Expedition into Taurine Biology: Structural Insights and Therapeutic Perspective of Taurine in Neurodegenerative Diseases


 , ,
, ,
Abstract
1. Introduction
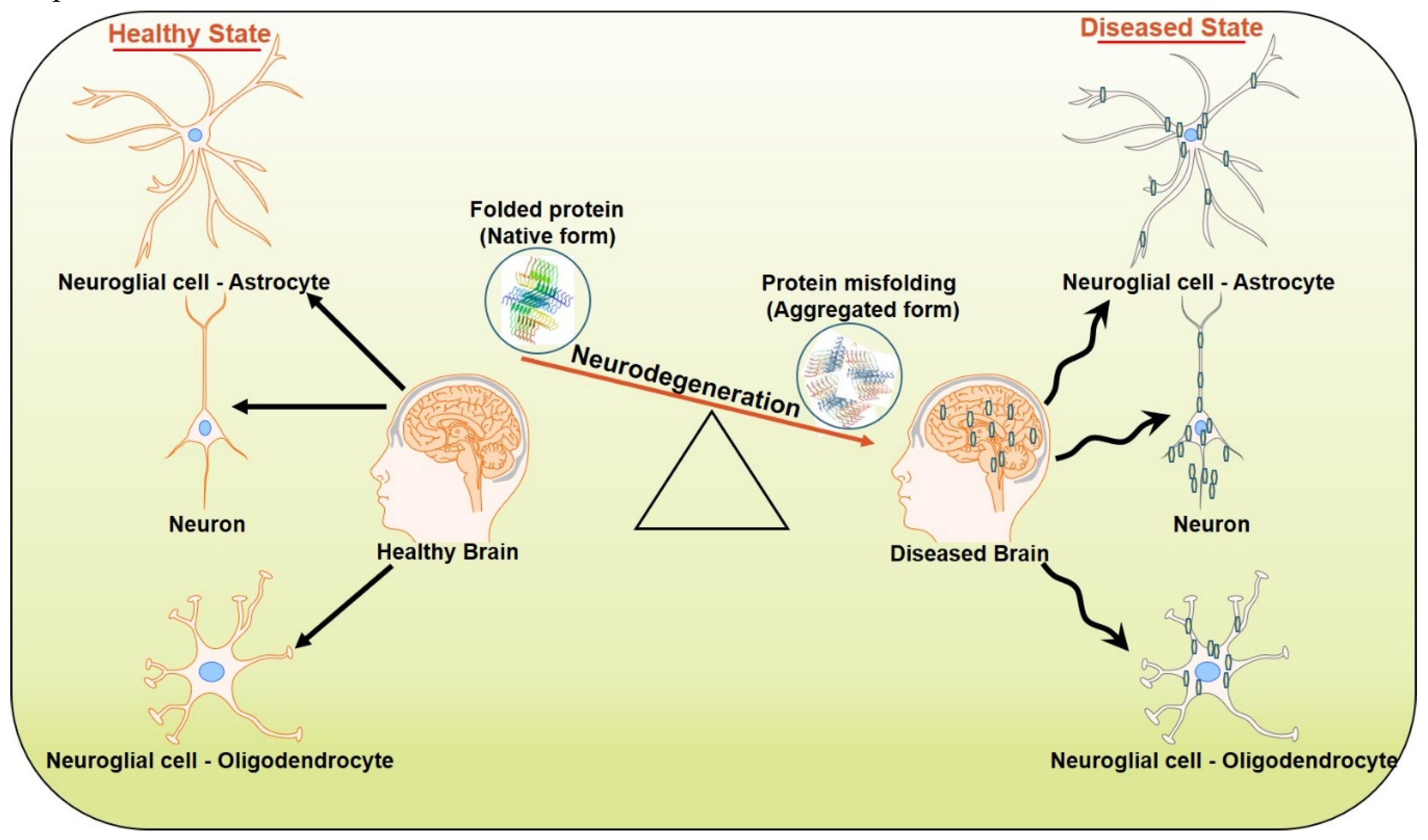
2. Neurodegeneration
2.1. Mitochondrial Dysfunction
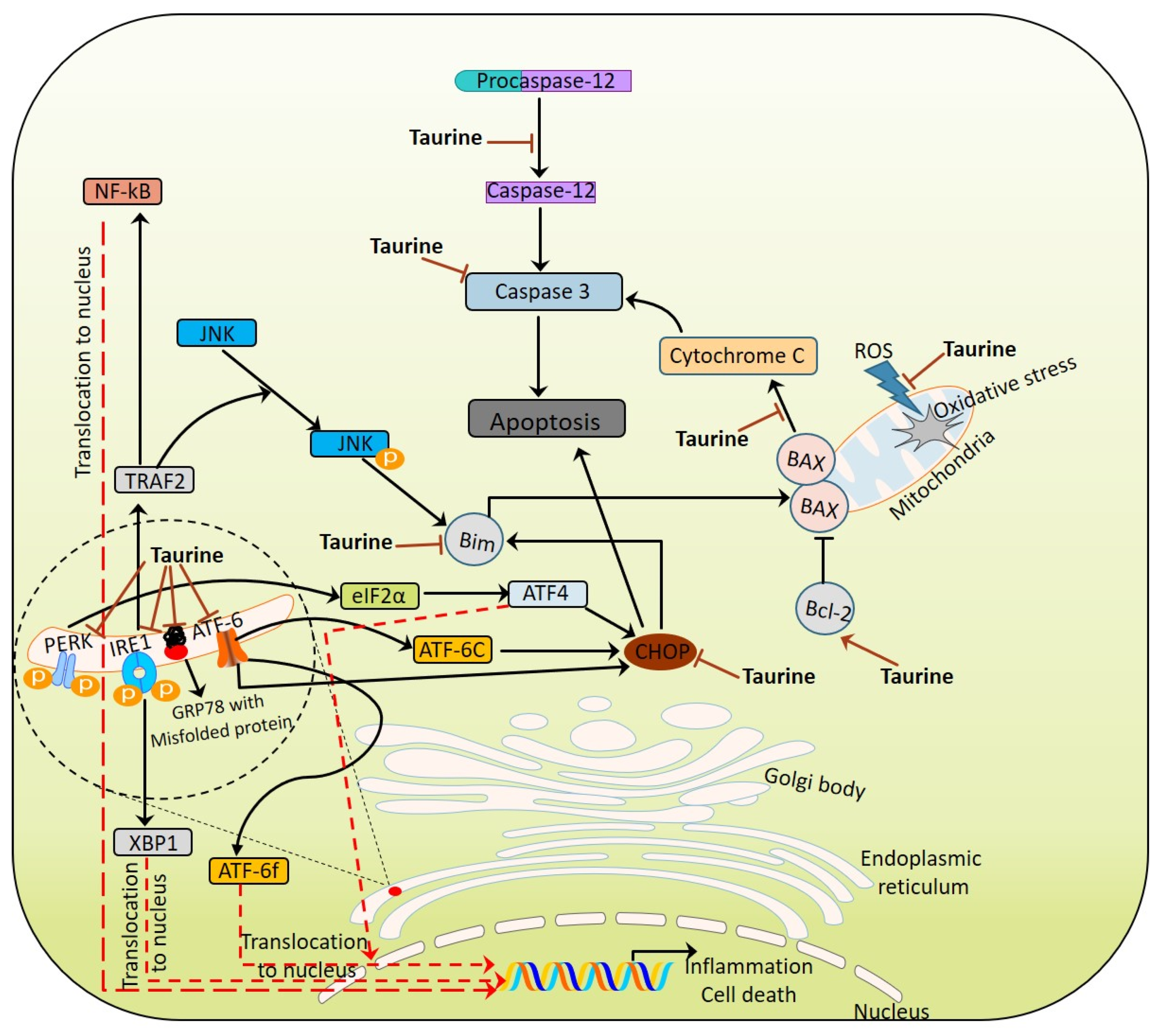
2.2. ER Stress
2.3. Neuroinflamation.
2.4. Synaptic Loss
3. Taurine—A Savior
4. Structure and Physiochemical Properties
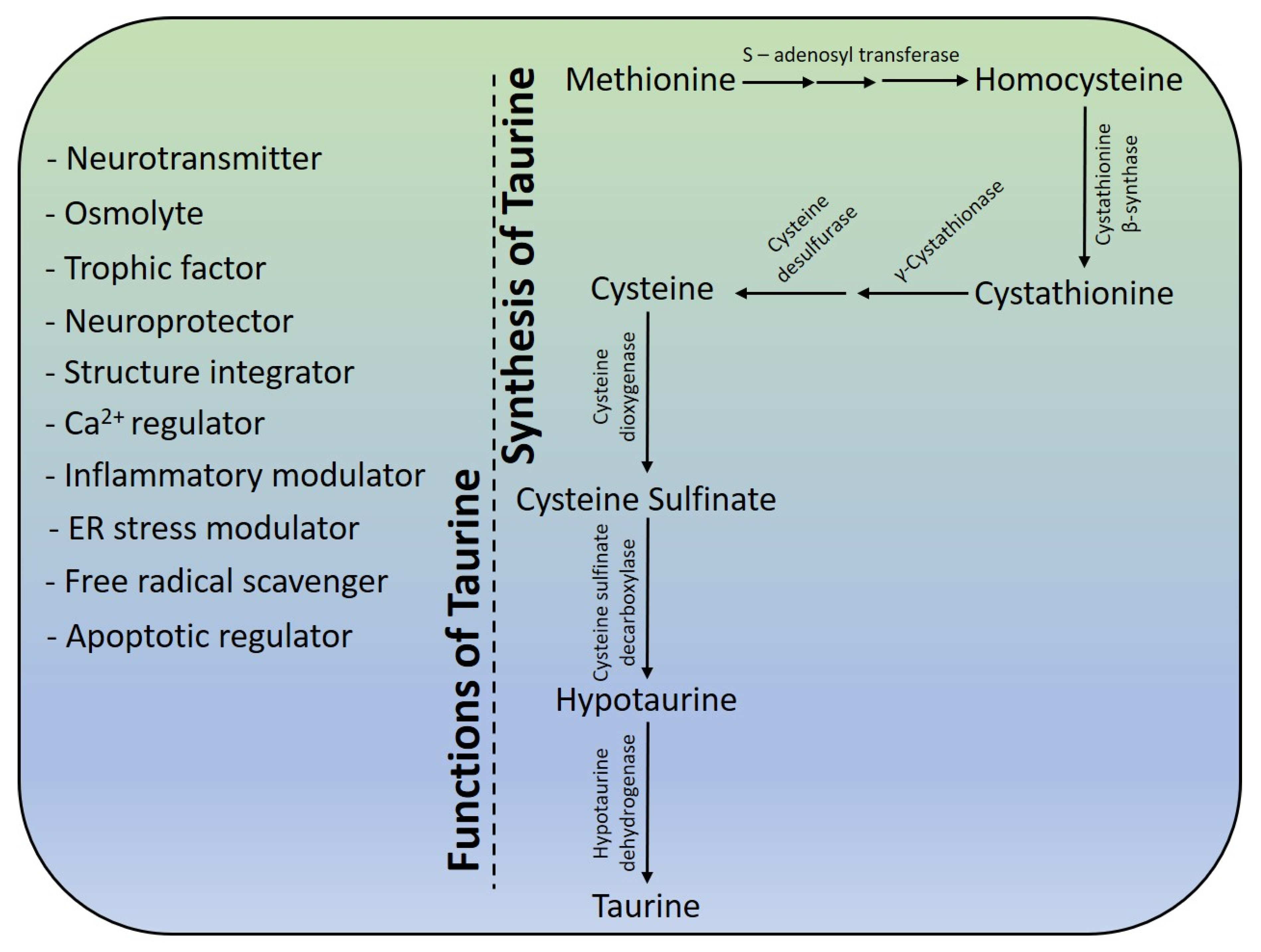
5. Taurine Biosynthesis
6. Neuroprotective Effects of Taurine
6.1. As Antioxidant Molecule
6.2. As Stabilizer in Regulating Protein Folding/Unfolding
6.3. As Inhibitory Neuromodulator
6.4. Energy Metabolism Modulator
6.5. As ER Stress Modulator
6.6. As Neuroinflamatory and Synaptic Loss Modulator
6.7. As Ca2+ Homeostasis and Apoptotic Modulator
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, I.P. Age-related neurodegenerative disease research needs aging models. Front. Aging Neurosci 2015, 7, 168. [Google Scholar] [CrossRef]
- Jan, A.T.; Azam, M.; Rahman, S.; Almigeiti, A.M.S.; Choi, D.H.; Lee, E.J.; Haq, Q.M.R.; Choi, I. Perspective Insights into Disease Progression, Diagnostics, and Therapeutic Approaches in Alzheimer’s Disease: A Judicious Update. Front. Aging Neurosci. 2017, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- J Jan, A.T.; Malik, M.A.; Rahman, S.; Yeo, H.R.; Lee, E.J.; Abdullah, T.S.; Choi, I. Perspective Insights of Exosomes in Neurodegenerative Diseases: A Critical Appraisal. Front. Aging Neurosci. 2017, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Jan, A.T.; Ayyagari, A.; Kim, J.; Kim, J.; Minakshi, R. Entanglement of UPRER in Aging Driven Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Archana, A.; Jan, A.T.; Minakshi, R. Dissecting Endoplasmic Reticulum Unfolded Protein Response (UPR(ER)) in Managing Clandestine Modus Operandi of Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 30. [Google Scholar] [CrossRef]
- Feigin, V.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abera, S.F.; Abyu, G.Y. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef]
- Sami, N.; Rahman, S.; Kumar, V.; Zaidi, S.; Islam, A.; Ali, S.; Ahmad, F.; Hassan, M.I. Protein aggregation, misfolding and consequential human neurodegenerative diseases. Int. J. Neurosci. 2017, 127, 1047–1057. [Google Scholar] [CrossRef]
- Moran, J.; Salazar, P.; Pasantes-Morales, H. Effect of tocopherol and taurine on membrane fluidity of retinal rod outer segments. Exp. Eye Res. 1987, 45, 769–776. [Google Scholar] [CrossRef]
- You, J.S.; Chang, K.J. Effects of taurine supplementation on lipid peroxidation, blood glucose and blood lipid metabolism in streptozotocin-induced diabetic rats. Adv. Exp. Med. Biol. 1998, 442, 163–168. [Google Scholar] [CrossRef]
- Roychoudhury, A.; Bieker, A.; Haussinger, D.; Oesterhelt, F. Membrane protein stability depends on the concentration of compatible solutes--a single molecule force spectroscopic study. Biol. Chem. 2013, 394, 1465–1474. [Google Scholar] [CrossRef]
- Taranukhin, A.G.; Taranukhina, E.Y.; Saransaari, P.; Pelto-Huikko, M.; Podkletnova, I.M.; Oja, S.S. Taurine protects cerebellar neurons of the external granular layer against ethanol-induced apoptosis in 7-day-old mice. Amino Acids 2012, 43, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Qu, J.; Li, Q.; Cui, M.; Wang, J.; Zhang, K.; Liu, X.; Feng, H.; Chen, Y. Taurine supplementation reduces neuroinflammation and protects against white matter injury after intracerebral hemorrhage in rats. Amino Acids 2018, 50, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Menzie, J.; Prentice, H.; Wu, J.Y. Neuroprotective Mechanisms of Taurine against Ischemic Stroke. Brain Sci. 2013, 3, 877–907. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhao, Y.; Gu, Y.; Xu, C. Anti-inflammatory mechanism of taurine against ischemic stroke is related to down-regulation of PARP and NF-kappaB. Amino Acids 2012, 42, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Schousboe, A. Taurine Interaction with Neurotransmitter Receptors in the CNS: An Update. Neurochem. Res. 2006, 30, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Chepkova, A.N.; Doreulee, N.; Yanovsky, Y.; Mukhopadhyay, D.; Haas, H.L.; Sergeeva, O.A. Long-lasting enhancement of corticostriatal neurotransmission by taurine. Eur. J. Neurosci. 2002, 16, 1523–1530. [Google Scholar] [CrossRef]
- Sergeeva, O.A.; Chepkova, A.N.; Doreulee, N.; Eriksson, K.S.; Poelchen, W.; Monnighoff, I.; Heller-Stilb, B.; Warskulat, U.; Haussinger, D.; Haas, H.L. Taurine-induced long-lasting enhancement of synaptic transmission in mice: Role of transporters. J. Physiol. 2003, 550, 911–919. [Google Scholar] [CrossRef]
- El Idrissi, A.; Shen, C.H.; L’Amoreaux, W.J. Neuroprotective role of taurine during aging. Amino Acids 2013, 45, 735–750. [Google Scholar] [CrossRef]
- El Idrissi, A. Taurine improves learning and retention in aged mice. Neurosci. Lett. 2008, 436, 19–22. [Google Scholar] [CrossRef]
- Neuwirth, L.S.; Volpe, N.P.; El Idrissi, A. Taurine effects on emotional learning and memory in aged mice: Neurochemical alterations and differentiation in auditory cued fear and context conditioning. Adv. Exp. Med. Biol. 2013, 775, 195–214. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimer’s Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef]
- Weggen, S.; Beher, D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimer’s Res. 2012, 4, 9. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer disease: Mechanistic understanding predicts novel therapies. Ann. Intern. Med. 2004, 140, 627–638. [Google Scholar] [CrossRef]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s disease: Treatments in discovery and development. Nat. Neurosci. 2002, 5, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Atwood, C.S.; Bowen, R.L. A Unified Hypothesis of Early- and Late-Onset Alzheimer’s Disease Pathogenesis. J. Alzheimer’s Dis. 2015, 47, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Chesselet, M.F. In vivo alpha-synuclein overexpression in rodents: A useful model of Parkinson’s disease? Exp. Neurol. 2008, 209, 22–27. [Google Scholar] [CrossRef]
- Kalia, L.V. Biomarkers for cognitive dysfunction in Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 46, S19–S23. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci. 1999, 22, 123–144. [Google Scholar] [CrossRef]
- Sohrab, S.S.; Suhail, M.; Ali, A.; Kamal, M.A.; Husen, A.; Ahmad, F.; Azhar, E.I.; Greig, N.H. Role of viruses, prions and miRNA in neurodegenerative disorders and dementia. Virusdisease 2018, 29, 419–433. [Google Scholar] [CrossRef]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Chakrabartty, A. Structure, folding, and misfolding of Cu, Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 2006, 1762, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Rahman, S.; Choudhry, H.; Zamzami, M.A.; Sarwar Jamal, M.; Islam, A.; Ahmad, F.; Hassan, M.I. Computing disease-linked SOD1 mutations: Deciphering protein stability and patient-phenotype relations. Sci. Rep. 2017, 7, 4678. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Estevez, A.G.; Crow, J.P.; Barbeito, L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001, 24, S15–S20. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Miranda, F.; Hetz, C. ER Stress and Neurodegenerative Disease: A Cause or Effect Relationship? Curr. Top. Microbiol. Immunol. 2018, 414, 131–157. [Google Scholar] [CrossRef]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Hoekstra, J.G.; Hipp, M.J.; Montine, T.J.; Kennedy, S.R. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann. Neurol. 2016, 80, 301–306. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Brys, M.; Switalski, R.; Mistur, R.; Glodzik, L.; Pirraglia, E.; Tsui, W.; De Santi, S.; de Leon, M.J. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc. Natl. Acad Sci. USA 2007, 104, 19067–19072. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Ratnaike, T.E.; De Gruyter, H.L.; Jaros, E.; Turnbull, D.M. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 2210–2214. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The Aβ Peptide of Alzheimer’s Disease Directly Produces Hydrogen Peroxide through Metal Ion Reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease. Redox Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.T.; Liu, X. Dual Perturbation of Electron Transport Chain (ETC) Complex and ATP Synthase Triggers PINK1/Parkin-dependent Mitophagy. FASEB J. 2018, 32, 543–549. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Moneim, A.E. Oxidant/Antioxidant imbalance and the risk of Alzheimer’s disease. Curr. Alzheimer. Res. 2015, 12, 335–349. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Lindquist, S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes. Dev. 2008, 22, 3308–3319. [Google Scholar] [CrossRef]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Park, K.W.; Eun Kim, G.; Morales, R.; Moda, F.; Moreno-Gonzalez, I.; Concha-Marambio, L.; Lee, A.S.; Hetz, C.; Soto, C. The Endoplasmic Reticulum Chaperone GRP78/BiP Modulates Prion Propagation in vitro and in vivo. Sci. Rep. 2017, 7, 44723. [Google Scholar] [CrossRef]
- Rahman, S.; Archana, A.; Jan, A.T.; Dutta, D.; Shankar, A.; Kim, J.; Minakshi, R. Molecular Insights Into the Relationship Between Autoimmune Thyroid Diseases and Breast Cancer: A Critical Perspective on Autoimmunity and ER Stress. Front. Immunol. 2019, 10, 344. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Archana, A.; Dutta, D.; Kumar, V.; Kim, J.; Jan, A.T.; Minakshi, R. The onus of cannabinoids in interrupting the molecular odyssey of breast cancer: A critical perspective on UPR(ER) and beyond. Saudi Pharm. J. 2019, 27, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Rahman, S.; Jan, A.T.; Archana, A.; Kim, J. Implications of aging and the endoplasmic reticulum unfolded protein response on the molecular modality of breast cancer. Exp. Mol. Med. 2017, 49, e389. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Okada, T.; Yoshida, H.; Akazawa, R.; Negishi, M.; Mori, K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366, 585–594. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Loewen, C.A.; Feany, M.B. The Unfolded Protein Response Protects from Tau Neurotoxicity In Vivo. PLoS ONE 2010, 5, e13084. [Google Scholar] [CrossRef]
- Prussing, K.; Voigt, A.; Schulz, J.B. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Ikeyama, S.; Wang, X.T.; Li, J.; Podlutsky, A.; Martindale, J.L.; Kokkonen, G.; van Huizen, R.; Gorospe, M.; Holbrook, N.J. Expression of the pro-apoptotic gene gadd153/chop is elevated in liver with aging and sensitizes cells to oxidant injury. J. Biol. Chem. 2003, 278, 16726–16731. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Larson, J.L.; Lauffer, B.E.; Goldstein, L.D.; Appling, L.L.; Borneo, J.; Poon, C.; Ho, T.; Cai, F.; et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 2016, 7, 11295. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. Embo. Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Curran, B.P.; Murray, H.J.; O’Connor, J.J. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience 2003, 118, 347–357. [Google Scholar] [CrossRef]
- Allan, S.M.; Tyrrell, P.J.; Rothwell, N.J. Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 2005, 5, 629–640. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Guadagno, J.; Xu, X.; Karajgikar, M.; Brown, A.; Cregan, S.P. Microglia-derived TNFalpha induces apoptosis in neural precursor cells via transcriptional activation of the Bcl-2 family member Puma. Cell Death Dis. 2013, 4, e538. [Google Scholar] [CrossRef] [PubMed]
- Neniskyte, U.; Vilalta, A.; Brown, G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.; Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004, 429, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M.; Weinberg, R.J. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 2012, 4, a005587. [Google Scholar] [CrossRef]
- Chever, O.; Dossi, E.; Pannasch, U.; Derangeon, M.; Rouach, N. Astroglial networks promote neuronal coordination. Sci. Signal. 2016, 9, ra6. [Google Scholar] [CrossRef]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Sideris, D.I.; Carroll, E.; Rotariu, S.; Salomonsson, S.; Tzioras, M.; McKenzie, C.A.; Smith, C.; von Arnim, C.A.F.; Ludolph, A.C.; et al. Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 213–226. [Google Scholar] [CrossRef]
- Crimins, J.L.; Rocher, A.B.; Luebke, J.I. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 2012, 124, 777–795. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, H.; Liu, Y.; Li, X.; Qin, L.; Lou, H.; Duan, S.; Wang, H. Astrocytes contribute to synapse elimination via type 2 inositol 1,4,5-trisphosphate receptor-dependent release of ATP. eLife 2016, 5, e15043. [Google Scholar] [CrossRef] [PubMed]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Tzioras, M.; Paolicelli, R.C. Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration. Front. Cell Neurosci. 2019, 13, 63. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Michailidou, I.; Naessens, D.M.; Hametner, S.; Guldenaar, W.; Kooi, E.J.; Geurts, J.J.; Baas, F.; Lassmann, H.; Ramaglia, V. Complement C3 on microglial clusters in multiple sclerosis occur in chronic but not acute disease: Implication for disease pathogenesis. Glia 2017, 65, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Sokolow, S.; Henkins, K.M.; Bilousova, T.; Miller, C.A.; Vinters, H.V.; Poon, W.; Cole, G.M.; Gylys, K.H. AD synapses contain abundant Abeta monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol. Aging 2012, 33, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Capetillo-Zarate, E.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Accumulation of intraneuronal beta-amyloid 42 peptides is associated with early changes in microtubule-associated protein 2 in neurites and synapses. PLoS ONE 2013, 8, e51965. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Wu, H.Y.; Hudry, E.; Hashimoto, T.; Kuchibhotla, K.; Rozkalne, A.; Fan, Z.; Spires-Jones, T.; Xie, H.; Arbel-Ornath, M.; Grosskreutz, C.L.; et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010, 30, 2636–2649. [Google Scholar] [CrossRef]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. Activation of mGluR1 Mediates C1q-Dependent Microglial Phagocytosis of Glutamatergic Synapses in Alzheimer’s Rodent Models. Mol. Neurobiol. 2019, 56, 5568–5585. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef]
- Tzioras, M.; Davies, C.; Newman, A.; Jackson, R.; Spires-Jones, T. Invited Review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2019, 45, 327–346. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Ouali Alami, N.; Schurr, C.; Olde Heuvel, F.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-kappaB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 2018, 37, e98697. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Huxtable, R.J. Physiological actions of taurine. Physiol. Rev. 1992, 72, 101–163. [Google Scholar] [CrossRef]
- Oja, S.S.; Saransaari, P. Properties of Taurine Release in Glucose-Free Media in Hippocampal Slices from Developing and Adult Mice. J. Amino Acids 2015, 2015, 254583. [Google Scholar] [CrossRef]
- Lambert, I.H.; Kristensen, D.M.; Holm, J.B.; Mortensen, O.H. Physiological role of taurine--from organism to organelle. Acta Physiol. (Oxf) 2015, 213, 191–212. [Google Scholar] [CrossRef]
- Demarcay, H. Ueber die natur der Galle. J. Für Prakt. Chem. 1838, 15, 193–212. [Google Scholar] [CrossRef]
- Nakashio, S.; Nakanishi, T.; Koshikawa, T.; Nishihara, T.; Ichikawa, T.; Kondo, M. Identification of taurine occurring sporulating cells of Bacillus subtilis. Microbios 1982, 33, 73–80. [Google Scholar]
- Bouckenooghe, T.; Remacle, C.; Reusens, B. Is taurine a functional nutrient? Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 728–733. [Google Scholar] [CrossRef]
- Murakami, Y.; Tsuyama, M.; Kobayashi, Y.; Kodama, H.; Iba, K. Trienoic fatty acids and plant tolerance of high temperature. Science 2000, 287, 476–479. [Google Scholar] [CrossRef]
- Zhang, M.; Izumi, I.; Kagamimori, S.; Sokejima, S.; Yamagami, T.; Liu, Z.; Qi, B. Role of taurine supplementation to prevent exercise-induced oxidative stress in healthy young men. Amino Acids 2004, 26, 203–207. [Google Scholar] [CrossRef]
- Park, E.; Park, S.Y.; Dobkin, C.; Schuller-Levis, G. Development of a novel cysteine sulfinic Acid decarboxylase knockout mouse: Dietary taurine reduces neonatal mortality. J. Amino Acids 2014, 2014, 346809. [Google Scholar] [CrossRef] [PubMed]
- Froger, N.; Moutsimilli, L.; Cadetti, L.; Jammoul, F.; Wang, Q.P.; Fan, Y.; Gaucher, D.; Rosolen, S.G.; Neveux, N.; Cynober, L.; et al. Taurine: The comeback of a neutraceutical in the prevention of retinal degenerations. Prog. Retin. Eye Res. 2014, 41, 44–63. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kimura, H.; Sakai, Y. Antagonistic action of 6-aminomethyl-3-methyl-4H, 1,2,4-benzothiadiazine-1, 1-dioxide (TAG), and evidence for a transmitter role of taurine in stellate interneurons in the cerebellum. Prog. Clin. Biol. Res. 1983, 125, 151–160. [Google Scholar]
- Lin, C.-T.; Song, G.-X.; Wu, J.-Y. Is taurine a neurotransmitter in rabbit retina? Brain Res. 1985, 337, 293–298. [Google Scholar] [CrossRef]
- Wade, J.V.; Olson, J.P.; Samson, F.E.; Nelson, S.R.; Pazdernik, T.L. A possible role for taurine in osmoregulation within the brain. J. Neurochem. 1988, 51, 740–745. [Google Scholar] [CrossRef]
- Schaffer, S.; Takahashi, K.; Azuma, J. Role of osmoregulation in the actions of taurine. Amino Acids 2000, 19, 527–546. [Google Scholar] [CrossRef]
- Lambert, I.H. Modulation of volume-sensitive taurine release from NIH3T3 mouse fibroblasts by reactive oxygen species. In Cell Volume and Signaling; Springer: Berlin/Heidelberg, Germany, 2004; pp. 369–378. [Google Scholar]
- Sturman, J.A. Taurine in development. Physiol. Rev. 1993, 73, 119–147. [Google Scholar] [CrossRef]
- Tang, X.; Deupree, D.; Sun, Y.; Wu, J.-Y. Biphasic Effect of Taurine on Excitatory Amino Acid-Induced Neurotoxicity. In Taurine 2; Springer: Berlin/Heidelberg, Germany, 1996; pp. 499–505. [Google Scholar]
- Ward, R.; Cirkovic-Vellichovia, T.; Ledeque, F.; Tirizitis, G.; Dubars, G.; Datla, K.; Dexter, D.; Heushling, P.; Crichton, R. Neuroprotection by taurine and taurine analogues. Adv. Exp. Med. Biol. 2006, 583, 299–306. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Kramer, J.; Chovan, J.P. Regulation of calcium homeostasis in the heart by taurine. Fed. Proc. 1980, 39, 2691–2694. [Google Scholar]
- El Idrissi, A. Taurine increases mitochondrial buffering of calcium: Role in neuroprotection. Amino Acids 2008, 34, 321–328. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Grabowska, A.; Chain, B.M. Modulation of antigen-specific T-cell activation in vitro by taurine chloramine. Immunology 1998, 94, 325–330. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Kontny, E. Taurine and inflammatory diseases. Amino Acids 2014, 46, 7–20. [Google Scholar] [CrossRef]
- Ghosh, S.; Chowdhury, S.; Das, A.K.; Sil, P.C. Taurine ameliorates oxidative stress induced inflammation and ER stress mediated testicular damage in STZ-induced diabetic Wistar rats. Food Chem. Toxicol. 2019, 124, 64–80. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Azuma, J.; Mozaffari, M. Role of antioxidant activity of taurine in diabetes. Can. J. Physiol. Pharm. 2009, 87, 91–99. [Google Scholar] [CrossRef]
- Li, J.; Zheng, L.; Wang, X.; Yao, K.; Shi, L.; Sun, X.; Yang, G.; Jiang, L.; Zhang, C.; Wang, Y.; et al. Taurine protects INS-1 cells from apoptosis induced by Di(2-ethylhexyl) phthalate via reducing oxidative stress and autophagy. Toxicol. Mech. Methods 2019, 29, 445–456. [Google Scholar] [CrossRef]
- Li, K.; Inam, U.L.; Shi, X.; Zhang, M.; Wu, P.; Li, S.; Suleman, R.; Nisar, A.; Piao, F. Anti-apoptotic Effect of Taurine on Schwann Cells Exposed to High Glucose In Vitro. Adv. Exp. Med. Biol. 2019, 1155, 787–799. [Google Scholar] [CrossRef]
- Jacobsen, J.G.; Smith, L.H. Biochemistry and physiology of taurine and taurine derivatives. Physiol. Rev. 1968, 48, 424–511. [Google Scholar] [CrossRef]
- Sturman, J.A.; Hayes, K.C. The biology of taurine in nutrition and development. In Advances in Nutritional Research; Springer: Berlin/Heidelberg, Germany, 1980; pp. 231–299. [Google Scholar]
- Munck, L.K.; Munck, B.G. Distinction between chloride-dependent transport systems for taurine and beta-alanine in rabbit ileum. Am. J. Physiol. 1992, 262, G609–G615. [Google Scholar] [CrossRef]
- Thwaites, D.T.; Ford, D.; Glanville, M.; Simmons, N.L. H(+)/solute-induced intracellular acidification leads to selective activation of apical Na(+)/H(+) exchange in human intestinal epithelial cells. J. Clin. Investig. 1999, 104, 629–635. [Google Scholar] [CrossRef]
- Anderson, C.M.; Howard, A.; Walters, J.R.; Ganapathy, V.; Thwaites, D.T. Taurine uptake across the human intestinal brush-border membrane is via two transporters: H+-coupled PAT1 (SLC36A1) and Na+- and Cl(-)-dependent TauT (SLC6A6). J. Physiol. 2009, 587, 731–744. [Google Scholar] [CrossRef]
- Hayes, K.C.; Sturman, J.A. Taurine in metabolism. Annu Rev. Nutr. 1981, 1, 401–425. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Shigehisa, S.; Sakakibara, S.; Hosokawa, Y.; Ueda, I. Cysteine metabolism in vivo of vitamin B6-deficient rats. Biochim. Biophys. Acta 1975, 381, 1–8. [Google Scholar] [CrossRef]
- Huxtable, R.J. Taurine in the central nervous system and the mammalian actions of taurine. Prog. Neurobiol. 1989, 32, 471–533. [Google Scholar] [CrossRef]
- Dupre, S.; De Marco, C. Activity of some animal tissues on the oxidation of cysteamine to hypotaurine in the presence of sulphide. Ital. J. Biochem. 1964, 13, 386–390. [Google Scholar]
- Wu, J.Y.; Prentice, H. Role of taurine in the central nervous system. J. Biomed. Sci. 2010, 17 (Suppl. 1), S1. [Google Scholar] [CrossRef]
- Sun, Q.; Hu, H.; Wang, W.; Jin, H.; Feng, G.; Jia, N. Taurine attenuates amyloid beta 1-42-induced mitochondrial dysfunction by activating of SIRT1 in SK-N-SH cells. Biochem. Biophys. Res. Commun. 2014, 447, 485–489. [Google Scholar] [CrossRef]
- Pan, C.; Prentice, H.; Price, A.L.; Wu, J.Y. Beneficial effect of taurine on hypoxia- and glutamate-induced endoplasmic reticulum stress pathways in primary neuronal culture. Amino Acids 2012, 43, 845–855. [Google Scholar] [CrossRef]
- Menzie, J.; Pan, C.; Prentice, H.; Wu, J.Y. Taurine and central nervous system disorders. Amino Acids 2014, 46, 31–46. [Google Scholar] [CrossRef]
- Louzada, P.R.; Paula Lima, A.C.; Mendonca-Silva, D.L.; Noel, F.; De Mello, F.G.; Ferreira, S.T. Taurine prevents the neurotoxicity of beta-amyloid and glutamate receptor agonists: Activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J. 2004, 18, 511–518. [Google Scholar] [CrossRef]
- Alkholifi, F.K.; Albers, D.S. Attenuation of rotenone toxicity in SY5Y cells by taurine and N-acetyl cysteine alone or in combination. Brain Res. 2015, 1622, 409–413. [Google Scholar] [CrossRef]
- Che, Y.; Hou, L.; Sun, F.; Zhang, C.; Liu, X.; Piao, F.; Zhang, D.; Li, H.; Wang, Q. Taurine protects dopaminergic neurons in a mouse Parkinson’s disease model through inhibition of microglial M1 polarization. Cell Death Dis. 2018, 9, 435. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Kang, Y.S. Taurine Protects Glutamate Neurotoxicity in Motor Neuron Cells. Adv. Exp. Med. Biol. 2017, 975 Pt 2, 887–895. [Google Scholar] [CrossRef]
- Jung, M.K.; Kim, K.Y.; Lee, N.Y.; Kang, Y.S.; Hwang, Y.J.; Kim, Y.; Sung, J.J.; McKee, A.; Kowall, N.; Lee, J.; et al. Expression of taurine transporter (TauT) is modulated by heat shock factor 1 (HSF1) in motor neurons of ALS. Mol. Neurobiol. 2013, 47, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Cozzi, R.; Ricordy, R.; Bartolini, F.; Ramadori, L.; Perticone, P.; De Salvia, R. Taurine and ellagic acid: Two differently-acting natural antioxidants. Env. Mol. Mutagen. 1995, 26, 248–254. [Google Scholar] [CrossRef]
- Timbrell, J.A.; Seabra, V.; Waterfield, C.J. The in vivo and in vitro protective properties of taurine. Gen. Pharmcol. 1995, 26, 453–462. [Google Scholar] [CrossRef]
- Redmond, H.P.; Wang, J.H.; Bouchier-Hayes, D. Taurine attenuates nitric oxide- and reactive oxygen intermediate-dependent hepatocyte injury. Arch. Surg. 1996, 131, 1280–1287. [Google Scholar] [CrossRef]
- Wright, C.E.; Lin, T.T.; Lin, Y.Y.; Sturman, J.A.; Gaull, G.E. Taurine scavenges oxidized chlorine in biological systems. Prog. Clin. Biol. Res. 1985, 179, 137–147. [Google Scholar]
- Hamaguchi, T.; Azuma, J.; Schaffer, S.W. Interaction of taurine with methionine: Inhibition of myocardial phospholipid methyltransferase. J. Cardiovasc. Pharm. 1991, 18, 224–230. [Google Scholar] [CrossRef]
- Vohra, B.P.S.; Hui, X. Taurine Protects against Carbon Tetrachloride Toxicity in the Cultured Neurons and In Vivo. Arch. Physiol. Biochem. 2001, 109, 90–94. [Google Scholar] [CrossRef]
- Flora, S.J.; Chouhan, S.; Kannan, G.M.; Mittal, M.; Swarnkar, H. Combined administration of taurine and monoisoamyl DMSA protects arsenic induced oxidative injury in rats. Oxid. Med. Cell. Longev. 2008, 1, 39–45. [Google Scholar] [CrossRef]
- Yildirim, Z.; Kilic, N.; Ozer, C.; Babul, A.; Take, G.; Erdogan, D. Effects of taurine in cellular responses to oxidative stress in young and middle-aged rat liver. Ann. N. Y. Acad. Sci. 2007, 1100, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, H.; Tsujino, T.; Watari, Y.; Emoto, N.; Yokoyama, M. Taurine prevents the decrease in expression and secretion of extracellular superoxide dismutase induced by homocysteine: Amelioration of homocysteine-induced endoplasmic reticulum stress by taurine. Circulation 2001, 104, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Compatible and counteracting solutes: Protecting cells from the Dead Sea to the deep sea. Sci. Prog. 2004, 87, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Nukina, N. A novel therapeutic strategy for polyglutamine diseases by stabilizing aggregation-prone proteins with small molecules. J. Mol. Med. 2005, 83, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Yip, C.M.; Huang, T.H.; Chakrabartty, A.; Fraser, P.E. Manipulating the amyloid-beta aggregation pathway with chemical chaperones. J. Biol. Chem. 1999, 274, 32970–32974. [Google Scholar] [CrossRef]
- Leandro, P.; Gomes, C.M. Protein misfolding in conformational disorders: Rescue of folding defects and chemical chaperoning. Mini-Rev. Med. Chem. 2008, 8, 901–911. [Google Scholar] [CrossRef]
- Yancey, P.H. Proteins and counteracting osmolytes. Biologist 2003, 50, 126–131. [Google Scholar]
- Yancey, P.H.; Clark, M.E.; Hand, S.C.; Bowlus, R.D.; Somero, G.N. Living with water stress: Evolution of osmolyte systems. Science 1982, 217, 1214–1222. [Google Scholar] [CrossRef]
- Gregersen, N.; Bross, P.; Vang, S.; Christensen, J.H. Protein misfolding and human disease. Annu Rev. Genom. Hum. Genet. 2006, 7, 103–124. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Herczenik, E.; Gebbink, M.F. Molecular and cellular aspects of protein misfolding and disease. FASEB J. 2008, 22, 2115–2133. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.R.; Dar, T.A.; Rahman, S.; Jamal, S.; Ahmad, F. Glycine betaine may have opposite effects on protein stability at high and low pH values. Biochim. Biophys. Acta 2009, 1794, 929–935. [Google Scholar] [CrossRef]
- Singh, L.R.; Poddar, N.K.; Dar, T.A.; Rahman, S.; Kumar, R.; Ahmad, F. Forty years of research on osmolyte-induced protein folding and stability. J. Iran. Chem. Soc. 2011, 8, 1–23. [Google Scholar] [CrossRef]
- Rahman, S.; Rehman, M.T.; Singh, L.R.; Warepam, M.; Ahmad, F.; Dar, T.A. Salt potentiates methylamine counteraction system to offset the deleterious effects of urea on protein stability and function. PLoS ONE 2015, 10, e0119597. [Google Scholar] [CrossRef]
- Rahman, S.; Warepam, M.; Singh, L.R.; Dar, T.A. A current perspective on the compensatory effects of urea and methylamine on protein stability and function. Prog. Biophys. Mol. Biol. 2015, 119, 129–136. [Google Scholar] [CrossRef]
- Chowhan, R.K.; Ali, F.; Bhat, M.Y.; Rahman, S.; Singh, L.R.; Ahmad, F.; Dar, T.A. Alanine Counteracts the Destabilizing Effect that Urea has on RNase-A. Protein Pept. Lett. 2016, 23, 795–799. [Google Scholar] [CrossRef]
- Rahman, S.; Ali, S.A.; Islam, A.; Hassan, M.I.; Ahmad, F. Testing the dependence of stabilizing effect of osmolytes on the fractional increase in the accessible surface area on thermal and chemical denaturations of proteins. Arch. Biochem. Biophys. 2016, 591, 7–17. [Google Scholar] [CrossRef]
- Rahman, S.; Ali, S.A.; Islam, A.; Hassan, M.I.; Ahmad, F. Data on the role of accessible surface area on osmolytes-induced protein stabilization. Data Brief. 2017, 10, 47–56. [Google Scholar] [CrossRef]
- Rahman, S.; Park, J.; Kim, J. Osmolytes Offset the Urea’s Effect on Protein Structure and Function. In Cellular Osmolytes: From Chaperoning Protein Folding to Clinical Perspectives; Rajendrakumar, S., Laishram, D., Tanveer, A., Eds.; Springer: Singapore, 2017; pp. 77–96. [Google Scholar] [CrossRef]
- Rahman, S.; Archana, A.; Azam, M.; Jan, A.T.; Dutta, D.; Minakshi, R. Role of Osmolytes and their Transporter Systems in Pathogen Survival and Pathogenicity. Curr. Drug Metab. 2018, 19, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Islam, A.; Hassan, M.I.; Kim, J.; Ahmad, F. Unfoldness of the denatured state of proteins determines urea: Methylamine counteraction in terms of Gibbs free energy of stabilization. Int. J. Biol. Macromol. 2019, 132, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Green, T.R.; Fellman, J.H.; Eicher, A.L.; Pratt, K.L. Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim. Biophys. Acta 1991, 1073, 91–97. [Google Scholar] [CrossRef]
- Jeon, S.H.; Lee, M.Y.; Rahman, M.M.; Kim, S.J.; Kim, G.B.; Park, S.Y.; Hong, C.U.; Kim, S.Z.; Kim, J.S.; Kang, H.S. The antioxidant, taurine reduced lipopolysaccharide (LPS)-induced generation of ROS, and activation of MAPKs and Bax in cultured pneumocytes. Pulm. Pharm. Ther. 2009, 22, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.W.; Minotto, J.B.; de Oliveira, M.R.; Zanotto-Filho, A.; Behr, G.A.; Rocha, R.F.; Moreira, J.C.; Klamt, F. Scavenging and antioxidant potential of physiological taurine concentrations against different reactive oxygen/nitrogen species. Pharm. Rep. 2010, 62, 185–193. [Google Scholar] [CrossRef]
- Schaffer, S.; Kim, H.W. Effects and Mechanisms of Taurine as a Therapeutic Agent. Biomol. Ther. (Seoul) 2018, 26, 225–241. [Google Scholar] [CrossRef]
- Santoro, M.M.; Liu, Y.F.; Khan, S.M.A.; Hou, L.X.; Bolen, D.W. Increased Thermal-Stability of Proteins in the Presence of Naturally-Occurring Osmolytes. Biochemistry 1992, 31, 5278–5283. [Google Scholar] [CrossRef]
- Taneja, S.; Ahmad, F. Increased thermal stability of proteins in the presence of amino acids. Biochem. J. 1994, 303 Pt 1, 147–153. [Google Scholar] [CrossRef]
- Xie, G.F.; Timasheff, S.N. Temperature dependence of the preferential interactions of ribonuclease A in aqueous co-solvent systems: Thermodynamic analysis. Protein Sci. 1997, 6, 222–232. [Google Scholar] [CrossRef]
- Xie, G.F.; Timasheff, S.N. Mechanism of the stabilization of ribonuclease A by sorbitol: Preferential hydration is greater for the denatured than for the native protein. Protein Sci. 1997, 6, 211–221. [Google Scholar] [CrossRef]
- Anjum, F.; Rishi, V.; Ahmad, F. Compatibility of osmolytes with Gibbs energy of stabilization of proteins. Biochim. Biophys. Acta 2000, 1476, 75–84. [Google Scholar] [CrossRef]
- Abe, Y.; Ohkuri, T.; Yoshitomi, S.; Murakami, S.; Ueda, T. Role of the osmolyte taurine on the folding of a model protein, hen egg white lysozyme, under a crowding condition. Amino Acids 2015, 47, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Timasheff, S.N. The stabilization of proteins by osmolytes. Biophys. J. 1985, 47, 411–414. [Google Scholar] [CrossRef]
- Bruździak, P.; Panuszko, A.; Kaczkowska, E.; Piotrowski, B.; Daghir, A.; Demkowicz, S.; Stangret, J. Taurine as a water structure breaker and protein stabilizer. Amino Acids 2018, 50, 125–140. [Google Scholar] [CrossRef]
- Khan, S.; Bano, Z.; Singh, L.R.; Hassan, M.I.; Islam, A.; Ahmad, F. Testing the ability of non-methylamine osmolytes present in kidney cells to counteract the deleterious effects of urea on structure, stability and function of proteins. PLoS ONE 2013, 8, e72533. [Google Scholar] [CrossRef]
- Santa-María, I.; Hernández, F.; Moreno, F.J.; Avila, J. Taurine, an inducer for tau polymerization and a weak inhibitor for amyloid-β-peptide aggregation. Neurosci. Lett. 2007, 429, 91–94. [Google Scholar] [CrossRef]
- Chaturvedi, S.K.; Alam, P.; Khan, J.M.; Siddiqui, M.K.; Kalaiarasan, P.; Subbarao, N.; Ahmad, Z.; Khan, R.H. Biophysical insight into the anti-amyloidogenic behavior of taurine. Int. J. Biol. Macromol. 2015, 80, 375–384. [Google Scholar] [CrossRef]
- Jang, H.; Lee, S.; Choi, S.L.; Kim, H.Y.; Baek, S.; Kim, Y. Taurine Directly Binds to Oligomeric Amyloid-beta and Recovers Cognitive Deficits in Alzheimer Model Mice. Adv. Exp. Med. Biol. 2017, 975 Pt 1, 233–241. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, H.V.; Yoon, J.H.; Kang, B.R.; Cho, S.M.; Lee, S.; Kim, J.Y.; Kim, J.W.; Cho, Y.; Woo, J.; et al. Taurine in drinking water recovers learning and memory in the adult APP/PS1 mouse model of Alzheimer’s disease. Sci. Rep. 2014, 4, 7467. [Google Scholar] [CrossRef]
- Macchi, F.; Eisenkolb, M.; Kiefer, H.; Otzen, D.E. The effect of osmolytes on protein fibrillation. Int. J. Mol. Sci. 2012, 13, 3801–3819. [Google Scholar] [CrossRef]
- Kamisaki, Y.; Wada, K.; Nakamoto, K.; Itoh, T. Release of taurine and its effects on release of neurotransmitter amino acids in rat cerebral cortex. Adv. Exp. Med. Biol. 1996, 403, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Dubin, A.E.; Mathur, J.; Tu, B.; Reddy, K.; Miraglia, L.J.; Reinhardt, J.; Orth, A.P.; Patapoutian, A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 2014, 157, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Saransaari, P.; Oja, S.S. Modulation of the ischemia-induced taurine release by adenosine receptors in the developing and adult mouse hippocampus. Neuroscience 2000, 97, 425–430. [Google Scholar] [CrossRef]
- Saransaari, P.; Oja, S.S. Taurine and neural cell damage. Amino Acids 2000, 19, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Saransaari, P.; Oja, S.S. Characteristics of taurine release in slices from adult and developing mouse brain stem. Amino Acids 2006, 31, 35–43. [Google Scholar] [CrossRef]
- El Idrissi, A.; L’Amoreaux, W.J. Selective resistance of taurine-fed mice to isoniazide-potentiated seizures: In vivo functional test for the activity of glutamic acid decarboxylase. Neuroscience 2008, 156, 693–699. [Google Scholar] [CrossRef]
- Chan, C.Y.; Sun, H.S.; Shah, S.M.; Agovic, M.S.; Ho, I.; Friedman, E.; Banerjee, S.P. Direct interaction of taurine with the NMDA glutamate receptor subtype via multiple mechanisms. Adv. Exp. Med. Biol. 2013, 775, 45–52. [Google Scholar] [CrossRef]
- Suarez, L.M.; Solis, J.M. Taurine potentiates presynaptic NMDA receptors in hippocampal Schaffer collateral axons. Eur. J. Neurosci. 2006, 24, 405–418. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Shimada-Takaura, K.; Jong, C.J.; Ito, T.; Takahashi, K. Impaired energy metabolism of the taurinedeficient heart. Amino Acids 2016, 48, 549–558. [Google Scholar] [CrossRef]
- Jeejeebhoy, F.; Keith, M.; Freeman, M.; Barr, A.; McCall, M.; Kurian, R.; Mazer, D.; Errett, L. Nutritional supplementation with MyoVive repletes essential cardiac myocyte nutrients and reduces left ventricular size in patients with left ventricular dysfunction. Am. Heart J. 2002, 143, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Sokka, A.L.; Putkonen, N.; Mudo, G.; Pryazhnikov, E.; Reijonen, S.; Khiroug, L.; Belluardo, N.; Lindholm, D.; Korhonen, L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J. Neurosci. 2007, 27, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Higo, T.; Hamada, K.; Hisatsune, C.; Nukina, N.; Hashikawa, T.; Hattori, M.; Nakamura, T.; Mikoshiba, K. Mechanism of ER stress-induced brain damage by IP (3) receptor. Neuron 2010, 68, 865–878. [Google Scholar] [CrossRef]
- Anand, S.S.; Babu, P.P. Endoplasmic reticulum stress and neurodegeneration in experimental cerebral malaria. Neurosignals 2013, 21, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yoshikawa, N.; Inui, T.; Miyazaki, N.; Schaffer, S.W.; Azuma, J. Tissue depletion of taurine accelerates skeletal muscle senescence and leads to early death in mice. PLoS ONE 2014, 9, e107409. [Google Scholar] [CrossRef]
- Pan, C.; Giraldo, G.S.; Prentice, H.; Wu, J.Y. Taurine protection of PC12 cells against endoplasmic reticulum stress induced by oxidative stress. J. Biomed. Sci. 2010, 17, S17. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Zheng, S.; Liu, H.; Li, S. Protective effects of taurine against inflammation, apoptosis, and oxidative stress in brain injury. Mol. Med. Rep. 2018, 18, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Behar, T.N.; Scott, C.A.; Greene, C.L.; Wen, X.; Smith, S.V.; Maric, D.; Liu, Q.Y.; Colton, C.A.; Barker, J.L. Glutamate acting at NMDA receptors stimulates embryonic cortical neuronal migration. J. Neurosci. 1999, 19, 4449–4461. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vockler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef]
- Wu, G.; Meier, S.A.; Knabe, D.A. Dietary glutamine supplementation prevents jejunal atrophy in weaned pigs. J. Nutr. 1996, 126, 2578–2584. [Google Scholar] [CrossRef]
- Chen, W.Q.; Jin, H.; Nguyen, M.; Carr, J.; Lee, Y.J.; Hsu, C.C.; Faiman, M.D.; Schloss, J.V.; Wu, J.Y. Role of taurine in regulation of intracellular calcium level and neuroprotective function in cultured neurons. J. Neurosci. Res. 2001, 66, 612–619. [Google Scholar] [CrossRef]
- El Idrissi, A.; Trenkner, E. Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J. Neurosci. 1999, 19, 9459–9468. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Jin, Y.; Wei, J.; Jin, H.; Sha, D.; Wu, J.Y. Mode of action of taurine as a neuroprotector. Brain Res. 2005, 1038, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Lazarewicz, J.W.; Noremberg, K.; Lehmann, A.; Hamberger, A. Effects of taurine on calcium binding and accumulation in rabbit hippocampal and cortical synaptosomes. Neurochem. Int. 1985, 7, 421–427. [Google Scholar] [CrossRef]
- Ramila, K.C.; Jong, C.J.; Pastukh, V.; Ito, T.; Azuma, J.; Schaffer, S.W. Role of protein phosphorylation in excitation-contraction coupling in taurine deficient hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 308, H232–H239. [Google Scholar] [CrossRef] [PubMed]
- Vesce, S.; Kirk, L.; Nicholls, D.G. Relationships between superoxide levels and delayed calcium deregulation in cultured cerebellar granule cells exposed continuously to glutamate. J. Neurochem. 2004, 90, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Prentice, H.; Modi, J.P.; Wu, J.Y. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid. Med. Cell Longev. 2015, 2015, 964518. [Google Scholar] [CrossRef] [PubMed]
- Leon, R.; Wu, H.; Jin, Y.; Wei, J.; Buddhala, C.; Prentice, H.; Wu, J.Y. Protective function of taurine in glutamate-induced apoptosis in cultured neurons. J. Neurosci. Res. 2009, 87, 1185–1194. [Google Scholar] [CrossRef]
- Jeong, J.E.; Kim, T.Y.; Park, H.J.; Lee, K.H.; Lee, K.H.; Choi, E.J.; Kim, J.K.; Chung, H.L.; Seo, E.S.; Kim, W.T. Taurine exerts neuroprotective effects via anti-apoptosis in hypoxic-ischemic brain injury in neonatal rats. Korean J. Pediatr. 2009, 52, 1337–1347. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease | Hallmark of Disease | Taurine Effect | Reference |
|---|---|---|---|
| Alzheimer’s disease | Cerebral plaques consisting of β-amyloid peptides (Aβs) and intracellular neurofibrillary tangles (NFTs), mainly composed of hyperphosphorylated tau | Induces the synaptic potentiation, antioxidant property, inhibits neuronal death by increasing inhibitory neuro transmission via GABAA and glycine receptor stimulation, suppresses mPTP opening and reverse mitochondrial function, attenuates Aβ-induced Ca2+ and ROS generation, pores open, reduces the mitochondrial membrane potential and increases ATP production, prevents mitochondrial dysfunction, shifts the ratio of Bcl-2:Bax in favor of cell survival, inhibits the formation of the Apaf-1/caspase-9 complex (apoptosome), suppresses upregulation of Caspase-12 and CHOP, suppresses ATF6 and IRE1 pathway, acts as GABA and the GABAA receptor agonists, inhibits the Na+/Ca2+ exchanger reverse mode, inhibits L-, P/Q-, N-type voltage-gated calcium channels, prevents Ca2+ influx through NMDA receptor calcium channels, inhibits calcium release | [148,149,150,151,152] |
| Parkinson’s disease | Loss of dopaminergic nigrostriatal neurons, intra-cytoplasmic Lewy bodies (LBs), intra-axonal Lewy neurites (LNs) | Scavenges ROS by inducing the activity of endogenous anti-oxidants, catalases and glutathione peroxidase (GSHPx), reduces mitochondrial ROS to promote normal functioning by increase in anti-oxidant protection, suppresses upregulation of Caspase-12 and CHOP, suppresses ATF6 and IRE1 pathway, suppresses microglial M1 polarization via NOX2-NF-κB pathway | [150,153,154] |
| Amyotrophic lateral sclerosis | Neuronal death (motor) in the nervous system, mutations in the protein SOD1 | Neuroprotective effects, against excitotoxicity induced by glutamate in motor neuronal cell lines, protects motor neuron from oxidative stress | [155,156] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhat, M.A.; Ahmad, K.; Khan, M.S.A.; Bhat, M.A.; Almatroudi, A.; Rahman, S.; Jan, A.T. Expedition into Taurine Biology: Structural Insights and Therapeutic Perspective of Taurine in Neurodegenerative Diseases. Biomolecules 2020, 10, 863. https://doi.org/10.3390/biom10060863
Bhat MA, Ahmad K, Khan MSA, Bhat MA, Almatroudi A, Rahman S, Jan AT. Expedition into Taurine Biology: Structural Insights and Therapeutic Perspective of Taurine in Neurodegenerative Diseases. Biomolecules. 2020; 10(6):863. https://doi.org/10.3390/biom10060863
Chicago/Turabian StyleBhat, Mujtaba Aamir, Khurshid Ahmad, Mohd Sajjad Ahmad Khan, Mudasir Ahmad Bhat, Ahmad Almatroudi, Safikur Rahman, and Arif Tasleem Jan. 2020. "Expedition into Taurine Biology: Structural Insights and Therapeutic Perspective of Taurine in Neurodegenerative Diseases" Biomolecules 10, no. 6: 863. https://doi.org/10.3390/biom10060863
APA StyleBhat, M. A., Ahmad, K., Khan, M. S. A., Bhat, M. A., Almatroudi, A., Rahman, S., & Jan, A. T. (2020). Expedition into Taurine Biology: Structural Insights and Therapeutic Perspective of Taurine in Neurodegenerative Diseases. Biomolecules, 10(6), 863. https://doi.org/10.3390/biom10060863