; Robinson, M.B. Identification of motifs involved in endoplasmic reticulum retention-forward trafficking of the GLT-1 subtype of glutamate transporter. J. Neurosci. 2004, 24, 5183–5192. [Google Scholar] [CrossRef]
- Spangaro, M.; Bosia, M.; Zanoletti, A.; Bechi, M.; Mariachiara, B.; Pirovano, A.; Lorenzi, C.; Bramanti, P.; Smeraldi, E.; Cavallaro, R. Exploring effects of EAAT polymorphisms on cognitive functions in schizophrenia. Pharmacogenomics 2014, 15, 925–932. [Google Scholar] [CrossRef]
- Dietz, A.G.; Goldman, S.A.; Nedergaard, M. Glial cells in schizophrenia: A unified hypothesis. Lancet Psychiatry 2020, 7, 272–281. [Google Scholar] [CrossRef]
- Homayoun, H.; Stefani, M.R.; Adams, B.W.; Tamagan, G.D.; Moghaddam, B. Functional interaction between NMDA and mGlu5 receptors: Effects on working memory, instrumental learning, motor behaviors, and dopamine release. Neuropsychopharmacology 2004, 29, 1259–1269. [Google Scholar] [CrossRef]
- Lipska, B.K.; Weinberger, D.R. Animal models of schizophrenia. In Schizophrenia; Hirsch, S.R., Weinberger, D.R., Eds.; Blackwell Science: Malden, UK, 2003; pp. 388–402. [Google Scholar]
- Sams-Dodd, F. Effect of novel antipsychotic drugs on phencyclidine-induced stereotyped behaviour and social isolation in the rat social interaction test. Behav. Pharmacol. 1997, 8, 196–215. [Google Scholar]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Tran, A.; Taylor, J.R.; Roth, R.H. Prefrontal cortical involvement in phencyclidine-induced activation of the mesolimbic dopamine system: Behavioral and neurochemical evidence. Psychopharmacology (Berlin) 1998, 138, 89–95. [Google Scholar] [CrossRef]
- Bakshi, V.P.; Tricklebank, M.; Neijt, H.C.; Lehmann-Masten, V.; Geyer, M.A. Disruption of prepulse inhibition and increases in locomotor activity by competitive N-methyl-D-aspartate receptor antagonists in rats. J. Pharmacol. Exp. Ther. 1999, 288, 643–652. [Google Scholar]
- Broberg, B.V.; Oranje, B.; Glenthøj, B.Y.; Fejgin, K.; Plath, N.; Bastlund, J.F. Assessment of auditory sensory processing in a neurodevelopmental animal model of schizophrenia--gating of auditory-evoked potentials and prepulse inhibition. Behav. Brain Res. 2010, 213, 142–147. [Google Scholar] [CrossRef]
- Suzuki, Y.; Jodo, E.; Takeuchi, S.; Niwa, S.; Kayama, Y. Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience 2002, 114, 769–779. [Google Scholar] [CrossRef]
- Jackson, M.E.; Homayoun, H.; Moghaddam, B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2004, 101, 8467–8472. [Google Scholar] [CrossRef]
- Kargieman, L.; Santana, N.; Mengod, G.; Celada, P.; Artigas, F. Antipsychotic drugs reverse the disruption in prefrontal cortex function produced by NMDA receptor blockade with phencyclidine. Proc. Natl. Acad. Sci. USA 2007, 104, 14843–14848. [Google Scholar] [CrossRef]
- López-Gil, X.; Jiménez-Sánchez, L.; Romón, T.; Campa, L.; Artigas, F.; Adell, A. Importance of inter-hemispheric prefrontal connection in the effects of non-competitive NMDA receptor antagonists. Int. J. Neuropsychopharmacol. 2012, 15, 945–956. [Google Scholar] [CrossRef]
- Lladó-Pelfort, L.; Celada, P.; Riga, M.S.; Troyano-Rodríguez, E.; Santana, N.; Artigas, F. Effects of hallucinogens on neuronal activity. Curr. Top. Behav. Neurosci. 2018, 36, 75–105. [Google Scholar]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Adams, B.W.; Moghaddam, B. Effect of clozapine, haloperidol, or M100907 on phencyclidine-activated glutamate efflux in the prefrontal cortex. Biol. Psychiatry 2001, 50, 750–757. [Google Scholar] [CrossRef]
- Lorrain, D.S.; Baccei, C.S.; Bristow, L.J.; Anderson, J.J.; Varney, M.A. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 2003, 117, 697–706. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.W. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 1998, 281, 1349–1352. [Google Scholar] [CrossRef]
- Mathé, J.M.; Nomikos, G.G.; Blakeman, K.H.; Svensson, T.H. Differential actions of dizocilpine (MK-801) on the mesolimbic and mesocortical dopamine systems: Role of neuronal activity. Neuropharmacology 1999, 38, 121–128. [Google Scholar] [CrossRef]
- Schmidt, C.J.; Fadayel, G.M. Regional effects of MK-801 on dopamine release: Effects of competitive NMDA or 5-HT2A receptor blockade. J. Pharmacol. Exp. Ther. 1996, 277, 1541–1549. [Google Scholar]
- Martin, P.; Carlsson, M.L.; Hjorth, S. Systemic PCP treatment elevates brain extracellular 5-HT: A microdialysis study in awake rats. Neuroreport 1998, 9, 2985–2988. [Google Scholar] [CrossRef]
- Millan, M.J.; Brocco, M.; Gobert, A.; Joly, F.; Bervoets, K.; Rivet, J.; Newman-Tancredi, A.; Audinot, V.; Maurel, S. Contrasting mechanisms of action and sensitivity to antipsychotics of phencyclidine versus amphetamine: Importance of nucleus accumbens 5-HT2A sites for PCP-induced locomotion in the rat. Eur. J. Neurosci. 1999, 11, 4419–4432. [Google Scholar] [CrossRef]
- Amargós-Bosch, M.; López-Gil, X.; Artigas, F.; Adell, A. Clozapine and olanzapine, but not haloperidol, suppress serotonin efflux in the medial prefrontal cortex elicited by phencyclidine and ketamine. Int. J. Neuropsychopharmacol. 2006, 9, 565–573. [Google Scholar] [CrossRef]
- Schiffer, W.K.; Gerasimov, M.; Hofmann, L.; Marsteller, D.; Ashby, C.R.; Brodie, J.D.; Alexoff, D.L.; Dewey, S.L. Gamma vinyl-GABA differentially modulates NMDA antagonist-induced increases in mesocortical versus mesolimbic DA transmission. Neuropsychopharmacology 2001, 25, 704–712. [Google Scholar] [CrossRef]
- Nelson, C.L.; Burk, J.A.; Bruno, J.P.; Sarter, M. Effects of acute and repeated systemic administration of ketamine on prefrontal acetylcholine release and sustained attention performance in rats. Psychopharmacology (Berl) 2002, 161, 168–179. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, L.; Castañé, A.; Pérez-Caballero, L.; Grifoll-Escoda, M.; López-Gil, X.; Campa, L.; Galofré, M.; Berrocoso, E.; Adell, A. Activation of AMPA receptors mediates the antidepressant action of deep brain stimulation of the infralimbic prefrontal cortex. Cereb. Cortex 2016, 26, 2778–2789. [Google Scholar] [CrossRef]
- López-Gil, X.; Jiménez-Sánchez, L.; Campa, L.; Castro, E.; Frago, C.; Adell, A. Role of serotonin and noradrenaline in the rapid antidepressant action of ketamine. ACS Chem. Neurosci. 2019, 10, 3318–3326. [Google Scholar] [CrossRef]
- Krystal, J.H.; D’Souza, D.C.; Mathalon, D.; Perry, E.; Belger, A.; Hoffman, R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: Toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003, 169, 215–233. [Google Scholar] [CrossRef]
- Nyíri, G.; Stephenson, F.A.; Freund, T.F.; Somogyi, P. Large variability in synaptic N-methyl-D-aspartate receptor density on interneurons and a comparison with pyramidal-cell spines in the rat hippocampus. Neuroscience 2003, 119, 347–363. [Google Scholar] [CrossRef]
- Freund, T.F.; Katona, I. Perisomatic inhibition. Neuron 2007, 56, 33–42. [Google Scholar] [CrossRef]
- Razoux, F.; Garcia, R.; Lena, I. Ketamine, at a dose that disrupts motor behavior and latent inhibition, enhances prefrontal cortex synaptic efficacy and glutamate release in the nucleus accumbens. Neuropsychopharmacology 2007, 32, 719–727. [Google Scholar] [CrossRef]
- Kim, S.H.; Price, M.T.; Olney, J.W.; Farber, N.B. Excessive cerebrocortical release of acetylcholine induced by NMDA antagonists is reduced by GABAergic and alpha2-adrenergic agonists. Mol. Psychiatry 1999, 4, 344–352. [Google Scholar] [CrossRef][Green Version]
- Hutson, P.H.; Hogg, J.E. Effects of and interactions between antagonists for different sites on the NMDA receptor complex on hippocampal and striatal acetylcholine efflux in vivo. Eur. J. Pharmacol. 1996, 295, 45–52. [Google Scholar] [CrossRef]
- Yan, Q.S.; Reith, M.E.; Jobe, P.C.; Dailey, J.W. Dizocilpine (MK-801) increases not only dopamine but also serotonin and norepinephrine transmissions in the nucleus accumbens as measured by microdialysis in freely moving rats. Brain Res. 1997, 765, 149–158. [Google Scholar] [CrossRef]
- Lorrain, D.S.; Schaffhauser, H.; Campbell, U.C.; Baccei, C.S.; Correa, L.D.; Rowe, B.; Rodriguez, D.E.; Anderson, J.J.; Varney, M.A.; Pinkerton, A.B.; et al. Group II mGlu receptor activation suppresses norepinephrine release in the ventral hippocampus and locomotor responses to acute ketamine challenge. Neuropsychopharmacology 2003, 28, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Schoepp, D.D. A role for noradrenergic transmission in the actions of phencyclidine and the antipsychotic and antistress effects of mGlu2/3 receptor agonists. Ann. N. Y. Acad. Sci. 2003, 1003, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Whitton, P.S.; Maione, S.; Biggs, C.S.; Fowler, L.J. N-methyl-d-aspartate receptors modulate extracellular dopamine concentration and metabolism in rat hippocampus and striatum in vivo. Brain Res. 1994, 635, 312–316. [Google Scholar] [CrossRef]
- Kretschmer, B.D. NMDA receptor antagonist-induced dopamine release in the ventral pallidum does not correlate with motor activation. Brain Res. 2000, 859, 147–156. [Google Scholar] [CrossRef]
- Greenslade, R.G.; Mitchell, S.N. Selective action of (-)-2-oxa-4-aminobicyclo [3.1.0] hexane-4,6-dicarboxylate (LY379268), a group II metabotropic glutamate receptor agonist, on basal and phencyclidine-induced dopamine release in the nucleus accumbens shell. Neuropharmacology 2004, 47, 1–8. [Google Scholar] [CrossRef]
- Fitzgerald, P.J.; Watson, B.O. In vivo electrophysiological recordings of the effects of antidepressant drugs. Exp. Brain Res. 2019, 237, 1593–1614. [Google Scholar] [CrossRef]
- Pitkänen, M.; Sirviö, J.; Ylinen, A.; Koivisto, E.; Riekkinen, P. Effects of NMDA receptor modulation on hippocampal type 2 theta activity in rats. Gen. Pharmacol. 1995, 26, 1065–1070. [Google Scholar] [CrossRef]
- Lazarewicz, M.T.; Ehrlichman, R.S.; Maxwell, C.R.; Gandal, M.J.; Finkel, L.H.; Siegel, S.J. Ketamine modulates theta and gamma oscillations. J. Cogn. Neurosci. 2010, 22, 1452–1464. [Google Scholar] [CrossRef]
- Troyano-Rodriguez, E.; Lladó-Pelfort, L.; Santana, N.; Teruel-Martí, V.; Celada, P.; Artigas, F. Phencyclidine inhibits the activity of thalamic reticular gamma-aminobutyric acidergic neurons in rat brain. Biol. Psychiatry 2014, 76, 937–945. [Google Scholar] [CrossRef]
- Adell, A.; Jiménez-Sánchez, L.; López-Gil, X.; Romón, T. Is the acute NMDA receptor hypofunction a valid model of schizophrenia? Schizophr. Bull. 2012, 38, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Egerton, A.; Reid, L.; McGregor, S.; Cochran, S.M.; Morris, B.J.; Pratt, J.A. Subchronic and chronic PCP treatment produces temporally distinct deficits in attentional set shifting and prepulse inhibition in rats. Psychopharmacology (Berl) 2008, 198, 37–49. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Roth, R.H. The neuropsychopharmacology of phencyclidine: From NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999, 20, 201–225. [Google Scholar] [CrossRef]
- Neill, J.C.; Harte, M.K.; Haddad, P.M.; Lydall, E.S.; Dwyer, D.M. Acute and chronic effects of NMDA receptor antagonists in rodents, relevance to negative symptoms of schizophrenia: A translational link to humans. Eur. Neuropsychopharmacol. 2014, 24, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Spielewoy, C.; Markou, A. Withdrawal from chronic phencyclidine treatment induces long-lasting depression in brain reward function. Neuropsychopharmacology 2003, 28, 1106–1116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomson, D.M.; McVie, A.; Morris, B.J.; Pratt, J.A. Dissociation of acute and chronic intermittent phencyclidine-induced performance deficits in the 5-choice serial reaction time task: Influence of clozapine. Psychopharmacology (Berl) 2011, 213, 681–695. [Google Scholar] [CrossRef]
- Mouri, A.; Koseki, T.; Narusawa, S.; Niwa, M.; Mamiya, T.; Kano, S.; Sawa, A.; Nabeshima, T. Mouse strain differences in phencyclidine-induced behavioural changes. Int. J. Neuropsychopharmacol. 2012, 15, 767–779. [Google Scholar] [CrossRef]
- Xu, X.; Domino, E.F. Genetic differences in the locomotor response to single and daily doses of phencyclidine in inbred mouse strains. Behav. Pharmacol. 1994, 5, 623–629. [Google Scholar] [CrossRef]
- Castañé, A.; Santana, N.; Artigas, F. PCP-based mice models of schizophrenia: Differential behavioral, neurochemical and cellular effects of acute and subchronic treatments. Psychopharmacology (Berl) 2015, 232, 4085–4097. [Google Scholar] [CrossRef]
- Kondziella, D.; Brenner, E.; Eyjolfsson, E.M.; Markinhuhta, K.R.; Carlsson, M.L.; Sonnewald, U. Glial-neuronal interactions are impaired in the schizophrenia model of repeated MK801 exposure. Neuropsychopharmacology 2006, 31, 1880–1887. [Google Scholar] [CrossRef]
- Karlsson, R.M.; Tanaka, K.; Heilig, M.; Holmes, A. Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: Rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol. Psychiatry 2008, 64, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, S.I.; Mastropaolo, J.; Schwartz, B.L.; Rosse, R.B.; Morihisa, J.M. A "glutamatergic hypothesis" of schizophrenia. Rationale for pharmacotherapy with glycine. Clin. Neuropharmacol. 1989, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Zylberman, I.; Zukin, S.R.; Heresco-Levy, U.; Lindenmayer, J.P. Amelioration of negative symptoms in schizophrenia by glycine. Am. J. Psychiatry 1994, 151, 1234–1236. [Google Scholar]
- Heresco-Levy, U.; Javitt, D.C.; Ermilov, M.; Mordel, C.; Horowitz, A.; Kelly, D. Double-blind, placebo-controlled, crossover trial of glycine adjuvant therapy for treatment-resistant schizophrenia. Br. J. Psychiatry 1996, 169, 610–617. [Google Scholar] [CrossRef]
- Buchanan, R.W.; Javitt, D.C.; Marder, S.R.; Schooler, N.R.; Gold, J.M.; McMahon, R.P.; Heresco-Levy, U.; Carpenter, W.T. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): The efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am. J. Psychiatry 2007, 164, 1593–1602. [Google Scholar] [CrossRef]
- Tsai, G.; Yang, P.; Chung, L.C.; Lange, N.; Coyle, J.T. D-serine added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 1998, 44, 1081–1089. [Google Scholar] [CrossRef]
- Tsai, G.E.; Yang, P.; Chung, L.C.; Tsai, I.C.; Tsai, C.W.; Coyle, J.T. D-serine added to clozapine for the treatment of schizophrenia. Am. J. Psychiatry 1999, 156, 1822–1825. [Google Scholar]
- Heresco-Levy, U.; Javitt, D.C.; Ebstein, R.; Vass, A.; Lichtenberg, P.; Bar, G.; Catinari, S.; Ermilov, M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol. Psychiatry 2005, 57, 577–585. [Google Scholar] [CrossRef]
- van Berckel, B.N.; Hijman, R.; van der Linden, J.A.; Westenberg, H.G.; van Ree, J.M.; Kahn, R.S. Efficacy and tolerance of D-cycloserine in drug-free schizophrenic patients. Biol. Psychiatry 1996, 40, 1298–1300. [Google Scholar] [CrossRef]
- Heresco-Levy, U.; Javitt, D.C.; Ermilov, M.; Silipo, G.; Shimoni, J. Double-blind, placebo-controlled, crossover trial of D-cycloserine adjuvant therapy for treatment-resistant schizophrenia. Int. J. Neuropsychopharmacol. 1998, 1, 131–135. [Google Scholar] [CrossRef]
- Heresco-Levy, U.; Ermilov, M.; Shimoni, J.; Shapira, B.; Silipo, G.; Javitt, D.C. Placebo-controlled trial of D-cycloserine added to conventional neuroleptics, olanzapine, or risperidone in schizophrenia. Am. J. Psychiatry 2002, 159, 480–482. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.J.; Szilagyi, S.; Schwartz, M.P.; Bugarski-Kirola, D.; Kunzova, A.; Negi, S.; Stephanides, M.; Efferen, T.R.; Angrist, B.; Peselow, E.; et al. Effects of D-cycloserine on negative symptoms in schizophrenia. Schizophr. Res. 2004, 71, 239–248. [Google Scholar] [CrossRef]
- Goff, D.C.; Herz, L.; Posever, T.; Shih, V.; Tsai, G.; Henderson, D.C.; Freudenreich, O.; Evins, A.E.; Yovel, I.; Zhang, H.; et al. A six-month, placebo-controlled trial of d-cycloserine co-administered with conventional antipsychotics in schizophrenia patients. Psychopharmacology (Berl) 2005, 179, 144–150. [Google Scholar] [CrossRef]
- Harvey, R.J.; Yee, B.K. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat. Rev. Drug Discov. 2013, 12, 866–885. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Liang, S.Y.; Chang, Y.C.; Ting, S.Y.; Kao, C.L.; Wu, Y.H.; Tsai, G.E.; Lane, H.Y. Adjunctive sarcosine plus benzoate improved cognitive function in chronic schizophrenia patients with constant clinical symptoms: A randomised, double-blind, placebo-controlled trial. World J. Biol. Psychiatry 2017, 18, 357–368. [Google Scholar] [CrossRef]
- Fone, K.C.F.; Watson, D.J.G.; Billiras, R.I.; Sicard, D.I.; Dekeyne, A.; Rivet, J.M.; Gobert, A.; Millan, M.J. Comparative pro-cognitive and neurochemical profiles of glycine modulatory site agonists and glycine reuptake inhibitors in the rat: Potential relevance to cognitive dysfunction and its management. Mol. Neurobiol. 2020, 57, 2144–2166. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, Y.M.; Lane, H.Y. Novel treatment for the most resistant schizophrenia: Dual activation of NMDA receptor and antioxidant. Curr. Drug Targets 2020, 21, 610–615. [Google Scholar] [CrossRef]
- Kapur, S.; Zipursky, R.; Jones, C.; Remington, G.; Houle, S. Relationship between dopamine D2 occupancy, clinical response, and side effects: A double-blind PET study of first-episode schizophrenia. Am. J. Psychiatry 2000, 157, 514–520. [Google Scholar] [CrossRef]
- Newcomer, J.W. Second-generation (atypical) antipsychotics and metabolic effects: A comprehensive literature review. CNS Drugs 2005, 19, 1–93. [Google Scholar] [CrossRef]
- López-Gil, X.; Artigas, F.; Adell, A. Role of different monoamine receptors controlling MK-801-induced release of serotonin and glutamate in the medial prefrontal cortex: Relevance for antipsychotic action. Int. J. Neuropsychopharmacol. 2009, 12, 487–499. [Google Scholar] [CrossRef]
- López-Gil, X.; Artigas, F.; Adell, A. Unraveling monoamine receptors involved in the action of typical and atypical antipsychotics on glutamatergic and serotonergic transmission in prefrontal cortex. Curr. Pharm. Des. 2010, 16, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Baviera, M.; Invernizzi, R.W.; Balducci, C. Dissociable contribution of 5-HT1A and 5-HT2A receptors in the medial prefrontal cortex to different aspects of executive control such as impulsivity and compulsive perseveration in rats. Neuropsychopharmacology 2006, 31, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, Z.; Wu, Z.; Chen, J.; Wang, Z.; Peng, D.; Hong, W.; Yuan, C.; Wang, Z.; Yu, S.; et al. A study of N-methyl-D-aspartate receptor gene (GRIN2B) variants as predictors of treatment-resistant major depression. Psychopharmacology (Berl) 2014, 231, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Niciu, M.J.; Ionescu, D.F.; Richards, E.M.; Zarate, C.A.J. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J. Neural Transm. 2014, 121, 907–924. [Google Scholar] [CrossRef]
- Kaut, O.; Schmitt, I.; Hofmann, A.; Hoffmann, P.; Schlaepfer, T.E.; Wüllner, U.; Hurlemann, R. Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 331–341. [Google Scholar] [CrossRef]
- Marsden, W.N. Stressor-induced NMDAR dysfunction as a unifying hypothesis of the aetiology, pathogenesis and comorbidity of clinical depression. Med. Hypotheses 2011, 77, 508–528. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Zarate, C.A.J.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef]
- Jaso, B.A.; Niciu, M.J.; Iadarola, N.D.; Lally, N.; Richards, E.M.; Park, M.; Ballard, E.D.; Nugent, A.C.; Machado-Vieira, R.; Zarate, C.A.J. Therapeutic modulation of glutamate receptors in major depressive disorder. Curr. Neuropharmacol. 2017, 15, 57–70. [Google Scholar] [CrossRef]
- Paul, I.A.; Nowak, G.; Layer, R.T.; Popik, P.; Skolnick, P. Adaptation of the NMDA receptor complex following chronic antidepressant treatments. J. Pharmacol. Ther. 1994, 269, 95–102. [Google Scholar]
- Nowak, G.; Li, Y.; Paul, I.A. Adaptation of cortical but not hippocampal NMDA receptors after chronic citalopram treatment. Eur. J. Pharmacol. 1996, 295, 75–85. [Google Scholar] [CrossRef]
- Skolnick, P.; Popik, P.; Trullas, R. Glutamate-based antidepressants: 20 years on. Trends Pharmacol. Sci. 2009, 30, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.C.; Henter, I.D.; Zarate, C.A. Targeting the glutamatergic system to treat major depressive disorder: Rationale and progress to date. Drugs 2012, 72, 1313–1333. [Google Scholar] [CrossRef]
- Ghasemi, M.; Phillips, C.; Trillo, L.; De Miguel, Z.; Das, D.; Salehi, A. The role of NMDA receptors in the pathophysiology and treatment of mood disorders. Neurosci. Biobehav. Rev. 2014, 47, 336–358. [Google Scholar] [CrossRef]
- Naughton, M.; Clarke, G.; O’Leary, O.F.; Cryan, J.F.; Dinan, T.G. A review of ketamine in affective disorders: Current evidence of clinical efficacy, limitations of use and pre-clinical evidence on proposed mechanisms of action. J. Affect. Disord. 2014, 156, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Deutschenbaur, L.; Beck, J.; Kiyhankhadiv, A.; Mühlhauser, M.; Borgwardt, S.; Walter, M.; Hasler, G.; Sollberger, D.; Lang, U.E. Role of calcium, glutamate and NMDA in major depression and therapeutic application. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Feyissa, A.M.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007, 32, 1888–1902. [Google Scholar] [CrossRef]
- Karolewicz, B.; Szebeni, K.; Gilmore, T.; Maciag, D.; Stockmeier, C.A.; Ordway, G.A. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int. J. Neuropsychopharmacol. 2009, 12, 143–153. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Callado, L.F.; Meana, J.J.; Garzón-Niño, J. Schizophrenia and depression, two poles of endocannabinoid system deregulation. Transl. Psychiatry 2017, 7, 1291. [Google Scholar] [CrossRef]
- Karolewicz, B.; Stockmeier, C.; Ordway, G.A. Elevated levels of the NR2C subunit of the NMDA receptor in the locus coeruleus in depression. Neuropsychopharmacology 2005, 30, 1557–1567. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Underwood, M.D.; Bakalian, M.J.; Johnson, V.L.; Kassir, S.A.; Ellis, S.P.; Mann, J.J.; Arango, V. Less NMDA receptor binding in dorsolateral prefrontal cortex and anterior cingulate cortex associated with reported early life adversity but not suicide. Int. J. Neuropsychopharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Alexander, R.; Smith, M.A.; Pathak, S.; Kanes, S.; Lee, C.M.; Sanacora, G. Glutamate-based depression GBD. Med. Hypotheses 2012, 78, 675–681. [Google Scholar] [CrossRef]
- Chandley, M.J.; Szebeni, A.; Szebeni, K.; Crawford, J.D.; Stockmeier, C.A.; Turecki, G.; Kostrzewa, R.M.; Ordway, G.A. Elevated gene expression of glutamate receptors in noradrenergic neurons from the locus coeruleus in major depression. Int. J. Neuropsychopharmacol. 2014, 17, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Gray, A.L.; Hyde, T.M.; Deep-Soboslay, A.; Kleinman, J.E.; Sodhi, M.S. Sex differences in glutamate receptor gene expression in major depression and suicide. Mol. Psychiatry 2015, 20, 1057–1068. [Google Scholar] [CrossRef]
- Lin, E.; Kuo, P.H.; Liu, Y.L.; Yu, Y.W.; Yang, A.C.; Tsai, S.J. A deep learning approach for predicting antidepressant response in major depression using clinical and genetic biomarkers. Front. Psychiatry 2018, 9, 290. [Google Scholar] [CrossRef]
- Weder, N.; Zhang, H.; Jensen, K.; Yang, B.Z.; Simen, A.; Jackowski, A.; Lipschitz, D.; Douglas-Palumberi, H.; Ge, M.; Perepletchikova, F.; et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 417–424. [Google Scholar] [CrossRef]
- Calabrese, F.; Guidotti, G.; Molteni, R.; Racagni, G.; Mancini, M.; Riva, M.A. Stress-induced changes of hippocampal NMDA receptors: Modulation by duloxetine treatment. PLoS ONE 2012, 7, e37916. [Google Scholar] [CrossRef]
- Wang, Q.; Jie, W.; Liu, J.H.; Yang, J.M.; Gao, T.M. An astroglial basis of major depressive disorder? An overview. Glia 2017, 65, 1227–1250. [Google Scholar] [CrossRef]
- Czéh, B.; Nagy, S.A. Clinical findings documenting cellular and molecular abnormalities of glia in depressive disorders. Front. Mol. Neurosci. 2018, 11, 56. [Google Scholar] [CrossRef]
- Ongür, D.; Drevets, W.C.; Price, J.L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. USA 1998, 95, 13290–13295. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.; Mackay, D.; Chana, G.; Beasley, C.; Landau, S.; Everall, I.P. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 2002, 12, 386–394. [Google Scholar] [CrossRef]
- Rajkowska, G.; Stockmeier, C.A. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.A.; Simpson, J.; Mahajan, G.J.; Overholser, J.C.; Jurjus, G.J.; Dieter, L.; Herbst, N.; May, W.; Rajkowska, G.; Stockmeier, C.A. Hippocampal volume and total cell numbers in major depressive disorder. J. Psychiatr. Res. 2013, 47, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Hidalgo, J.J.; Baucom, C.; Dilley, G.; Overholser, J.C.; Meltzer, H.Y.; Stockmeier, C.A.; Rajkowska, G. Glial fibrillary acidic protein immunoreactivity in the prefrontal cortex distinguishes younger from older adults in major depressive disorder. Biol. Psychiatry 2000, 48, 861–873. [Google Scholar] [CrossRef]
- Moghaddam, B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: Comparison to hippocampus and basal ganglia. J. Neurochem. 1993, 60, 1650–1657. [Google Scholar] [CrossRef]
- Bartanusz, V. Stress-induced changes in messenger RNA levels of N-methyl-d-aspartate and AMPA receptor subunits in selected regions of the rat hippocampus and hypothalamus. Neuroscience 1995, 66, 247–252. [Google Scholar] [CrossRef]
- Fitzgerald, L.W.; Ortiz, J.; Hamedani, A.G.; Nestler, E.J. Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: Common adaptations among cross-sensitizing agents. J. Neurosci. 1996, 16, 274–282. [Google Scholar] [CrossRef]
- Shepard, R.; Page, C.E.; Coutellier, L. Sensitivity of the prefrontal GABAergic system to chronic stress in male and female mice: Relevance for sex differences in stress-related disorders. Neuroscience 2016, 332, 1–12. [Google Scholar] [CrossRef]
- Page, C.E.; Shepard, R.; Heslin, K.; Coutellier, L. Prefrontal parvalbumin cells are sensitive to stress and mediate anxiety-related behaviors in female mice. Sci. Rep. 2019, 9, 19772. [Google Scholar] [CrossRef]
- Ji, M.H.; Zhang, L.; Mao, M.J.; Zhang, H.; Yang, J.J.; Qiu, L.L. Overinhibition mediated by parvalbumin interneurons might contribute to depression-like behavior and working memory impairment induced by lipopolysaccharide challenge. Behav. Brain Res. 2020, 383, 112509. [Google Scholar] [CrossRef] [PubMed]
- Fogaça, M.V.; Duman, R.S. Cortical GABAergic dysfunction in stress and depression: New insights for therapeutic interventions. Front. Cell. Neurosci. 2019, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Masrour, F.F.; Peeri, M.; Azarbayjani, M.A.; Hosseini, M.J. Voluntary exercise during adolescence mitigated negative the effects of maternal separation stress on the depressive-like behaviors of adult male rats: Role of NMDA receptors. Neurochem. Res. 2018, 43, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Sathyanesan, M.; Haiar, J.M.; Watt, M.J.; Newton, S.S. Restraint stress differentially regulates inflammation and glutamate receptor gene expression in the hippocampus of C57BL/6 and BALB/c mice. Stress 2017, 20, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.; Aguayo, F.I.; Aliaga, E.; Muñoz, M.; García-Rojo, G.; Olave, F.A.; Parra-Fiedler, N.A.; García-Pérez, A.; Tejos-Bravo, M.; Rojas, P.S.; et al. Chronic stress triggers expression of immediate early genes and differentially affects the expression of AMPA and NMDA subunits in dorsal and ventral hippocampus of rats. Front. Mol. Neurosci. 2017, 10, 244. [Google Scholar] [CrossRef]
- Weiland, N.G.; Orchinik, M.; Tanapat, P. Chronic corticosterone treatment induces parallel changes in N-methyl-D-aspartate receptor subunit messenger RNA levels and antagonist binding sites in the hippocampus. Neuroscience 1997, 78, 653–662. [Google Scholar] [CrossRef]
- Webster, H.H.; Flores, G.; Marcotte, E.R.; Cecyre, D.; Quirion, R.; Srivastava, L.K. Olfactory bulbectomy alters NMDA receptor levels in the rat prefrontal cortex. Synapse 2000, 37, 159–162. [Google Scholar] [CrossRef]
- Ho, Y.J.; Liu, T.M.; Tai, M.Y.; Wen, Z.H.; Chow, R.S.S.; Tsai, Y.F.; Wong, C.S. Effects of olfactory bulbectomy on NMDA receptor density in the rat brain: [3H] MK-801 binding assay. Brain Res. 2001, 900, 214–218. [Google Scholar] [CrossRef]
- Dong, B.E.; Chen, H.; Sakata, K. BDNF deficiency and enriched environment treatment affect neurotransmitter gene expression differently across ages. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Boyce-Rustay, J.M.; Holmes, A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology 2006, 31, 2405–2414. [Google Scholar] [CrossRef]
- Salimando, G.J.; Hyun, M.; Boyt, K.M.; Winder, D.G. BNST GluN2D-containing NMDA receptors influence anxiety- and depressive-like behaviors and modulate cell-specific excitatory/inhibitory synaptic balance. J. Neurosci. 2020, 40, 3949–3968. [Google Scholar] [CrossRef] [PubMed]
- Forrest, D.; Yuzaki, M.; Soares, H.D.; Ng, L.; Luk, D.C.; Sheng, M.; Stewart, C.L.; Morgan, J.I.; Connor, J.A.; Curran, T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 1994, 13, 325–338. [Google Scholar] [CrossRef]
- Kutsuwada, T.; Sakimura, K.; Manabe, T.; Takayama, C.; Katakura, N.; Kushiya, E.; Natsume, R.; Watanabe, M.; Inoue, Y.; Yagi, T.; et al. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 1996, 16, 333–344. [Google Scholar] [CrossRef]
- Montalvo-Ortiz, J.L.; Bordner, K.A.; Carlyle, B.C.; Gelernter, J.; Simen, A.A.; Kaufman, J. The role of genes involved in stress, neural plasticity, and brain circuitry in depressive phenotypes: Convergent findings in a mouse model of neglect. Behav. Brain Res. 2016, 315, 71–74. [Google Scholar] [CrossRef]
- Tordera, R.M.; Garcia-García, A.L.; Elizalde, N.; Segura, V.; Aso, E.; Venzala, E.; Ramírez, M.J.; Del Rio, J. Chronic stress and impaired glutamate function elicit a depressive-like phenotype and common changes in gene expression in the mouse frontal cortex. Eur. Neuropsychopharmacol. 2011, 21, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Banasr, M. From pathophysiology to novel antidepressant drugs: Glial contributions to the pathology and treatment of mood disorders. Biol. Psychiatry 2013, 73, 1172–1179. [Google Scholar] [CrossRef]
- Banasr, M.; Chowdhury, G.M.; Terwilliger, R.; Newton, S.S.; Duman, R.S.; Behar, K.L.; Sanacora, G. Glial pathology in an animal model of depression: Reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol. Psychiatry 2010, 15, 501–511. [Google Scholar] [CrossRef]
- Gong, Y.; Sun, X.L.; Wu, F.F.; Su, C.J.; Ding, J.H.; Hu, G. Female early adult depression results in detrimental impacts on the behavioral performance and brain development in offspring. CNS Neurosci. Ther. 2012, 18, 461–470. [Google Scholar] [CrossRef]
- Banasr, M.; Duman, R.S. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 2008, 64, 863–870. [Google Scholar] [CrossRef]
- Fullana, M.N.; Ruiz-Bronchal, E.; Ferrés-Coy, A.; Juárez-Escoto, E.; Artigas, F.; Bortolozzi, A. Regionally selective knockdown of astroglial glutamate transporters in infralimbic cortex induces a depressive phenotype in mice. Glia 2019, 67, 1122–1137. [Google Scholar] [CrossRef]
- Fava, M. Diagnosis and definition of treatment-resistant depression. Biol. Psychiatry 2003, 53, 649–659. [Google Scholar] [CrossRef]
- Ionescu, D.F.; Rosenbaum, J.F.; Alpert, J.E. Pharmacological approaches to the challenge of treatment-resistant depression. Dialogues Clin. Neurosci. 2015, 17, 111–126. [Google Scholar] [PubMed]
- Gaynes, B.N.; Lux, L.; Gartlehner, G.; Asher, G.; Forman-Hoffman, V.; Green, J.; Boland, E.; Weber, R.P.; Randolph, C.; Bann, C.; et al. Defining treatment-resistant depression. Depress. Anxiety 2020, 37, 134–145. [Google Scholar] [CrossRef]
- Trullas, R.; Skolnick, P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur. J. Pharmacol. 1990, 185, 1–10. [Google Scholar] [CrossRef]
- DiazGranados, N.; Ibrahim, L.A.; Brutsche, N.E.; Ameli, R.; Henter, I.D.; Luckenbaugh, D.A.; Machado-Vieira, R.; Zarate, C.A., Jr. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J. Clin. Psychiatry 2010, 71, 1605–1611. [Google Scholar] [CrossRef]
- Singh, J.B.; Fedgchin, M.; Daly, E.J.; De Boer, P.; Cooper, K.; Lim, P.; Pinter, C.; Murrough, J.W.; Sanacora, G.; Shelton, R.C.; et al. A double-blind, randomized, placebo-controlled, dose-frequency study of intravenous ketamine in patients with treatment-resistant depression. Am. J. Psychiatry 2016, 173, 816–826. [Google Scholar] [CrossRef]
- Maeng, S.; Zarate, C.A.J.; Du, J.; Schloesser, R.J.; McCammon, J.; Chen, G.; Manji, H.K. Cellular mechanisms underlying the antidepressant effects of ketamine: Role of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol. Psychiatry 2008, 63, 349–352. [Google Scholar] [CrossRef]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef]
- Koike, H.; Iijima, M.; Chaki, S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav. Brain Res. 2011, 224, 107–111. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef]
- Hashimoto, K. Role of the mTOR signaling pathway in the rapid antidepressant action of ketamine. Expert Rev. Neurother. 2011, 11, 33–36. [Google Scholar] [CrossRef]
- Koike, H.; Chaki, S. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav. Brain Res. 2014, 271, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yamaki, V.N.; Wei, Z.; Zheng, Y.; Cai, X. Differential regulation of GluA1 expression by ketamine and memantine. Behav. Brain Res. 2017, 316, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Shinohara, R.; Fogaça, M.V.; Hare, B. Neurobiology of rapid-acting antidepressants: Convergent effects on GluA1-synaptic function. Mol. Psychiatry 2019, 24, 1816–1832. [Google Scholar] [CrossRef] [PubMed]
- Klann, E.; Antion, M.D.; Banko, J.L.; Hou, L. Synaptic plasticity and translation initiation. Learn. Mem. 2004, 11, 365–372. [Google Scholar] [CrossRef]
- Abe, N.; Borson, S.H.; Gambello, M.J.; Wang, F.; Cavalli, V. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J. Biol. Chem. 2010, 285, 28034–28043. [Google Scholar] [CrossRef]
- Jernigan, C.S.; Goswami, D.B.; Austin, M.C.; Iyo, A.H.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 774–1779. [Google Scholar] [CrossRef]
- Chandran, A.; Iyo, A.H.; Jernigan, C.S.; Legutko, B.; Austin, M.C.; Karolewicz, B. Reduced phosphorylation of the mTOR signaling pathway components in the amygdala of rats exposed to chronic stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 40, 240–245. [Google Scholar] [CrossRef]
- Fang, Z.H.; Lee, C.H.; Seo, M.K.; Cho, H.; Lee, J.G.; Lee, B.J.; Park, S.W.; Kim, Y.H. Effect of treadmill exercise on the BDNF-mediated pathway in the hippocampus of stressed rats. Neurosci. Res. 2013, 76, 187–194. [Google Scholar] [CrossRef]
- Zhong, P.; Wang, W.; Pan, B.; Liu, X.; Zhang, Z.; Long, J.Z.; Zhang, H.T.; Cravatt, B.F.; Liu, Q.S. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology 2014, 39, 1763–1776. [Google Scholar] [CrossRef]
- Fuchikami, M.; Thomas, A.; Liu, R.; Wohleb, E.S.; Land, B.B.; DiLeone, R.J.; Aghajanian, G.K.; Duman, R.S. Optogenetic stimulation of infralimbic PFC reproduces ketamine’s rapid and sustained antidepressant actions. Proc. Natl. Acad. Sci. USA 2015, 112, 8106–8111. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, K.; Iijima, M.; Chaki, S. The antidepressant effects of an mGlu2/3 receptor antagonist and ketamine require AMPA receptor stimulation in the mPFC and subsequent activation of the 5-HT neurons in the DRN. Neuropsychopharmacology 2016, 41, 1046–1056. [Google Scholar] [CrossRef]
- Covington, H.E., III; Lobo, M.K.; Maze, I.; Vialou, V.; Hyman, J.M.; Zaman, S.; LaPlant, Q.; Mouzon, E.; Ghose, S.; Tamminga, C.A.; et al. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J. Neurosci. 2010, 30, 16082–16090. [Google Scholar] [CrossRef]
- Warden, M.R.; Selimbeyoglu, A.; Mirzabekov, J.J.; Lo, M.; Thompson, K.R.; Kim, S.Y.; Adhikari, A.; Tye, K.M.; Frank, L.M.; Deisseroth, K. A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 2012, 492, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Challis, C.; Beck, S.G.; Berton, O. Optogenetic modulation of descending prefrontocortical inputs to the dorsal raphe bidirectionally bias socioaffective choices after social defeat. Front. Behav. Neurosci. 2014, 8, 43. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Guo, W. Emotional roles of mono-aminergic neurotransmitters in major depressive disorder and anxiety disorders. Front. Psychol. 2018, 9, 2201. [Google Scholar] [CrossRef] [PubMed]
- Lucki, I.; Singh, A.; Kreiss, D.S. Antidepressant-like behavioral effects of serotonin receptor agonists. Neurosci. Biobehav. Rev. 1994, 18, 85–95. [Google Scholar] [CrossRef]
- Mccauley, E.; Carlson, G.A.; Calderon, R. The role of somatic complaints in the diagnosis of depression in children and adolescents. J. Am. Acad. Child Adolesc. Psychiatry 1991, 30, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Gerber, P.D.; Barrett, J.E.; Barrett, J.A.; Oxman, T.E.; Manheimer, E.; Smith, R.; Whiting, R.D. The relationship of presenting physical complaints to depressive symptoms in primary care patients. J. Gen. Intern. Med. 1992, 7, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Fan, X.; Guo, W. Dysfunction in serotonergic and noradrenergic systems and somatic symptoms in psychiatric disorders. Front. Psychiatry 2019, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Fava, M. The role of the serotonergic and noradrenergic neurotransmitter systems in the treatment of psychological and physical symptoms of depression. J. Clin. Psychiatry 2003, 64, 26–29. [Google Scholar] [PubMed]
- Stahl, S.M. The psychopharmacology of painful physical symptoms in depression. J. Clin. Psychiatry. 2002, 63, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Ruhé, H.G.; Mason, N.S.; Schene, A.H. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: A meta-analysis of monoamine depletion studies. Mol. Psychiatry 2007, 12, 331–359. [Google Scholar] [CrossRef] [PubMed]
- Shopsin, B.; Gershon, S.; Goldstein, M.; Friedman, E.; Wilk, S. Use of synthesis inhibitors in defining a role for biogenic amines during imipramine treatment in depressed patients. Psychopharmacol. Commun. 1975, 1, 239–249. [Google Scholar] [PubMed]
- Shopsin, B.; Friedman, E.; Gershon, S. Parachlorophenylalanine reversal of tranylcypromine effects in depressed patients. Arch. Gen. Psychiatry 1976, 33, 811–819. [Google Scholar] [CrossRef]
- Domino, E.F. Taming the ketamine tiger. Anesthesiology 2010, 113, 678–684. [Google Scholar] [CrossRef]
- Muller, J.; Pentyala, S.; Dilger, J.; Pentyala, S. Ketamine enantiomers in the rapid and sustained antidepressant effects. Ther. Adv. Psychopharmacol. 2016, 6, 185–192. [Google Scholar] [CrossRef]
- Zhang, J.C.; Li, S.X.; Hashimoto, K. R (−)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine. Pharmacol. Biochem. Behav. 2014, 116, 137–141. [Google Scholar] [CrossRef]
- Hashimoto, K. The R-stereoisomer of ketamine as an alternative for ketamine for treatment-resistant major depression. Clin. Psychopharmacol. Neurosci. 2014, 12, 72–73. [Google Scholar] [CrossRef]
- Yang, C.; Shirayama, Y.; Zhang, J.C.; Ren, Q.; Yao, W.; Ma, M.; Dong, C.; Hashimoto, K. R-ketamine: A rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl. Psychiatry 2015, 5, e632. [Google Scholar] [CrossRef]
- Fukumoto, K.; Toki, H.; Iijima, M.; Hashihayata, T.; Yamaguchi, J.I.; Hashimoto, K.; Chaki, S. Antidepressant potential of (R)-ketamine in rodent models: Comparison with (S)-ketamine. J. Pharmacol. Exp. Ther. 2017, 361, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.C.; Bandeira, I.D.; Correia-Melo, F.S.; Telles, M.; Mello, R.P.; Vieira, F.; Lima, C.S.; Jesus-Nunes, A.P.; Guerreiro-Costa, L.N.F.; Marback, R.F.; et al. Intravenous arketamine for treatment-resistant depression: Open-label pilot study. Eur. Arch. Psychiatry Clin. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Preskorn, S.H.; Baker, B.; Kolluri, S.; Menniti, F.S.; Krams, M.; Landen, J.W. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101, 606, in patients with treatment refractory major depressive disorder. J. Clin. Psychopharmacol. 2008, 28, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, L.; Diaz Granados, N.; Jolkovsky, L.; Brutsche, N.; Luckenbaugh, D.A.; Herring, W.J.; Potter, W.Z.; Zarate, C.A.J. A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J. Clin. Psychopharmacol. 2012, 32, 551–557. [Google Scholar] [CrossRef]
- Burgdorf, J.; Zhang, X.L.; Nicholson, K.L.; Balster, R.L.; Leander, J.D.; Stanton, P.K.; Gross, A.L.; Kroes, R.A.; Moskal, J.R. GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology 2013, 38, 729–742. [Google Scholar] [CrossRef]
- Sanacora, G.; Smith, M.A.; Pathak, S.; Su, H.L.; Boeijinga, P.H.; McCarthy, D.J.; Quirk, M.C. Lanicemine: A low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol. Psychiatry 2014, 19, 978–985. [Google Scholar] [CrossRef]
- Zarate, C.A.J.; Mathews, D.; Ibrahim, L.; Chaves, J.F.; Marquardt, C.; Ukoh, I.; Jolkovsky, L.; Brutsche, N.E.; Smith, M.A.; Luckenbaugh, D.A. A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol. Psychiatry 2013, 74, 257–264. [Google Scholar] [CrossRef]
- Auberson, Y.P.; Allgeier, H.; Bischoff, S.; Lingenhoehl, K.; Moretti, R.; Schmutz, M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg. Med. Chem. Lett. 2002, 12, 1099–1102. [Google Scholar] [CrossRef]
- Neyton, J.; Paoletti, P. Relating NMDA receptor function to receptor subunit composition: Limitations of the pharmacological approach. J. Neurosci. 2006, 36, 1331–1333. [Google Scholar] [CrossRef]
- Fischer, G.; Mutel, V.; Trube, G.; Malherbe, P.; Kew, J.N.; Mohacsi, E.; Heitz, M.P.; Kemp, J.A. Ro 25-6981, a highly potent and selective blocker of N-methyl-D-aspartate receptors containing the NR2B subunit. Characterization in vitro. J. Pharmacol. Exp. Ther. 1997, 283, 1285–1292. [Google Scholar]
- Jiménez-Sánchez, L.; Campa, L.; Auberson, Y.P.; Adell, A. The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modeling schizophrenia and treating refractory depression. Neuropsychopharmacology 2014, 39, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Ide, S.; Ikekubo, Y.; Mishina, M.; Hashimoto, K.; Ikeda, K. Role of NMDA receptor GluN2D subunit in the antidepressant effects of enantiomers of ketamine. J. Pharmacol. Sci. 2017, 135, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Ide, S.; Ikekubo, Y.; Mishina, M.; Hashimoto, K.; Ikeda, K. Cognitive impairment that is induced by (R)-ketamine is abolished in NMDA GluN2D receptor subunit knockout mice. Int. J. Neuropsychopharmacol. 2019, 22, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.A.; Li, P. Effects of 5-HT (6) receptor blockade on the neurochemical outcome of antidepressant treatment in the frontal cortex of the rat. J. Neural Transm. (Vienna) 2003, 110, 577–590. [Google Scholar] [CrossRef]
- Gören, M.Z.; Küçükibrahimoglu, E.; Berkman, K.; Terzioglu, B. Fluoxetine partly exerts its actions through GABA: A neurochemical evidence. Neurochem. Res. 2007, 32, 1559–1565. [Google Scholar] [CrossRef]
- Murray, J.D.; Anticevic, A.; Gancsos, M.; Ichinose, M.; Corlett, P.R.; Krystal, J.H.; Wang, X.-J. Linking microcircuit dysfunction to cognitive impairment: Effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb. Cortex 2014, 24, 859–872. [Google Scholar] [CrossRef]
- Gerhard, D.M.; Pothula, S.; Liu, R.J.; Wu, M.; Li, X.Y.; Girgenti, M.J.; Taylor, S.R.; Duman, C.H.; Delpire, E.; Picciotto, M.; et al. GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. J. Clin. Invest. 2020, 130, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Miller, O.H.; Yang, L.; Wang, C.C.; Hargroder, E.A.; Zhang, Y.; Delpire, E.; Hall, B.J. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife 2014, 3, e03581. [Google Scholar] [CrossRef] [PubMed]
- López-Gil, X.; Babot, Z.; Amargós-Bosch, M.; Suñol, C.; Artigas, F.; Adell, A. Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology 2007, 32, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Akinfiresoye, L.; Tizabi, Y. Antidepressant effects of AMPA and ketamine combination: Role of hippocampal BDNF, synapsin, and mTOR. Psychopharmacology (Berl) 2013, 230, 291–298. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adell, A. Brain NMDA Receptors in Schizophrenia and Depression. Biomolecules 2020, 10, 947. https://doi.org/10.3390/biom10060947
Adell A. Brain NMDA Receptors in Schizophrenia and Depression. Biomolecules. 2020; 10(6):947. https://doi.org/10.3390/biom10060947
Chicago/Turabian StyleAdell, Albert. 2020. "Brain NMDA Receptors in Schizophrenia and Depression" Biomolecules 10, no. 6: 947. https://doi.org/10.3390/biom10060947
APA StyleAdell, A. (2020). Brain NMDA Receptors in Schizophrenia and Depression. Biomolecules, 10(6), 947. https://doi.org/10.3390/biom10060947