PSIONplusm Server for Accurate Multi-Label Prediction of Ion Channels and Their Types





Abstract

1. Introduction
2. Materials and Methods
2.1. Benchmark Dataset and Annotation of Ion Channel Types and Subtypes
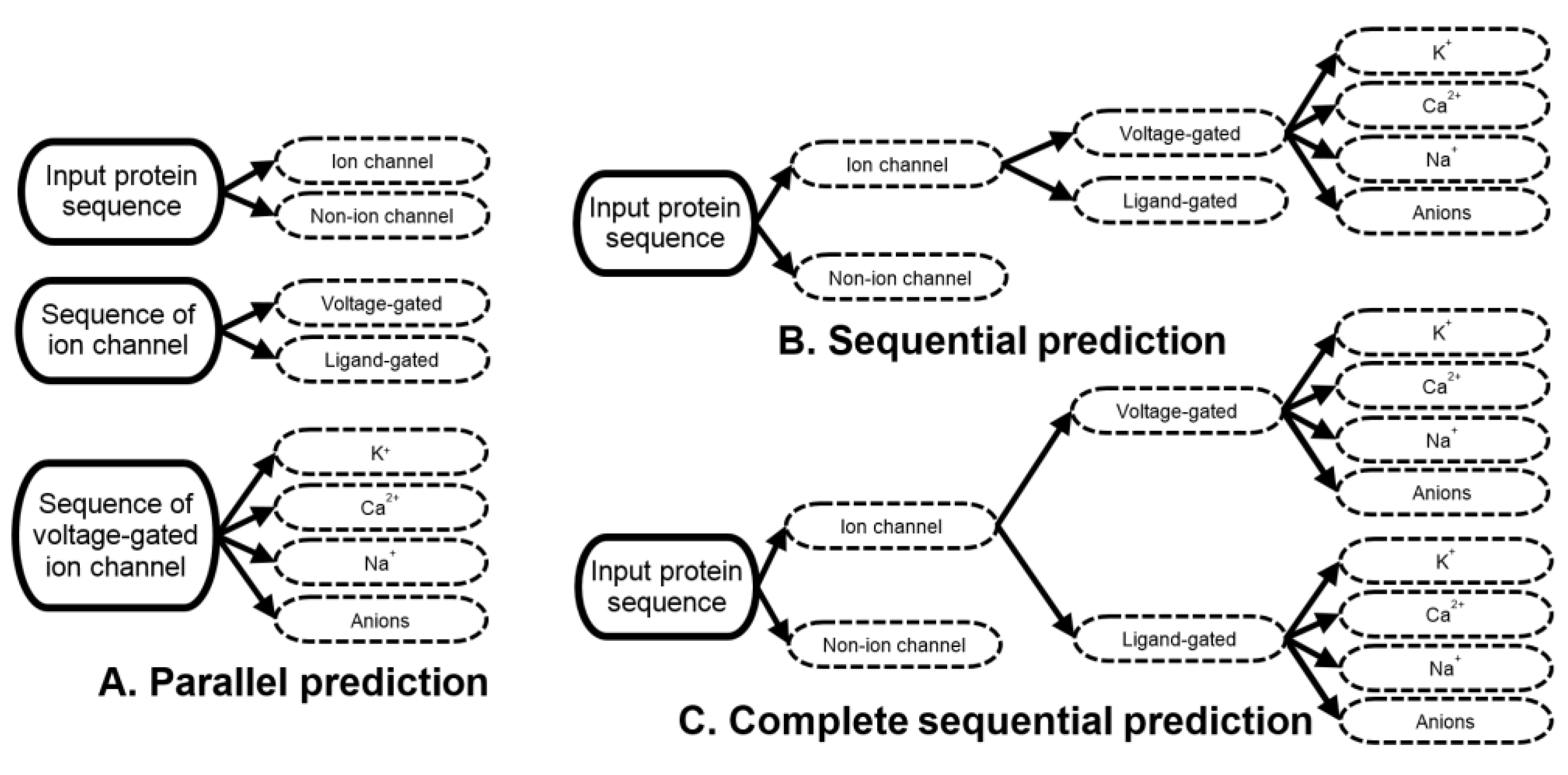
2.2. Sequential Multi-Label Prediction
2.3. Evaluation of the Predictive Performance
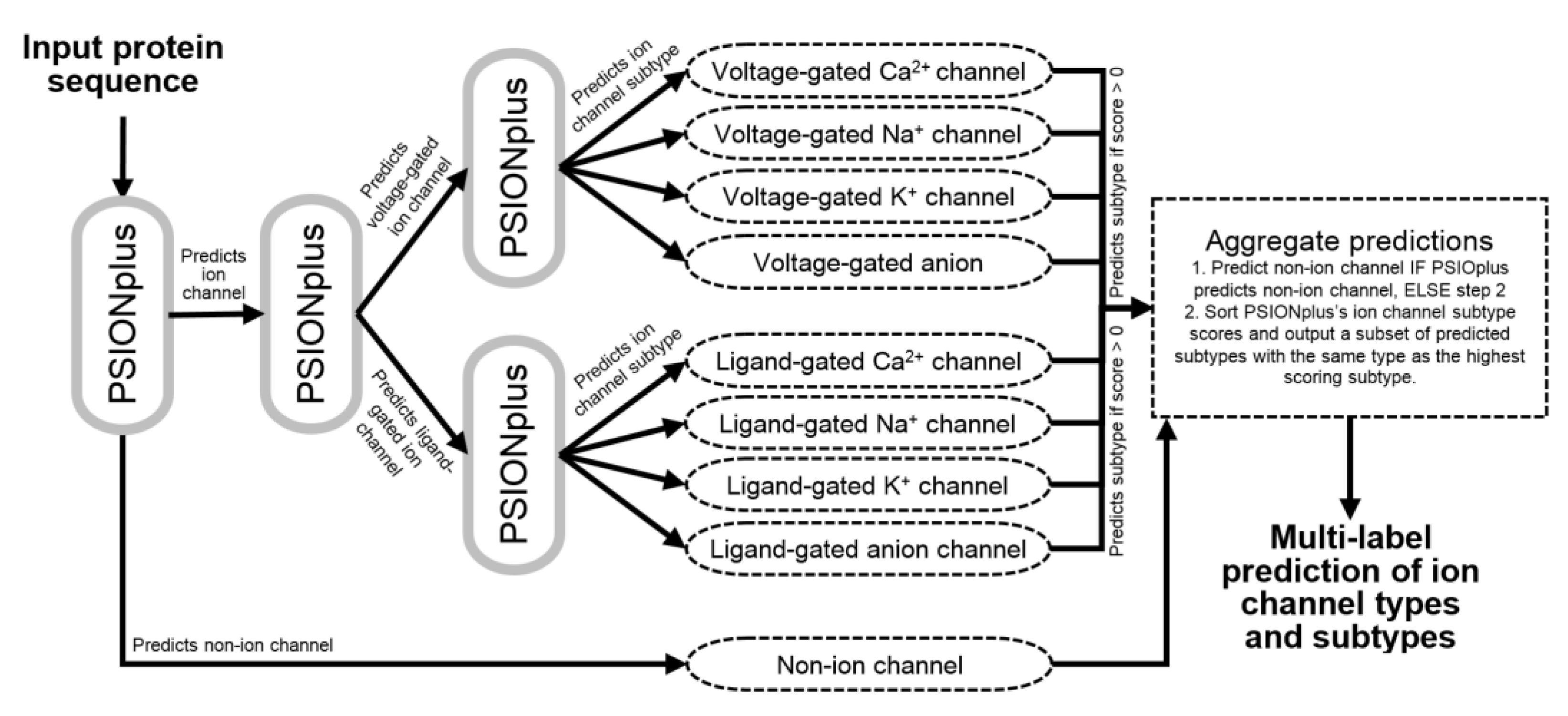
2.4. Architecture of the PSIONplusm Predictor
3. Results
3.1. Ion Channels and Their Types and Subtypes Are Hard to Predict Directly from the Sequence
3.2. Comparative Assessment of PSIONplusm
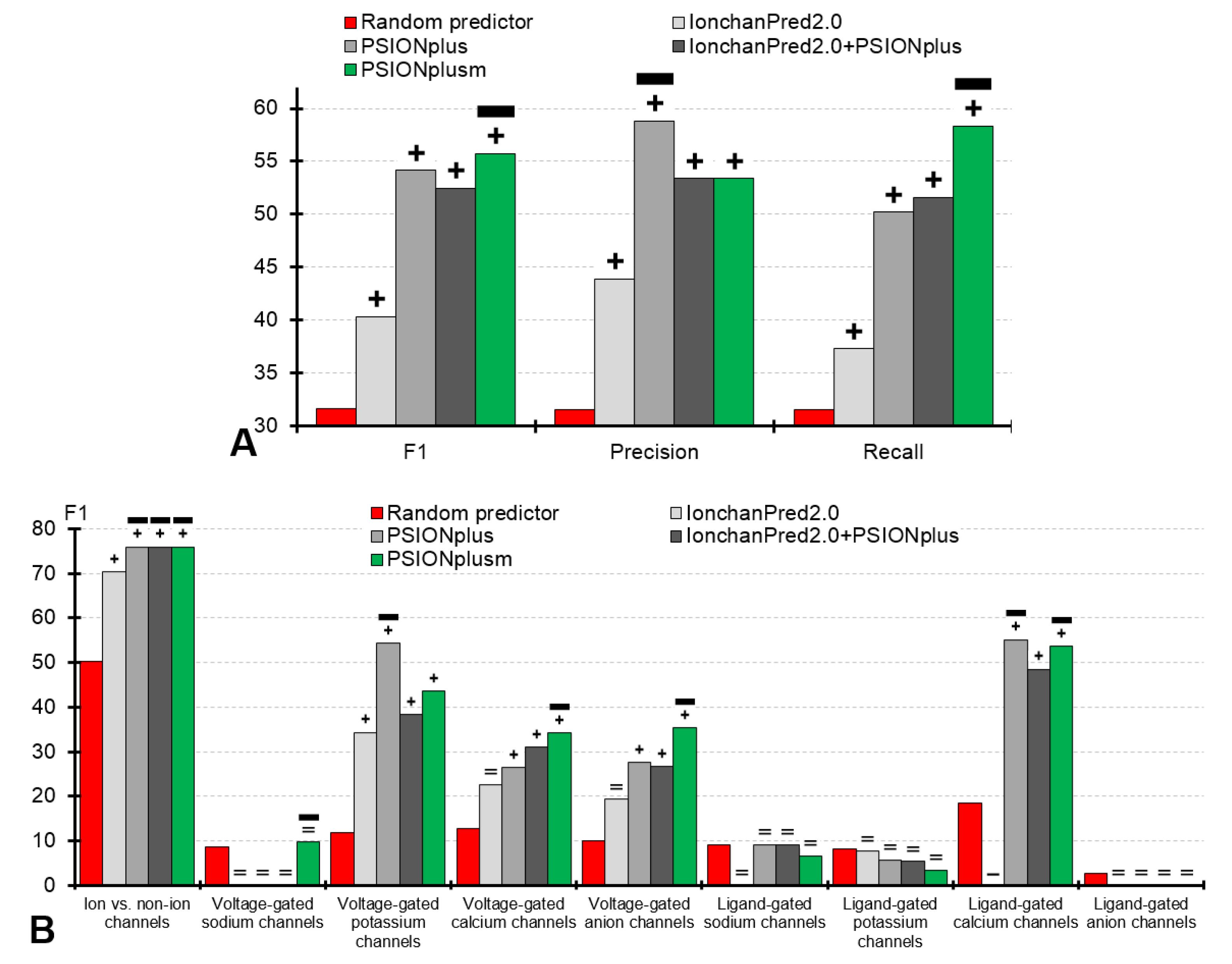
3.2.1. Assessment of the Overall Multi-label Prediction of the Ion Channels and Their Types and Subtypes
3.2.2. Assessment of the Prediction of the Ion Channels
3.2.3. Assessment of the Prediction of the Ion Channel Subtypes
4. PSIONplusm Webserver
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sands, Z.; Grottesi, A.; Sansom, M.S. Voltage-gated ion channels. Curr. Biol. 2005, 15, R44–R47. [Google Scholar] [CrossRef] [PubMed]
- Hucho, F.; Weise, C. Ligand-gated ion channels. Angew. Chem. Int. Ed. Engl. 2001, 40, 3100–3116. [Google Scholar] [CrossRef]
- Tabassum, N.; Ahmed, F. Ion channels and their modulation. Eur. J. Pharm. Sci. 2011, 1, 20–25. [Google Scholar]
- Bockenhauer, D. Ion channels in disease. Curr. Opin. Pediatr. 2001, 13, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef]
- Panyi, G.; Beeton, C.; Felipe, A. Ion channels and anti-cancer immunity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130106. [Google Scholar] [CrossRef]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef]
- Kaczorowski, G.J.; McManus, O.B.; Priest, B.T.; Garcia, M.L. Ion channels as drug targets: The next gpcrs. J. Gen. Physiol. 2008, 131, 399–405. [Google Scholar] [CrossRef]
- Waszkielewicz, A.M.; Gunia, A.; Szkaradek, N.; Sloczynska, K.; Krupinska, S.; Marona, H. Ion channels as drug targets in central nervous system disorders. Curr. Med. Chem. 2013, 20, 1241–1285. [Google Scholar] [CrossRef]
- McGivern, J.G. Advantages of voltage-gated ion channels as drug targets. Expert Opin. Ther. Targets 2007, 11, 265–271. [Google Scholar] [CrossRef]
- Bagal, S.K.; Brown, A.D.; Cox, P.J.; Omoto, K.; Owen, R.M.; Pryde, D.C.; Sidders, B.; Skerratt, S.E.; Stevens, E.B.; Storer, R.I.; et al. Ion channels as therapeutic targets: A drug discovery perspective. J. Med. Chem. 2013, 56, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Miao, Z.; Zhang, Z.; Wei, H.; Kurgan, L. Prediction of ion channels and their types from protein sequences: Comprehensive review and comparative assessment. Curr. Drug Targets 2019, 20, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. Uniprot: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Liu, L.X.; Li, M.L.; Tan, F.Y.; Lu, M.C.; Wang, K.L.; Guo, Y.Z.; Wen, Z.N.; Jiang, L. Local sequence information-based support vector machine to classify voltage-gated potassium channels. Acta Biochim. Biophys. Sin. 2006, 38, 363–371. [Google Scholar] [CrossRef]
- Chen, W.; Lin, H. Identification of voltage-gated potassium channel subfamilies from sequence information using support vector machine. Comput. Biol. Med. 2012, 42, 504–507. [Google Scholar] [CrossRef]
- Liu, W.; Deng, E.; Chen, W.; Lin, H. Identifying the subfamilies of voltage-gated potassium channels using feature selection technique. Int. J. Mol. Sci. 2014, 15, 12940–12951. [Google Scholar] [CrossRef]
- Saha, S.; Zack, J.; Singh, B.; Raghava, G. Vgichan: Prediction and classification of voltage-gated ion channels. Genom. Proteom. Bioinform. 2006, 4, 253–258. [Google Scholar] [CrossRef]
- Lin, H.; Ding, H. Predicting ion channels and their types by the dipeptide mode of pseudo amino acid composition. J. Theor. Biol. 2011, 269, 64–69. [Google Scholar] [CrossRef]
- Zhao, Y.W.; Su, Z.D.; Yang, W.; Lin, H.; Chen, W.; Tang, H. Ionchanpred 2.0: A tool to predict ion channels and their types. Int. J. Mol. Sci. 2017, 18, 1838. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Srivastava, R. An efficient approach for the prediction of ion channels and their subfamilies. Comput. Biol. Chem. 2015, 58, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Wang, M.; Zhang, L.; Wang, Y.; Guo, M.; Zhao, M.; Zhao, Q.; Zhang, Y.; Zeng, N.; Wang, C. Predicting ion channels genes and their types with machine learning techniques. Front. Genet. 2019, 10, 399. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cui, W.; Sheng, Y.; Ruan, J.; Kurgan, L. Psionplus: Accurate sequence-based predictor of ion channels and their types. PLoS ONE 2016, 11, e0152964. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.G.O. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. Cd-hit: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. Cd-hit suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Miao, Z.; Westhof, E. A large-scale assessment of nucleic acids binding site prediction programs. PLoS Comput. Biol. 2015, 11, e1004639. [Google Scholar] [CrossRef]
- Peng, Z.; Kurgan, L. High-throughput prediction of rna, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015, 43, e121. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, Z.; Kurgan, L. Comprehensive review and empirical analysis of hallmarks of DNA-, rna- and protein-binding residues in protein chains. Brief. Bioinform. 2019, 20, 1250–1268. [Google Scholar] [CrossRef]
- Yan, J.; Friedrich, S.; Kurgan, L. A comprehensive comparative review of sequence-based predictors of DNA- and rna-binding residues. Brief. Bioinform. 2016, 17, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, Y.; Zhou, Y. Prediction of rna binding proteins comes of age from low resolution to high resolution. Mol. Biosyst. 2013, 9, 2417–2425. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.M.; Pan, X.Y.; Xu, C.; Shen, H.B. Computational prediction of DNA-protein interactions: A review. Curr. Comput. Aided Drug Des. 2010, 6, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Walia, R.R.; EL-Manzalawy, Y.; Honavar, V.G.; Dobbs, D. Sequence-based prediction of rna-binding residues in proteins. Predict. Protein Second. Struct. 2017, 1484, 205–235. [Google Scholar]
- Yan, J.; Kurgan, L. Drnapred, fast sequence-based method that accurately predicts and discriminates DNA- and rna-binding residues. Nucleic Acids Res. 2017, 45, e84. [Google Scholar] [CrossRef]
- Zhang, J.; Kurgan, L. Review and comparative assessment of sequence-based predictors of protein-binding residues. Brief. Bioinform. 2018, 19, 821–837. [Google Scholar] [CrossRef]
- Zhang, M.L.; Zhou, Z.H. A review on multi-label learning algorithms. IEEE Trans. Knowl. Data Eng. 2014, 26, 1819–1837. [Google Scholar] [CrossRef]
- Cerri, R.; Barros, R.C.; de Carvalho, A.C.; Jin, Y. Reduction strategies for hierarchical multi-label classification in protein function prediction. BMC Bioinform. 2016, 17, 373. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. Mem-adsvm: A two-layer multi-label predictor for identifying multi-functional types of membrane proteins. J. Theor. Biol. 2016, 398, 32–42. [Google Scholar] [CrossRef]
- Stojanova, D.; Ceci, M.; Malerba, D.; Dzeroski, S. Using ppi network autocorrelation in hierarchical multi-label classification trees for gene function prediction. BMC Bioinform. 2013, 14, 285. [Google Scholar] [CrossRef]
- Guo, X.; Liu, F.; Ju, Y.; Wang, Z.; Wang, C. Human protein subcellular localization with integrated source and multi-label ensemble classifier. Sci. Rep. 2016, 6, 28087. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-Y.; Yang, F.; Shen, H.-B. Incorporating organelle correlations into semi-supervised learning for protein subcellular localization prediction. Bioinformatics (Oxf. Engl.) 2016, 32, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Duan, Y.; Zou, Q. Hpslpred: An ensemble multi-label classifier for human protein subcellular location prediction with imbalanced source. Proteomics 2017, 17, 1700262. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Amidi, S.; Amidi, A.; Vlachakis, D.; Paragios, N.; Zacharaki, E.I. Automatic single- and multi-label enzymatic function prediction by machine learning. PeerJ 2017, 5, e3095. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Type | Ion Channel Type | Ion Channel Subtype | Number of Proteins |
|---|---|---|---|
| Ion channels | Voltage-gated | Sodium (Na+) | 19 |
| Potassium (K+) | 26 | ||
| Calcium (Ca2+) | 28 | ||
| Anions | 22 | ||
| Ligand-gated | Sodium (Na+) | 20 | |
| Potassium (K+) | 18 | ||
| Calcium (Ca2+) | 41 | ||
| Anions | 6 | ||
| Non-ion channels (other types of membrane proteins) | 111 | ||
| Total number of proteins | 221 | ||
| Prediction Target/Label | % of Proteins with at Least One Pfam Domain | Average Rate of Correct Predictions on Benchmark Dataset | |
|---|---|---|---|
| PSIONplus Training Dataset | Benchmark Dataset | ||
| Non-ion channels | 93.0% | 95.5% | 92.8% |
| Voltage-gated sodium channels | 100.0% | 94.7% | 58.9% |
| Voltage-gated potassium channels | 100.0% | 96.2% | 49.4% |
| Voltage-gated calcium channels | 96.6% | 96.4% | 14.3% |
| Voltage-gated anion channels | 90.9% | 77.3% | 13.6% |
| Ligand-gated sodium channels | 100.0% | 100.0% | 64.8% |
| Ligand-gated potassium channels | 100.0% | 100.0% | 71.7% |
| Ligand-gated calcium channels | 100.0% | 100.0% | 4.9% |
| Ligand-gated anion channels | 100.0% | 100.0% | 0.0% |
| Prediction Target/Label | Measure | Predictors | ||||
|---|---|---|---|---|---|---|
| Random | IonchanPred2.0 | PSIONplus | IonchanPred2.0+ PSIONplus | PSIONplusm | ||
| Overall (multi-label prediction of ion channels and their types) | F1 | 31.6 | 40.3 +/− a | 54.1 +/− | 52.5 +/− | 55.7 + |
| Accuracy | 30.6 | 37.3 +/− | 50.2 +/+ | 46.6 +/− | 47.1 + | |
| Precision | 31.6 | 43.9 +/− | 58.8 +/+ | 53.4 +/= | 53.4 + | |
| Recall | 31.6 | 37.3 +/− | 50.2 +/− | 51.6 +/− | 58.3 + | |
| Ion vs. Non-ion channels | F1 | 50.2 | 70.4 +/− | 76.0+/= | 76.0+/= | 76.0+ |
| Precision | 0.2 | 81.2 +/= | 81.4+/= | 81.4+/= | 81.4+ | |
| Recall | 50.2 | 62.2 +/− | 71.2+/= | 71.2+/= | 71.2+ | |
| Voltage-gated sodium channels | F1 | 8.6 | 0.0 =/- | 0.0 =/- | 0.0 =/- | 9.7= |
| Precision | 8.6 | 0.0 =/- | 0.0 =/- | 0.0 =/- | 7.0 = | |
| Recall | 8.6 | 0.0 =/- | 0.0 =/- | 0.0 =/- | 15.8= | |
| Voltage-gated potassium channels | F1 | 11.8 | 34.3 +/− | 54.3+/+ | 38.3 +/− | 43.6 + |
| Precision | 11.8 | 22.8 =/- | 40.0+/+ | 24.5 +/− | 29.3 + | |
| Recall | 11.8 | 69.2 +/− | 84.6 +/= | 88.5+/+ | 84.6 + | |
| Voltage-gated calcium channels | F1 | 12.7 | 22.6 =/- | 26.4 +/− | 31.0 +/− | 34.2+ |
| Precision | 12.7 | 24.0 =/= | 28.0+/= | 25.6 +/− | 27.1 + | |
| Recall | 12.7 | 21.4 =/- | 25.0 =/- | 39.3 +/− | 46.4+ | |
| Voltage-gated anion channels | F1 | 10.0 | 19.4 =/- | 27.6 +/− | 26.7 +/− | 35.3+ |
| Precision | 10.0 | 33.3 +/= | 57.1+/+ | 50.0 +/+ | 26.1 + | |
| Recall | 10.0 | 13.6 =/- | 18.2 =/- | 18.2 =/- | 54.5+ | |
| Ligand-gated sodium channels | F1 | 9.0 | 0.0 =/= | 9.1=/= | 9.1=/= | 6.6 = |
| Precision | 9.0 | 0.0 =/- | 50.0+/+ | 50.0+/+ | 4.9 = | |
| Recall | 9.0 | 0.0 =/- | 5.0 =/- | 5.0 =/- | 10.0= | |
| Ligand-gated potassium channels | F1 | 8.1 | 7.7 =/+ | 5.7 =/+ | 5.4 =/+ | 3.4 = |
| Precision | 8.1 | 12.5=/+ | 5.9 =/+ | 5.3 =/+ | 2.4 = | |
| Recall | 8.1 | 5.6 =/= | 5.6 =/= | 5.6 =/= | 5.6 = | |
| Ligand-gated calcium channels | F1 | 18.5 | 0.0 −/− | 55.2+/= | 48.5 +/− | 53.7 + |
| Precision | 18.5 | 0.0 −/− | 94.1+/+ | 64.0 +/+ | 53.7 + | |
| Recall | 18.5 | 0.0 −/− | 39.0 +/− | 39.0 +/− | 53.7+ | |
| Ligand-gated anion channels | F1 | 2.7 | 0.0 =/= | 0.0 =/= | 0.0 =/= | 0.0 = |
| Precision | 2.7 | 0.0 =/= | 0.0 =/= | 0.0 =/= | 0.0 = | |
| Recall | 2.7 | 0.0 =/= | 0.0 =/= | 0.0 =/= | 0.0 = | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Wei, H.; Cano, A.; Kurgan, L. PSIONplusm Server for Accurate Multi-Label Prediction of Ion Channels and Their Types. Biomolecules 2020, 10, 876. https://doi.org/10.3390/biom10060876
Gao J, Wei H, Cano A, Kurgan L. PSIONplusm Server for Accurate Multi-Label Prediction of Ion Channels and Their Types. Biomolecules. 2020; 10(6):876. https://doi.org/10.3390/biom10060876
Chicago/Turabian StyleGao, Jianzhao, Hong Wei, Alberto Cano, and Lukasz Kurgan. 2020. "PSIONplusm Server for Accurate Multi-Label Prediction of Ion Channels and Their Types" Biomolecules 10, no. 6: 876. https://doi.org/10.3390/biom10060876
APA StyleGao, J., Wei, H., Cano, A., & Kurgan, L. (2020). PSIONplusm Server for Accurate Multi-Label Prediction of Ion Channels and Their Types. Biomolecules, 10(6), 876. https://doi.org/10.3390/biom10060876