Role of Lipopolysaccharide in Protecting OmpT from Autoproteolysis during In Vitro Refolding

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Recombinant OmpT Expression
2.3. Refolding of OmpT IBs
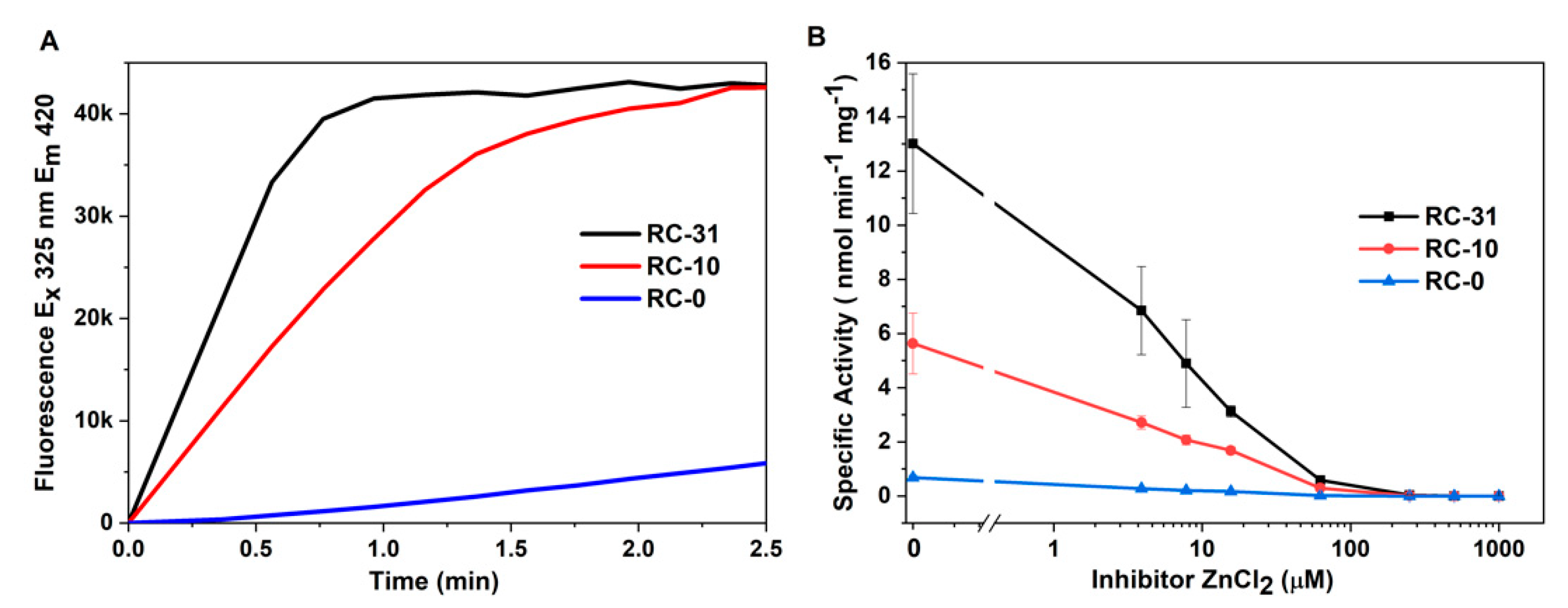
2.4. OmpT Protease Activity in the Presence of an Inhibitor
2.5. OmpT Protease Activity Assay
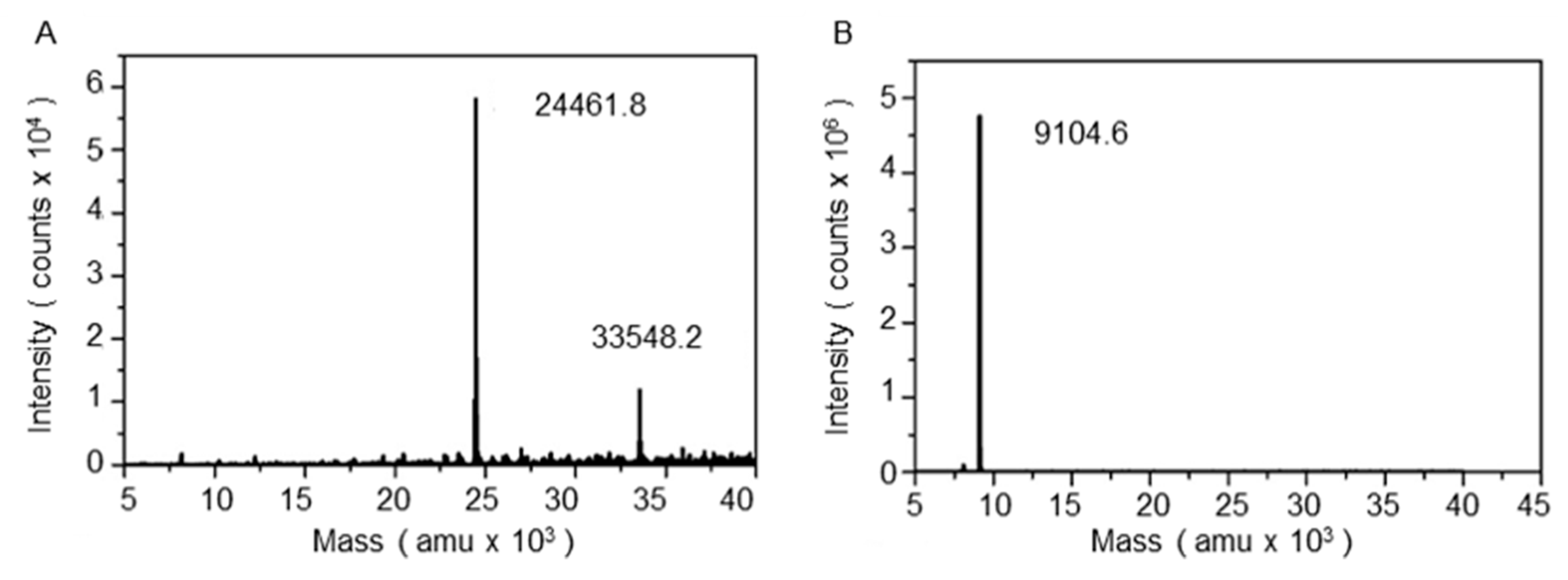
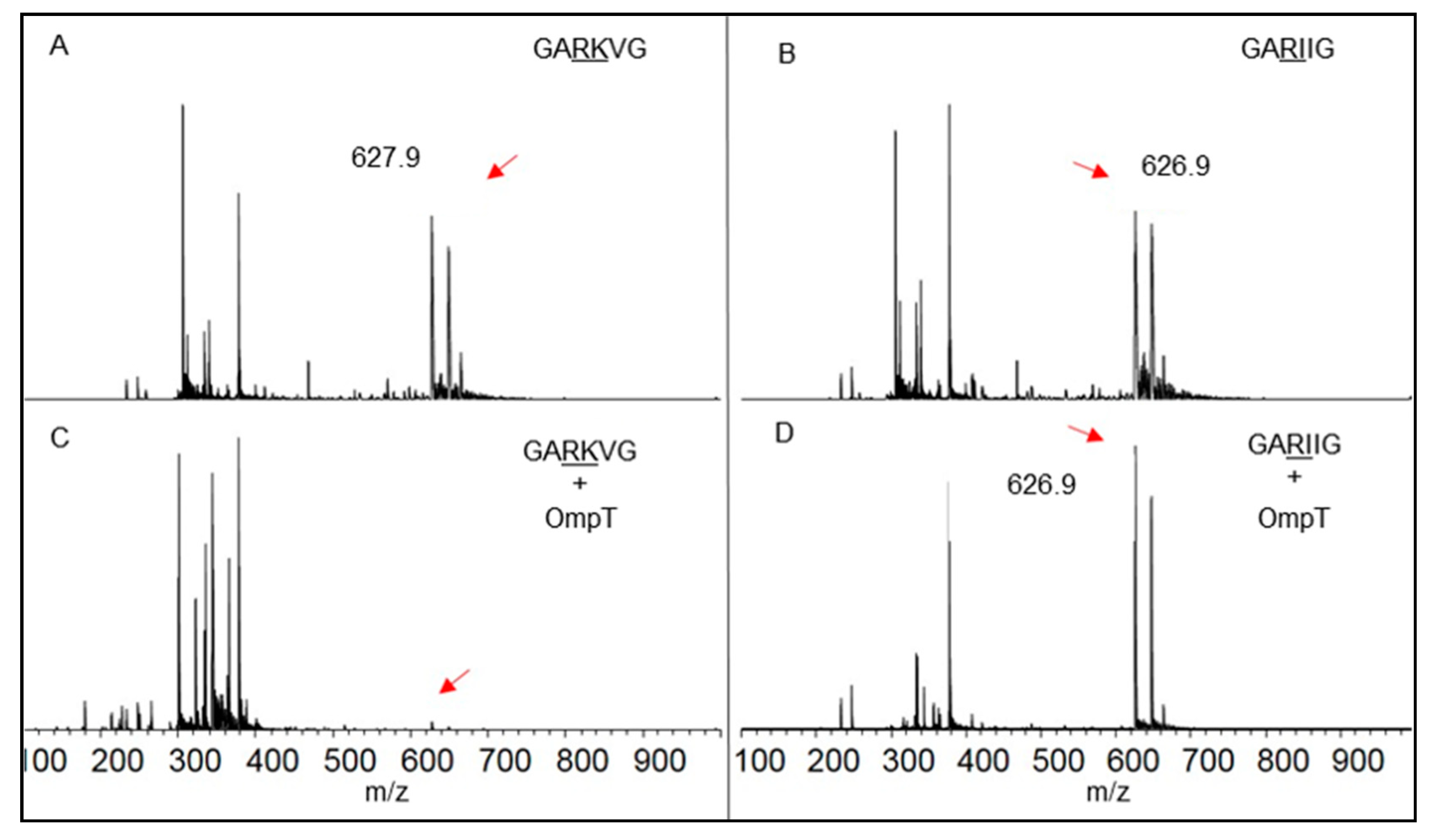
2.6. Liquid Chromatography-Mass Spectrometry (LC-MS) and MALDI-TOF
3. Results and Discussion
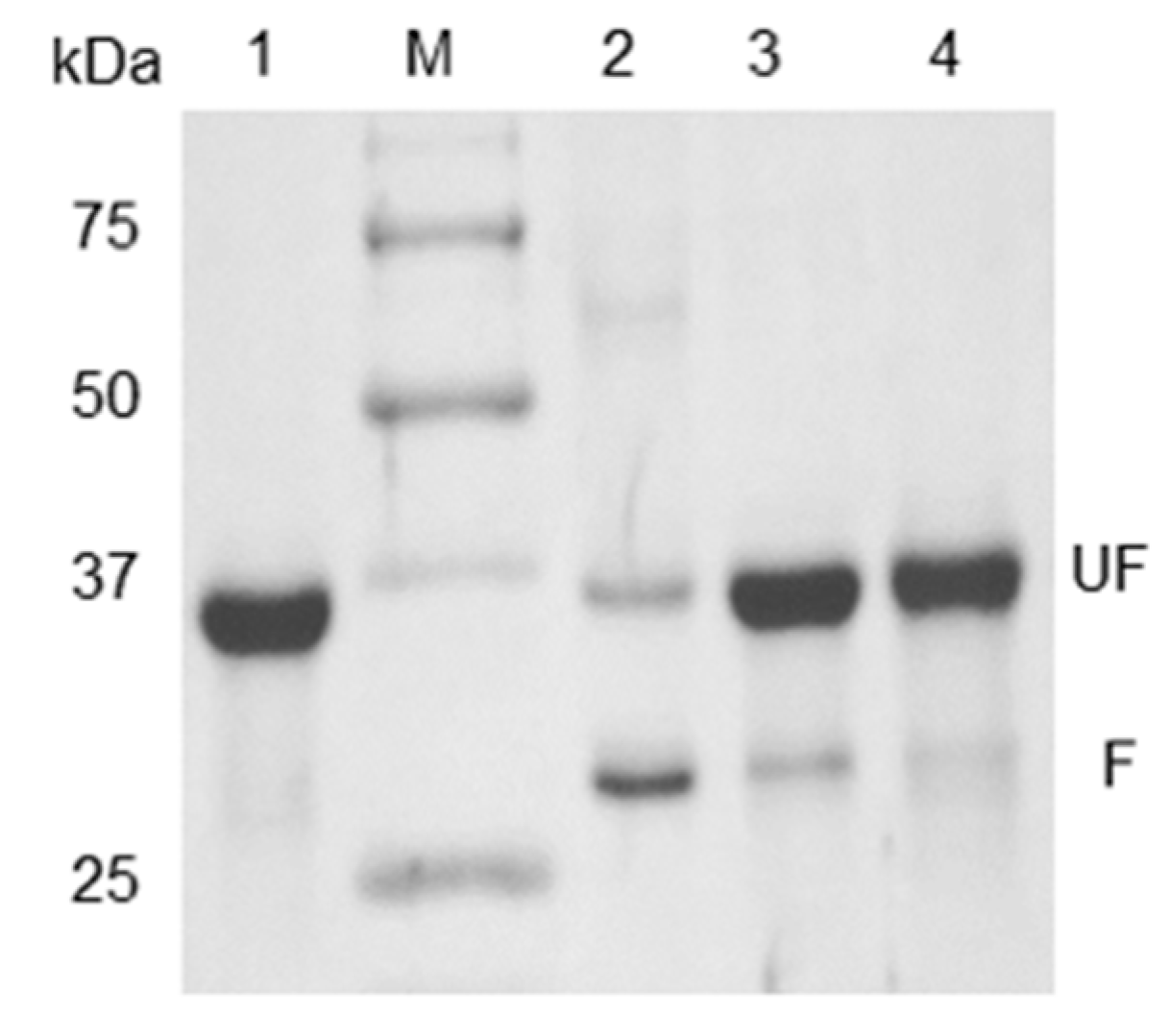
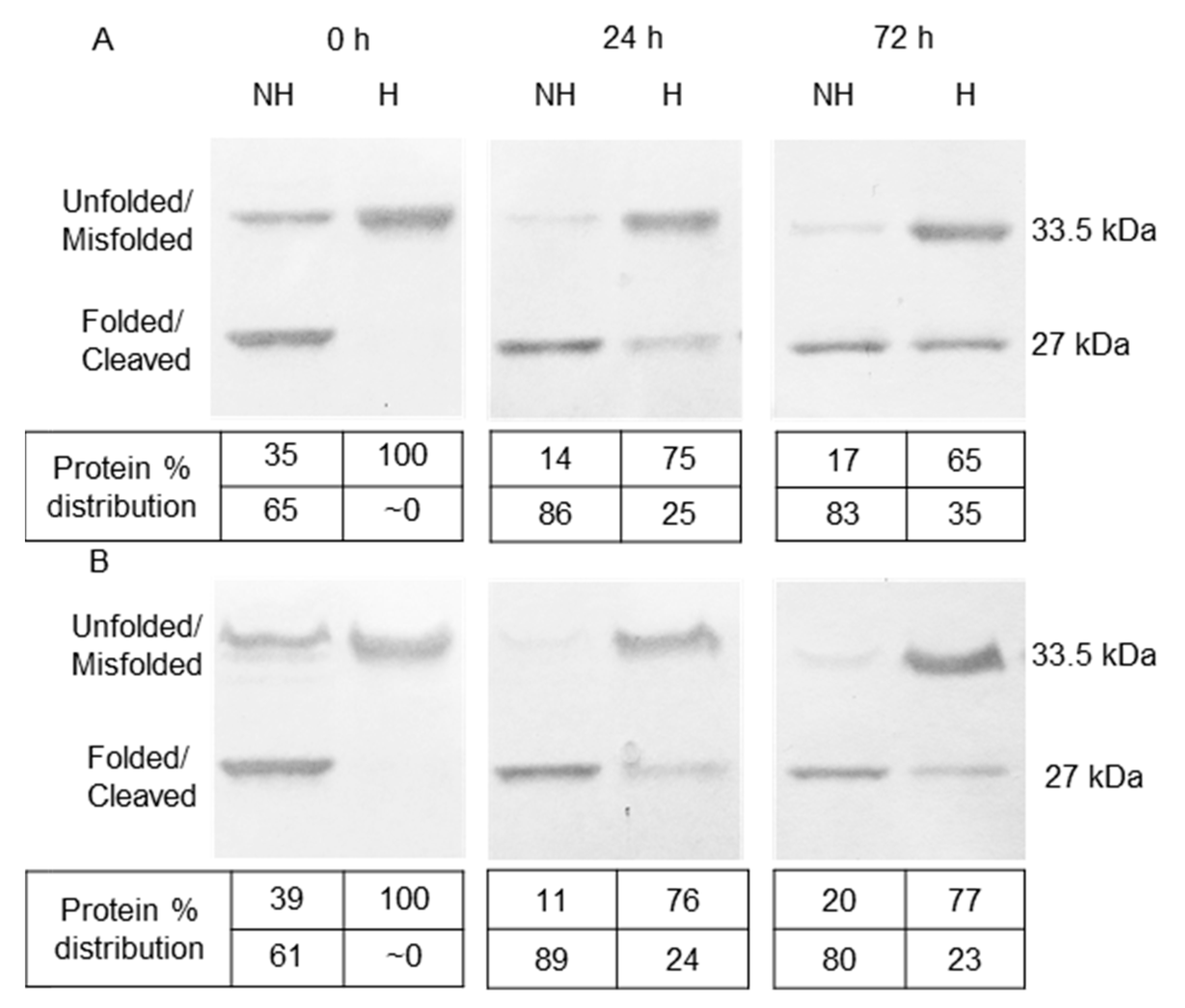
3.1. Refolding and Heat-Modifiable Migration of OmpT
3.2. Autoproteolysis of OmpT
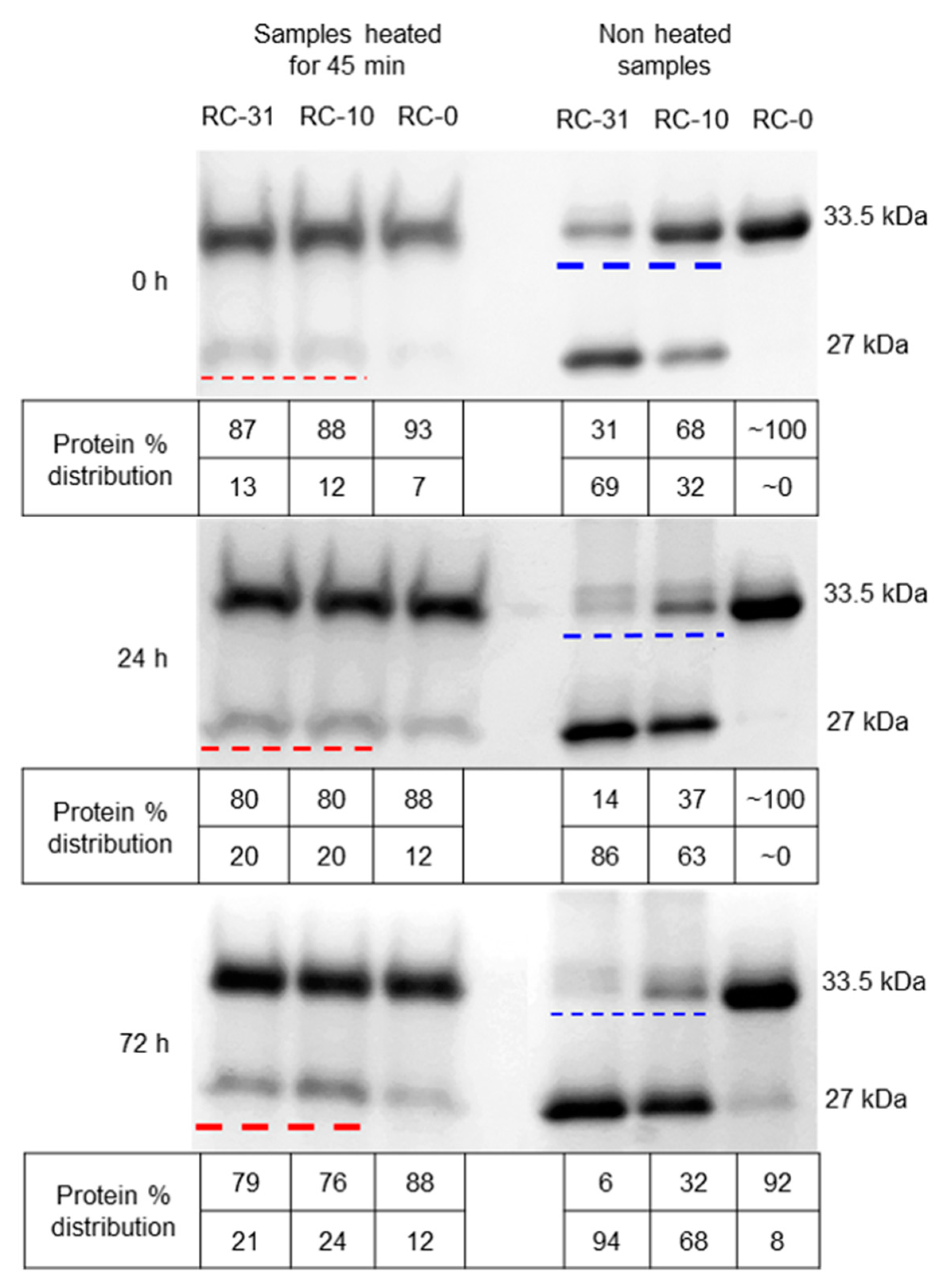
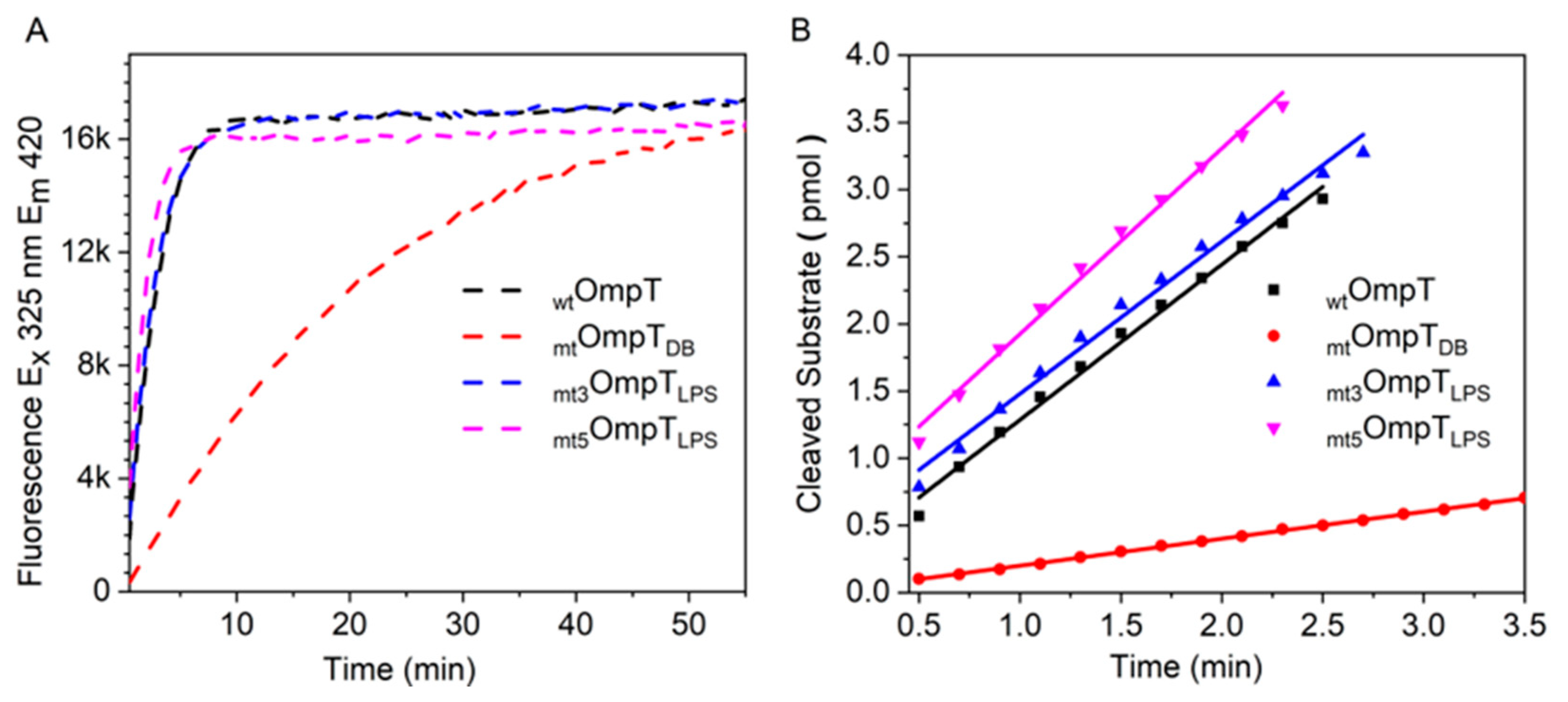
3.3. Role of LPS in Autoproteolysis of Unfolded OmpT
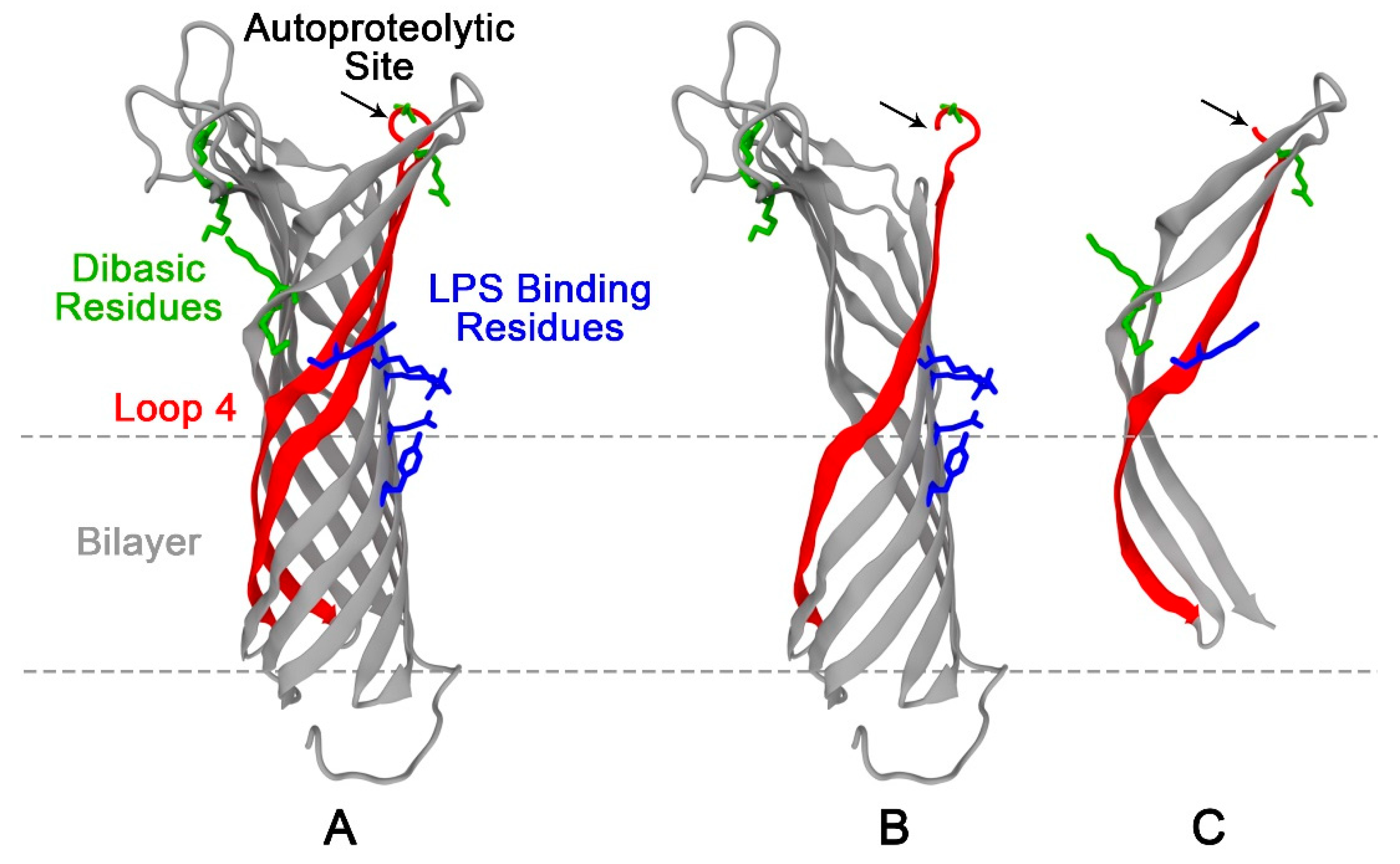
3.4. Mutation of the Predicted LPS Biding Residues in OmpT
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vandeputte-Rutten, L.; Kramer, R.A.; Kroon, J.; Dekker, N.; Egmond, M.R.; Gros, P. Crystal structure of the outer membrane protease OmpT from Escherichia coli suggests a novel catalytic site. EMBO J. 2001, 20, 5033–5039. [Google Scholar] [CrossRef] [PubMed]
- Hritonenko, V.; Stathopoulos, C. Omptin proteins: An expanding family of outer membrane proteases in Gram-negative Enterobacteriaceae. Mol. Membr. Biol. 2007, 24, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Stumpe, S.; Schmid, R.; Stephens, D.L.; Georgiou, G.; Bakker, E.P. Identification of OmpT as the protease that hydrolyzes the antimicrobial peptide protamine before it enters growing cells of Escherichia coli. J. Bacteriol. 1998, 180, 4002–4006. [Google Scholar] [CrossRef] [PubMed]
- Brannon, J.R.; Thomassin, J.-L.; Desloges, I.; Gruenheid, S.; Le Moual, H. Role of uropathogenic Escherichia coli OmpT in the resistance against human cathelicidin LL-37. FEMS Microbiol. Lett. 2013, 345, 64–71. [Google Scholar] [CrossRef]
- Hui, C.-Y.; Guo, Y.; He, Q.-S.; Peng, L.; Wu, S.-C.; Cao, H.; Huang, S.-H. Escherichia coli outer membrane protease OmpT confers resistance to urinary cationic peptides. Microbiol. Immunol. 2010, 54, 452–459. [Google Scholar] [CrossRef]
- Nielubowicz, G.R.; Mobley, H.L.T. Host-pathogen interactions in urinary tract infection. Nat. Rev. Urol. 2010, 7, 430–441. [Google Scholar] [CrossRef]
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary tract infections: Epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef]
- Thomassin, J.-L.; Brannon, J.R.; Gibbs, B.F.; Gruenheid, S.; Le Moual, H. OmpT outer membrane proteases of enterohemorrhagic and enteropathogenic Escherichia coli contribute differently to the degradation of human LL-37. Infect. Immun. 2012, 80, 483–492. [Google Scholar] [CrossRef]
- Montero, D.; Orellana, P.; Gutiérrez, D.; Araya, D.; Salazar, J.C.; Prado, V.; Oñate, Á.; Canto, F.D.; Vidal, R. Immunoproteomic Analysis To Identify Shiga Toxin-Producing Escherichia coli Outer Membrane Proteins Expressed during Human Infection. Infect Immun. 2014, 82, 4767–4777. [Google Scholar] [CrossRef]
- Hartland, E.L.; Leong, J.M. Enteropathogenic and enterohemorrhagic E. Coli: Ecology, pathogenesis, and evolution. Front. Cell. Infect. Microbiol. 2013, 3, 1–3. [Google Scholar] [CrossRef]
- Thomassin, J.L.; Brannon, J.R.; Kaiser, J.; Gruenheid, S.; Le Moual, H. Enterohemorrhagic and enteropathogenic Escherichia coli evolved different strategies to resist antimicrobial peptides. Gut Microbes. 2012, 3, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Urashima, A.; Sanou, A.; Yen, H.; Tobe, T. Enterohaemorrhagic Escherichia coli produces outer membrane vesicles as an active defence system against antimicrobial peptide LL-37. Cell. Microbiol. 2017, 19, e12758. [Google Scholar] [CrossRef] [PubMed]
- Premjani, V.; Tilley, D.; Gruenheid, S.; Le Moual, H.; Samis, J.A. Enterohemorrhagic Escherichia coli OmpT regulates outer membrane vesicle biogenesis. FEMS Microbiol. Lett. 2014, 355, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.A.; Vandeputte-Rutten, L.; De Roon, G.J.; Gros, P.; Dekker, N.; Egmond, M.R. Identification of essential acidic residues of outer membrane protease OmpT supports a novel active site. FEBS Lett. 2001, 505, 426–430. [Google Scholar] [CrossRef]
- Kramer, R.A.; Brandenburg, K.; Vandeputte-Rutten, L.; Werkhoven, M.; Gros, P.; Dekker, N.; Egmond, M.R. Lipopolysaccharide regions involved in the activation of Escherichia coli outer membrane protease OmpT. Eur. J. Biochem. 2002, 269, 1746–1752. [Google Scholar] [CrossRef]
- Sugimura, K.; Nishihara, T. Purification, characterization, and primary structure of Escherichia coli protease VII with specificity for paired basic residues: Identity of protease VII and OmpT. J. Bacteriol. 1988, 170, 5625–5632. [Google Scholar] [CrossRef]
- Dekker, N.; Cox, R.C.; Kramer, R.A.; Egmond, M.R. Substrate Specificity of the Integral Membrane Protease OmpT Determined by Spatially Addressed Peptide Libraries. Biochemistry. 2001, 40, 1694–1701. [Google Scholar] [CrossRef]
- Rollauer, S.E.; Sooreshjani, M.A.; Noinaj, N.; Buchanan, S.K. Outer membrane protein biogenesis in Gram-negative bacteria. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20150023. [Google Scholar] [CrossRef]
- Kramer, R.A.; Zandwijken, D.; Egmond, M.R.; Dekker, N. In vitro folding, purification and characterization of Escherichia coli outer membrane protease OmpT. Eur. J. Biochem. 2000, 267, 885–893. [Google Scholar] [CrossRef]
- Wood, S.E.; Sinsinbar, G.; Gudlur, S.; Nallani, M.; Huang, C.-F.F.; Liedberg, B.; Mrksich, M. A Bottom-Up Proteomic Approach to Identify Substrate Specificity of Outer-Membrane Protease OmpT. Angew. Chemie Int. Ed. 2017, 56, 16531–16535. [Google Scholar] [CrossRef]
- Wu, C.; Tran, J.C.; Zamdborg, L.; Durbin, K.R.; Li, M.; Ahlf, D.R.; Early, B.P.; Thomas, P.M.; Sweedler, J.V.; Kelleher, N.L. A protease for “middle-down” proteomics. Nat. Methods. 2012, 9, 822–824. [Google Scholar] [CrossRef] [PubMed]
- WU, C. Development and Application of Ompt Based Middle down Proteomics. Ph.D. Thesis, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 2013. [Google Scholar]
- Kudva, R.; Denks, K.; Kuhn, P.; Vogt, A.; Müller, M.; Koch, H.-G. Protein translocation across the inner membrane of Gram-negative bacteria: The Sec and Tat dependent protein transport pathways. Res. Microbiol. 2013, 164, 505–534. [Google Scholar] [CrossRef] [PubMed]
- Hirel, P.H.; Schmitter, M.J.; Dessen, P.; Fayat, G.; Blanquet, S. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc. Natl. Acad. Sci. USA 1989, 86, 8247–8251. [Google Scholar] [CrossRef] [PubMed]
- Wessel, D.; Flügge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Carrió, M.M.; Villaverde, A. Localization of chaperones DnaK and GroEL in bacterial inclusion bodies. J. Bacteriol. 2005, 187, 3599–3601. [Google Scholar] [CrossRef]
- Noinaj, N.; Kuszak, A.J.; Buchanan, S.K. Heat Modifiability of Outer Membrane Proteins from Gram-Negative Bacteria. Methods Mol. Biol. 2015, 1329, 51–56. [Google Scholar]
- Koebnik, R. Structural and Functional Roles of the Surface-Exposed Loops of the β-Barrel Membrane Protein OmpA from Escherichia coli. J. Bacteriol. 1999, 181, 3688–3694. [Google Scholar] [CrossRef]
- Sánchez, S.; Arenas, J.; Abel, A.; Criado, M.-T.; Ferreirós, C.M. Analysis of Outer Membrane Protein Complexes and Heat-Modifiable Proteins in Neisseria Strains Using Two-Dimensional Diagonal Electrophoresis. J. Proteome Res. 2005, 4, 91–95. [Google Scholar] [CrossRef]
- Minetti, C.A.; Tai, J.Y.; Blake, M.S.; Pullen, J.K.; Liang, S.M.; Remeta, D.P. Structural and functional characterization of a recombinant PorB class 2 protein from Neisseria meningitidis. Conformational stability and porin activity. J. Biol. Chem. 1997, 272, 10710–10720. [Google Scholar] [CrossRef]
- Visudtiphole, V.; Thomas, M.B.; Chalton, D.A.; Lakey, J.H. Refolding of Escherichia coli outer membrane protein F in detergent creates LPS-free trimers and asymmetric dimers. Biochem. J. 2005, 392, 375–381. [Google Scholar] [CrossRef]
- Hong, H.; Tamm, L.K. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc. Natl. Acad. Sci. USA 2004, 101, 4065–4070. [Google Scholar] [CrossRef] [PubMed]
- Tulumello, D.V.; Deber, C.M. Efficiency of detergents at maintaining membrane protein structures in their biologically relevant forms. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Anandan, A.; Vrielink, A. Detergents in membrane protein purification and crystallisation. Adv. Exp. Med. Biol. 2016, 922, 13–28. [Google Scholar] [PubMed]
- Kleinschmidt, J.H.; Wiener, M.C.; Tamm, L.K. Outer membrane protein A of E. coli folds into detergent micelles, but not in the presence of monomeric detergent. Protein Sci. 1999, 8, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Garidel, P.; Schromm, A.B.; Andrae, J.; Kramer, A.; Egmond, M.; Wiese, A.; Andra, J.; Kramer, A.; Egmond, M.; et al. Investigation into the interaction of the bacterial protease OmpT with outer membrane lipids and biological activity of OmpT:lipopolysaccharide complexes. Eur. Biophys. J. 2005, 34, 28–41. [Google Scholar] [CrossRef]
- Ebbensgaard, A.; Mordhorst, H.; Aarestrup, F.M.; Hansen, E.B. The Role of Outer Membrane Proteins and Lipopolysaccharides for the Sensitivity of Escherichia coli to Antimicrobial Peptides. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Eren, E.; Van Den Berg, B. Structural basis for activation of an integral membrane protease by lipopolysaccharide. J. Biol. Chem. 2012, 287, 23971–23976. [Google Scholar] [CrossRef]
- Kukkonen, M.; Suomalainen, M.; Kyllönen, P.; Lähteenmäki, K.; Lång, H.; Virkola, R.; Helander, I.M.; Holst, O.; Korhonen, T.K.; Kyllonen, P.; et al. Lack of O-antigen is essential for plasminogen activation by Yersinia pestis and Salmonella enterica. Mol. Microbiol. 2004, 51, 215–225. [Google Scholar] [CrossRef]
- Ongkudon, C.M.; Chew, J.H.; Liu, B.; Danquah, M.K. Chromatographic Removal of Endotoxins: A Bioprocess Engineer’s Perspective. ISRN Chromatogr. 2012, 2012, 1–9. [Google Scholar] [CrossRef][Green Version]
- Ferguson, A.D.; Welte, W.; Hofmann, E.; Lindner, B.; Holst, O.; Coulton, J.W.; Diederichs, K. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure 2000, 8, 585–592. [Google Scholar] [CrossRef]
- Kucharska, I.; Liang, B.; Ursini, N.; Tamm, L.K. Molecular Interactions of Lipopolysaccharide with an Outer Membrane Protein from Pseudomonas aeruginosa Probed by Solution NMR. Biochemistry 2016, 55, 5061–5072. [Google Scholar] [CrossRef] [PubMed]
- Edrington, T.C.; Kintz, E.; Goldberg, J.B.; Tamm, L.K. Structural basis for the interaction of lipopolysaccharide with outer membrane protein H (OprH) from Pseudomonas aeruginosa. J. Biol. Chem. 2011, 286, 39211–39223. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OmpT I.D. | Residues Mutated * | Specific Activity (pmol min−1 mg−1) |
|---|---|---|
| OmpT | None | 1.3 × 104 |
| mtOmpTDB | G216K and K217G | 1.6 × 103 |
| mt3OmpTLPS | R138E, R175E, K226E | 1.5 × 104 |
| mt5OmpTLPS | Y134A, E136A, R138E, R175E, K226E | 1.6 × 104 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinsinbar, G.; Gudlur, S.; Metcalf, K.J.; Mrksich, M.; Nallani, M.; Liedberg, B. Role of Lipopolysaccharide in Protecting OmpT from Autoproteolysis during In Vitro Refolding. Biomolecules 2020, 10, 922. https://doi.org/10.3390/biom10060922
Sinsinbar G, Gudlur S, Metcalf KJ, Mrksich M, Nallani M, Liedberg B. Role of Lipopolysaccharide in Protecting OmpT from Autoproteolysis during In Vitro Refolding. Biomolecules. 2020; 10(6):922. https://doi.org/10.3390/biom10060922
Chicago/Turabian StyleSinsinbar, Gaurav, Sushanth Gudlur, Kevin J Metcalf, Milan Mrksich, Madhavan Nallani, and Bo Liedberg. 2020. "Role of Lipopolysaccharide in Protecting OmpT from Autoproteolysis during In Vitro Refolding" Biomolecules 10, no. 6: 922. https://doi.org/10.3390/biom10060922
APA StyleSinsinbar, G., Gudlur, S., Metcalf, K. J., Mrksich, M., Nallani, M., & Liedberg, B. (2020). Role of Lipopolysaccharide in Protecting OmpT from Autoproteolysis during In Vitro Refolding. Biomolecules, 10(6), 922. https://doi.org/10.3390/biom10060922