1. Introduction
Numerous studies have indicated the limited capacity of the commonly used planar plastic and glass substrates to ensure physiologically relevant responses in cell cultures. It is well-known that the physicochemical properties of these materials are essentially different from those of the living tissues [
1,
2,
3,
4]. Therefore, there is a rapidly increasing need for establishing realistic tissue-like cell culture systems by employing both scaffold-free and scaffold-based approaches [
1,
2,
5,
6]. Hydrogels are among the favorable scaffolds because of their tissue-resembling properties such as a fibrous structure and softness, high content of bound water, and extracellular matrix (ECM) mimicking biochemistry [
7,
8,
9,
10]. However, there is still a shortage of hydrogel scaffold-based cell culture models that could be standardized, easily manipulated, and compatible with specific media or with high throughput workflows [
11]. In regard to the brain cells, the challenge lies in providing a robust and repeatable assay, ensuring tissue-like cell composition and interaction and the efficient maturation of the functional network characterized by spontaneous neuronal firing activity [
12]. The ECM components responsible for the cell attachment via focal adhesions are important players in cell differentiation and functional tissue formation [
13]. Collagen and fibronectin have changing expression patterns in the developing cerebellum to control cell differentiation and migration [
14,
15]. Recently, different peptide fragments of collagen (CLPs) and the integrin-recognizing fragment of fibronectin (RGD) have been reported to mimic the effect of the entire protein molecules by enhancing neuronal adhesion, neurite outgrowth, and branching [
16,
17,
18]. Such functional peptide building blocks are attractive tools for tissue engineering because they are relatively easy to synthesize and can be integrated into more complex synthetic materials. However, not only the presence of bioactive ECM-mimicking peptides but also other cues, such as the elastomechanical properties of the cellular environment, are important. For example, the stiffness plays a significant role in mediating focal adhesion formation by neural cells via mechanosensitive cellular membrane-embedded receptors such as integrins and integrin-regulating proteins [
19].
The present study aimed to explore synthetic ECM-mimicking hydrogels based on CLP and CLP-RGD blocks conjugated to a poly(ethylene glycol) template (PEG-CLP and PEG-CLP-RGD, respectively), as well as to investigate how these artificial, all-synthetic substrates affect primary cerebellar cell organization, composition, motility and function. Recently, chimeric peptides have emerged as a promising synthetic biological strategy for medical applications [
20], and we have already reported the application of PEG-CLP-RGD hydrogels for cancer cell models ([
21], in revision). Additionally, we aimed to elucidate the role of the viscoelasticity of the ECM-mimicking substrate. Our study reveals a completely different but closer to the in vivo-like nature of the primary cerebellar assemblies obtained on the ECM-mimetic hydrogels as compared to the poly-L-lysine-coated laboratory plastic and glass substrates that are commonly used for neuronal-glial cultures.
2. Materials and Methods
2.1. Fabrication and Characterization of the Hydrogel Membranes
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich.
Peptide synthesis, the conjugation of the peptides to PEG, and the synthesis of hydrogels were carried out following the protocols described elsewhere [
21,
22]. The peptides CLP (Cys-Gly-(Pro-Lys-Gly)
4(Pro-Hyp-Gly)
4(Asp-Hyp-Gly)
4) and CLP-RGD (-Arg-Gly-Asp-Ser-Pro-Gly) were synthesized by UAB Ferentis (Vilnius, Lithuania). For the conjugation of the peptides to PEG, 40 kDa 8-arm PEG-maleimide (hexa-glycerol core, Creative PEGWorks, NC, USA or JenKem, TX, USA) in dimethyl sulfoxide (DMSO) was mixed with an aqueous solution of CLP or CLP-RGD peptide at a molar ratio of PEG-maleimide:peptide 1:2.5 at room temperature. After 2 days of continuous stirring, a homogeneous solution of the peptide-PEG conjugate was dialyzed for 2 days using 12–14 kD MW cut-off tubing (Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA), lyophilized, and stored at 4 °C until further use.
For hydrogel synthesis, 12% (
w/w) PEG-CLP and 12% (
w/w) PEG-CLP-RGD aqueous stock solutions were prepared. Then, 500 mg of the stock solutions were taken into a T-piece 2 mL glass syringe mixing system [
23]. The pH values of the solutions were adjusted to 4.5 by the addition of 2 M sodium hydroxide. After this step, calculated volumes of N-hydroxysuccinimide (NHS) and N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) were added to the mixing system as 10% solutions in 0.625 M 2-(N-morpholino)ethanesulfonic acid buffer (MES). The molar equivalent for peptide-NH
2:NHS:EDC was 1:1:1. All the reagents were thoroughly mixed, and then the hydrogel was cast and kept between two glass slides with a 500 µm-thick spacer to control the thickness of the resulting membrane. It was left for curing for 16 h in a humidified chamber. The composition of peptides and the conjugation of CLP or CLP-RGD with 8-arm PEG-maleimide were characterized using
1H NMR on a Bruker Ascend 400 MHz spectrometer at room temperature. Briefly, 2% solutions of CLP, CLP-RGD, PEG-CLP, and PEG-CLP-RGD were prepared in D
2O (Deuterium oxide). The resonance of deuterated solvent (D
2O, δ = 4.79) was used as the internal standard.
2.2. Glass Functionalization by ECM Peptides
As model solid substrates, 10 mm diameter and 150 ± 10 µm thickness glass slides and 10 × 10 mm
2 silicon substrates were employed. The latter substrates were cut from a 4′ silicon wafer (Topsil, Denmark). The substrates first were rinsed in ethanol and dried in a stream of N
2 gas. Then, they were washed using the so-called SC-1 cleaning procedure: in a 5:1:1 mixture of ultrapure water, 30% H
2O
2, and 25% NH
4OH solutions at 85 °C temperature for 10 min. Subsequently, they were treated in a plasma dry cleaner at the 20 W power (Femto, Diener Electronic GmbH, Ebhausen, Germany) for 3 min. After the plasma treatment, the glass substrates were silanized with (3-aminopropyl)trimethoxysilane (APTMS) (Thermo Fisher Scientific) [
24]. Prior to the PEG-CLP and PEG-CLP-RGD coating process, a drop of 5%
w/
w glutaraldehyde solution in a 0.1M PB pH = 8.0 buffer was applied onto a surface for 20 min to convert the amine groups into aldehydes. After this step, the samples were rinsed with water and dried in the N
2 gas. A solution of 2% PEG-CLP or 2% PEG-CLP-RGD in a PB buffer (0.1 M pH = 5.7) was applied onto the glass slides functionalized with the aldehyde groups and kept for 40 min at 37 °C temperature. After incubation with the respective peptide-PEG conjugate solutions, the samples were rinsed in water and dried in the N
2 gas stream. The samples were stored dry in 4–8 °C until further use. The functionalized silicon substrates were investigated by using both imagining ellipsometry and atomic force microscopy (AFM). In contrast, the glass substrates were analyzed solely by AFM (see the respective Methods section below and the SI file for details). In total, 9 samples for each peptide-PEG conjugate were prepared and characterized.
2.3. Spectral Characterization of the Peptide Assembly
The three-dimensional structure of the peptides and the respective PEG-peptide conjugates was estimated using a J-815 circular dichroism (CD) spectropolarimeter (Jasco, MD, USA) equipped with a Peltier temperature-control system. Briefly, 1% (w/w) sample solutions in Milli-Q water with an adjusted pH = 5 were measured in a quartz cuvette with a 1 mm path length. Three spectra were collected and averaged for each type of peptide/conjugates from 190 to 250 nm at 25 °C at the 50 nm/min scanning speed.
2.4. Quantitative Characterization of the Elastomechanical Properties of Hydrogels
The mechanics of the fabricated hydrogel membranes was analyzed by measuring the two elastic parameters: shear storage modulus G’ and Young modulus E*. For G’ determination, oscillatory rheology was performed on a DHR-2 discovery hybrid rheometer (TA instruments, Sweden). All frequency sweep experiments were performed using 8 mm diameter stainless steel parallel plate geometry at 25 °C under an axial load of 150 mN. Circular discs of an 8 mm diameter were cut out from 0.5 mm-thick hydrogel membranes using a regular biopsy punch. Hydrogel samples were kept in 10 mM PBS. All experiments were performed using a shear strain amplitude of 0.27% at a frequency range of 0.02–50 Hz. To enable direct comparison with the Young modulus values obtained at a specific loading rate, storage modulus values obtained at 10 Hz were selected for presentation.
The AFM nanoindentation technique was employed to measure the elastic (Young) modulus E* values of the hydrogel samples [
25,
26]. The nanoindentation probes were prepared as follows. The spring constant of the tipless AFM cantilevers (NSC36-B, MicroMasch) was calibrated before the attachment of the microsphere particles using the thermal noise method [
27] and was found to be 4.0 ± 0.6 N/m. The probes from one fabrication batch, which showed a low variation of the spring constant, were used. To ensure a controlled geometry of the contact, silicon dioxide microspheres (Microparticles GMbH) with a manufacturer-reported diameter of 6.65 ± 0.28 μm were attached to the AFM cantilevers using UV-curable glue (NOA68, Norland) while taking care to control the amount of glue on the cantilever. The attachment of the microspheres and their diameter was inspected by optical microscopy (x50 lens, BX51, Olympus, Japan), yielding the expected diameter of 6.65 ± 0.15 μm.
Cutouts of the hydrogel membranes with a typical width of 3 mm and a length of 5 mm were attached to the (3-aminopropyl)trimethoxysilane-treated glass substrates cut from regular carrier slides via glutaraldehyde chemistry (see above). In a typical experiment, one glass substrate had 2–5 hydrogel pieces mounted.
The elastic modulus measurement procedure was carried out as follows. The hydrogel sample was placed in a small Petri dish filled with PBS (pH = 7.4) and mounted onto the stage of an inverted optical microscope (IX73, Olympus, Japan), which served as a base for the AFM instrument (Nanowizard 3, JPK, Germany). The nanoindentation probe was approached to the hard glass surface, and the detection sensitivity factor was determined from the force-displacement curve. Typical detection sensitivity values were 10–20 nm/V. Then, the probe was repositioned over the hydrogel sample, and 1024 force curves were collected over the 50 × 50 µm area using the Quantitative Imaging™ (QI, JPK) mode with a force setpoint of 120 nN and a loading rate of 50 µm/s. For each sample analyzed, this procedure was repeated at 4 different locations.
The obtained curves were analyzed, and the values of the elastic modulus of the hydrogel samples extracted by fitting the approach part of the force-displacement curve with the Hertz sphere-on-plane elastic contact model using the JPK data processing software (version spm-5.0.84). The elastic modulus values were calculated from measurements of 4–12 different samples for every hydrogel formulation.
To compare the stiffness of the fabricated hydrogels, we estimated the compressive modulus (i.e., Young modulus) from the storage modulus at 10 Hz, as described in [
28]. This was done by using the following equation:
where E is the compressive modulus, G’ is the storage modulus, and ν is the Poisson’s ratio; for hydrogels, ν = 0.5.
2.5. Surface Topography and Thickness Measurements of Membranes and Coatings
Surface topography images of the PEG-CLP and PEG-CLP-RGD hydrogel membranes and their respective coatings on glass were acquired using the same AFM instrument (NanoWizard 3, JPK) with the MLCT-A (Bruker) and NSC35-C (MicroMasch) AFM probes in a PBS pH = 7.4 buffer solution. Imaging was done using the QI mode at the 0.4–5 nN force setpoint; different regions of each sample were scanned. The scan size ranged from 50 × 50 to 1 × 1 µm2 at the 256 × 256 pixel resolution. Three samples from three different fabrication batches were analyzed for each hydrogel type.
The thickness of the organic layers on the silicon substrates was measured with an imaging null ellipsometer (Nanofilm_ep3, Accurion GmbH, Goettingen, Germany), using a laser emitting at the 658 nm wavelength and a 10× objective. The measurements were carried out at the 70° angle of incidence unless otherwise stated. Thickness modeling was performed with the Nanofilm_ep4 Model software (Accurion GmbH, Goettingen, Germany). The actual substrate layer thicknesses were fitted from the angle of incidence spectrum, measured on a substrate freshly cleaned by the SCI-1 procedure (see above). The coatings of APTMS, GA, and the PEG-peptide conjugates analyzed in air were modeled as organic layers with a refractive index of n = 1.5 and k = 0. The measurements were performed after each step of coating. For statistics, 9 samples of the conjugates bound to glass were measured.
2.6. Preparation of Neuronal-Glial Cell Culture and Viability Assessment
All experimental procedures were performed according to the Guide for the Care and Use of Laboratory Animals. The rats were maintained at Lithuanian University of Health Sciences animal house in agreement with the Guide for the Care and Use of Laboratory Rats. Cerebella were isolated from euthanized postnatal 5–7-day-old Wistar rats and minced into small cubes (ca. 0.5 mm3). The minced tissue was transferred into a Falcon tube containing 7 mL of a Versene solution (1:5000; Gibco, Thermo Fisher Scientific) and incubated at 37 °C for 5 min. The tissue was further triturated with a Pasteur pipette to obtain a single-cell suspension. The incubation and trituration procedures were repeated until any undissociated tissue remains became invisible. The single-cell suspension was centrifuged at 270 g for 5 min and resuspended in a DMEM medium with Glutamax (Thermo Fisher Scientific) supplemented with 5% horse serum, 5% fetal calf serum, 38 mM glucose, 25 mM KCl, and antibiotic-antimycotic (Thermo Fisher Scientific). The cells were plated at a density of 0.25 × 106 cells/cm2 in 96-well plates (VWR) on disc-shaped hydrogel membranes or on the plate bottom coated with 0.0001% poly-L-lysine. The cells were kept at 37 °C in a humidified incubator containing 5% CO2. On in vitro days 4 and 7, the viability of the cultures was assessed by double nuclear staining with fluorescent dyes Hoechst33342 (6 µg/mL) and propidium iodide (3 µg/mL) for 10 min and imaging in a fluorescent microscope Olympus IX71 (Olympus Corporation, Tokyo, Japan) equipped with a ×20 objective. The images were taken by a 01-Exi-AQA-R-F-M-14-C camera (QImaging, Surrey, Canada). The image analysis was performed by the ImageJ software.
2.7. Preparation of Pure Microglial Cell Culture
Primary mixed astrocyte and microglial cultures were used for pure microglial culture preparation. First, mixed glial cultures were prepared from the cerebral cortices of 5–7-day-old Wistar rats. After the dissection of the cerebral hemispheres, the meninges were removed, and the tissue was dissociated in a solution of EBSS containing 0.3% BSA, 103.2 Kunitz units/mL DNase I, and 3800 BAEE units/mL trypsin. The cells were plated at 2 × 105 cells/cm2 in 75 cm2 flasks coated with 0.0001% poly-L-lysine. The cultures were maintained in DMEM supplemented with 10% fetal calf serum and 1 mg/mL antibiotic-antimycotic (Gibco). The cells were kept at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. After the mixed glial cultures reached confluence (on the 6th–8th days in vitro), the 15 microglial cells were isolated by shaking and tapping the flasks. The medium from the mixed glial cultures, containing separated microglial cells, was removed and centrifuged at 135 × g for 5 min. The supernatant was discarded, and the cells were resuspended in DMEM with the same supplements as those of the mixed glial cultures and plated at a density of 2 × 105 cells/cm2 in the uncoated flat bottom 96 well plates (VWR) with or without hydrogel inserts (membranes). The cells were kept at 37 °C in a humidified incubator containing 5% CO2 and were later used for the proliferation assessment.
2.8. Evaluation of Cell Number, Composition, and Neuritogenesis
All nuclei were stained with Hoechst33342 (6 µg/mL, 15 min at 37 °C). Neurons were identified by immunostaining for microtubule-associated protein 2 (MAP2) and astrocytes by immunostaining for glial fibrillary acidic protein (GFAP). The cultures were fixed in 4% paraformaldehyde in PBS for 20 min, permeabilized in 0.3% Triton X-100 in PBS, blocked with 10% BSA in PBS, and incubated for 1h with primary antibodies: 1 µg/mL of rabbit polyclonal anti-MAP2 (Abcam) and 4 µg/mL of mouse monoclonal anti-GFAP (Thermo Fisher Scientific), as well as 30 min with the secondary antibody AlexaFluor®555 conjugated goat anti-mouse IgG (Invitrogen) and AlexaFluor®647 conjugated chicken anti-rabbit IgG (Thermo Fisher Scientific), both diluted in PBS 1:200. Fixed microglial cells in the cultures were detected by isolectin GS-IB4 from Griffonia simplificolia, Alexa Fluor® 488 conjugate (10 ng/mL for 15 min at 37 °C, Molecular Probes). The cells in cultures were detected using laser scanning confocal microscopes: Zeiss Axio Observer LSM 700 (Carl Zeiss Microimaging Inc., Jena, Germany) and Olympus Fluoview FV1000 (Olympus Corporation, Tokyo, Japan). Each image displayed a maximum intensity projection of a stack of 5–12 z-sections of 3–6 different cultures. The total cell number on the hydrogel, glass, and plastic samples was evaluated by counting the nuclei in the 400 × 500 μm2 area of the confocal micrographs. For neurite evaluation, the anti-MAP2-positive area in micrographs was calculated by the ImageJ software and expressed as the percentage of the total image area per neuron.
2.9. Evaluation of Microglial Proliferation
Microglial proliferation in mixed neuronal-glial cultures was assessed by the daily cell counting of isolectin GS-IB4 positive cells in fluorescent images and in pure microglial cultures in phase-contrast images by the use of the inverted OlympusIX71 microscope (Olympus Corporation, Tokyo, Japan). Cells in the micrographs were counted by ImageJ software.
2.10. Cytokine Detection
The cell culture medium for cytokine detection was collected each day starting from the 1st day and finishing on the 6th day in vitro. The levels of pro-inflammatory cytokines in the neuronal-glial cell medium were detected by ELISA kits for rat TNF-α (Thermo Fisher Scientific) and IL-1β (Abcam) according to manufacturer’s protocol. A lipopolysaccharide (LPS)-activated cell culture medium was used as the positive control. The optical density in the samples was measured in a MultiskanFC plate reader (Thermo Fisher Scientific).
2.11. Neuronal Activity Monitoring by Ca2+-Sensitive Fluorescence
For Ca
2+ oscillation measurement, the neuronal-glial cells on days 4 and 7 in culture were pre-loaded with the cell-permeable Ca
2+-sensitive dye Oregon Green™ 488 BAPTA-1, AM (OGB-AM, Thermo Fisher Scientific). A solution of OGB-AM (5 µM) was added to the cell culture medium, and the cultures were placed to the incubator for 15 min. To wash out the dye that had not penetrated into the cells, the medium was replaced with fresh DMEM with all the supplements used for the mixed neuronal-glial cell cultures (as described in 2.6 above), and the cells were further incubated for 10 min. For Ca
2+ signal registration, we applied the methods used in [
29]. The LED light was limited to the recording site by a diaphragm. Images were acquired with the Solis software (Andor Technology Ltd., Belfast, UK) and stored on the disk for further analysis. Image analysis was performed with the Solis analysis software, as well as by employing the image analysis package ImageJ and custom written subroutines. The cultured cells stained with calcium-sensitive dye OGB-AM were registered in an area of 640 × 250 pixels. In every culture, 4–6 different regions were registered. The fluorescence images were acquired for 20 s with a rate of 30 Hz. In order to identify the cells with large spontaneous changes in calcium concentration, the image stacks were analyzed by custom-written Python subroutines by using the SciPy open-source software package. The fluorescence traces from single pixels were selected if the peaks were larger than 5% of the time-averaged fluorescence and the amplitude of the peak was 4 times larger than the smallest standard deviation in pixels of the corresponding image stack. Since the cells were imaged by a number of pixels, the detected signals were summed and averaged if they were all present in the neighboring pixels in the area smaller than 20 × 20 pixels. The areas with the detected signals in the image stacks were also inspected by using the ImageJ software package. The time course of the detected fluorescence signals was corrected for bleaching by the simple ratio method [
30]. In this procedure, the decaying mean intensity of fluorescence in the region outside the detected signals was used to calculate the decay ratio for each image and to compensate for the bleaching in the time course of the signals. The time constant, τ, of the exponential decay for fluorescence signals was calculated by using the curve_fit procedure from the SciPy package, which uses the non-linear least-squares method to fit the first-order exponential decay. The average square error at each time moment was smaller than 0.05%.
2.12. Statistical Analysis
The statistical significance of the elastic modulus (E*) was evaluated by ANOVA (SPSS software) [F(1, 61) = 29.108, p = 0.000]. All quantitative data in the graphs are presented as means of 4–7 experiments and standard error. The graphs were made by and statistical significance was evaluated by the SigmaPlot v13 software by a one-way ANOVA Tukey test. The statistical analysis for the fluorescence data was conducted using the procedures from the SciPy package. The normality of data distribution was assessed by using D’Agostino and Pearson’s normality test. The statistical significance of the difference between the averages was assessed using Student’s t-test for normally distributed data. The surface roughness and layer thickness data are represented as mean values ± SDV.
4. Discussion
The materials synthesized as the mimetics of the natural ECM that were analyzed in this study differ from most of the hydrogels reported in the literature [
37] and/or those available commercially. While the peptide fragments of collagen have been used as substrates for cell cultures [
38,
39], in our PEG-peptide hydrogels, the native-like assemblies of the peptides are facilitated by the covalent attachment to an eight-armed PEG-maleimide. The overall architecture is further locked by chemical crosslinking, as described in [
22]. Besides that, such a synthetic strategy is advantageous and flexible in terms of (bio)chemical variation by introducing and combining different active ECM elements. For example, in our case, we employed the CLP peptide sequence and the C-terminus extended with a different functionality—the cell adhesion peptide RGD—as the building blocks of the hydrogel [
21].
The circular dichroism analysis of CLP, CLP-RGD, and their respective PEG conjugates showed the triple helix signature at a wavelength of 225 nm (
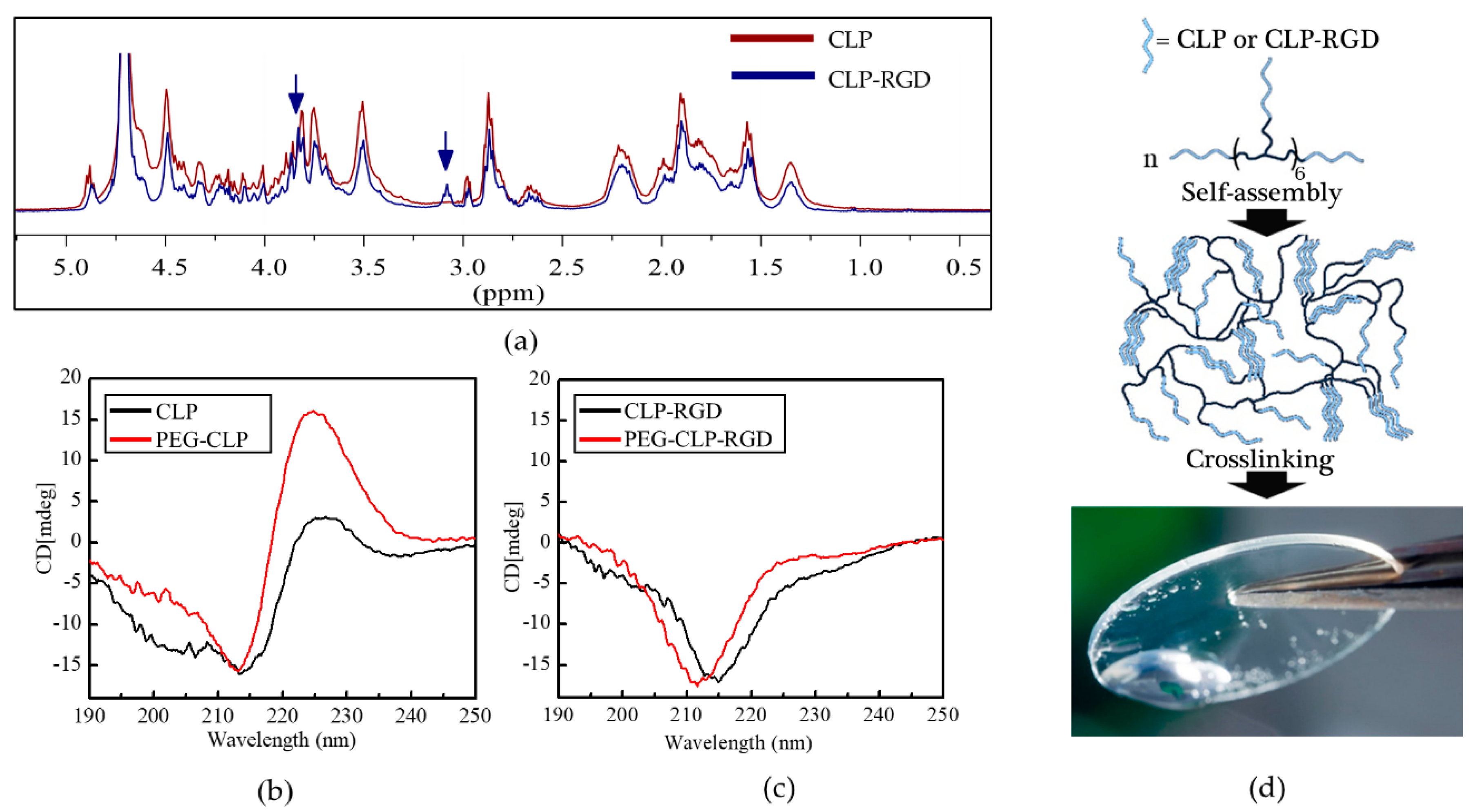
Figure 1b). Conjugation to eight-arm-PEG further promoted the self-assembly for PEG-CLP, as was observed before [
22]. However, we found that for CLP-RGD, this signal was significantly weaker compared to the unmodified CLP block, thus suggesting that the introduction of RGD partially hindered the formation of the triple helical structure.
From the elastomechanical analysis (
Table 1), one can judge that both shear storage and elastic moduli showed the same tendency, with PEG-CLP having the higher stiffness than PEG-CLP-RGD. The decrease in stiffness for PEG-CLP-RGD hydrogel compared to PEG-CLP could be partly attributed to the reduced helicity of these conjugates compared to PEG-CLP. Nevertheless, the moduli values obtained for both synthesized hydrogels were within the range of those measured for brain tissues [
40] and at least by four orders of magnitude lower than those of polystyrene [
41] commonly used as a substrate for in vitro cell cultures. The calculated elastic modulus (E’) from the shear modulus (G’) data yielded similar values to the elastic modulus (E*) obtained by the AFM nanoindentation-based technique (E*). The AFM nanoindentation assay measured deformation force in submicrometer-sized surface regions, and it was limited by the low depth of indentation and the actual dimensions (6.65 ± 0.28 µm diameter) of the silicon dioxide microsphere used as the indenter. As such, this parameter is arguably a better indicator of the mechanical properties of the substrate as sensed by a living cell. On the other hand, dynamic oscillatory rheology revealed the mechanical properties that are associated more with the macroscopic properties of the bulk of the material. These data are more relevant for technical operations with the hydrogel membranes such as handling during the cell culture, post-culture analysis, medical techniques like trepanning, and suturing.
The RGD peptide promotes cellular adhesion and acts as a neuritogenic fragment [
16,
31,
32]. However, the different stiffnesses of the PEG-CLP and PEG-CLP-RGD hydrogels may have also affected the cell organization on the hydrogels surface. As an attempt to separate the biochemical and the mechanical cues, we performed a comparison of the cell culture on both the elastic hydrogel substrates and the more rigid coating of the PEG-peptide conjugates on a solid surface. We expected that in the latter case, the interaction between the peptides and the cellular receptors would be more sterically hindered. Indeed, the attachment of cells to the PEG-CLP and PEG-CLP-RGD coatings on glass was poor compared to the attachment to the respective peptide hydrogels. One has to bear in mind that the surface concentration of the peptide moieties available for interaction with the cells could be lower on the glass substrates as compared to the hydrogels. Upon making the coating, the PEG-peptide conjugates were chemically coupled to the glutaraldehyde-modified glass substrates via the amine groups present on the peptides. Additionally, the peptides tethered to the glass surface had less steric freedom to form the triple-helical assemblies, and this could also negatively affect molecular recognition. Nevertheless, the number of cells attached to the PEG-CLP-RGD coating was two times higher than that on the PEG-CLP coating. This was in favor of a receptor-mediated interaction between the cells and the peptide molecules on the surface. However, for revealing a detailed picture of these interactions, one should design a separate study including synthesis of model surfaces with a controlled and tunable presentation of the peptides (also scrambled sequences) and the performing of molecular characterization.
We also observed the same effect of the RGD motif promoting the cell adhesion upon comparing the cultures on the PEG-CLP-RGD and PEG-CLP hydrogels. The RGD motif promoted cellular attachment despite at the same time restricting the formation of the helical assemblies of the peptides (see above). Thus, the above observations correlate with the previous studies, which demonstrated that RGD-functionalized membranes promote neural cell adhesion and aggregation [
42,
43].
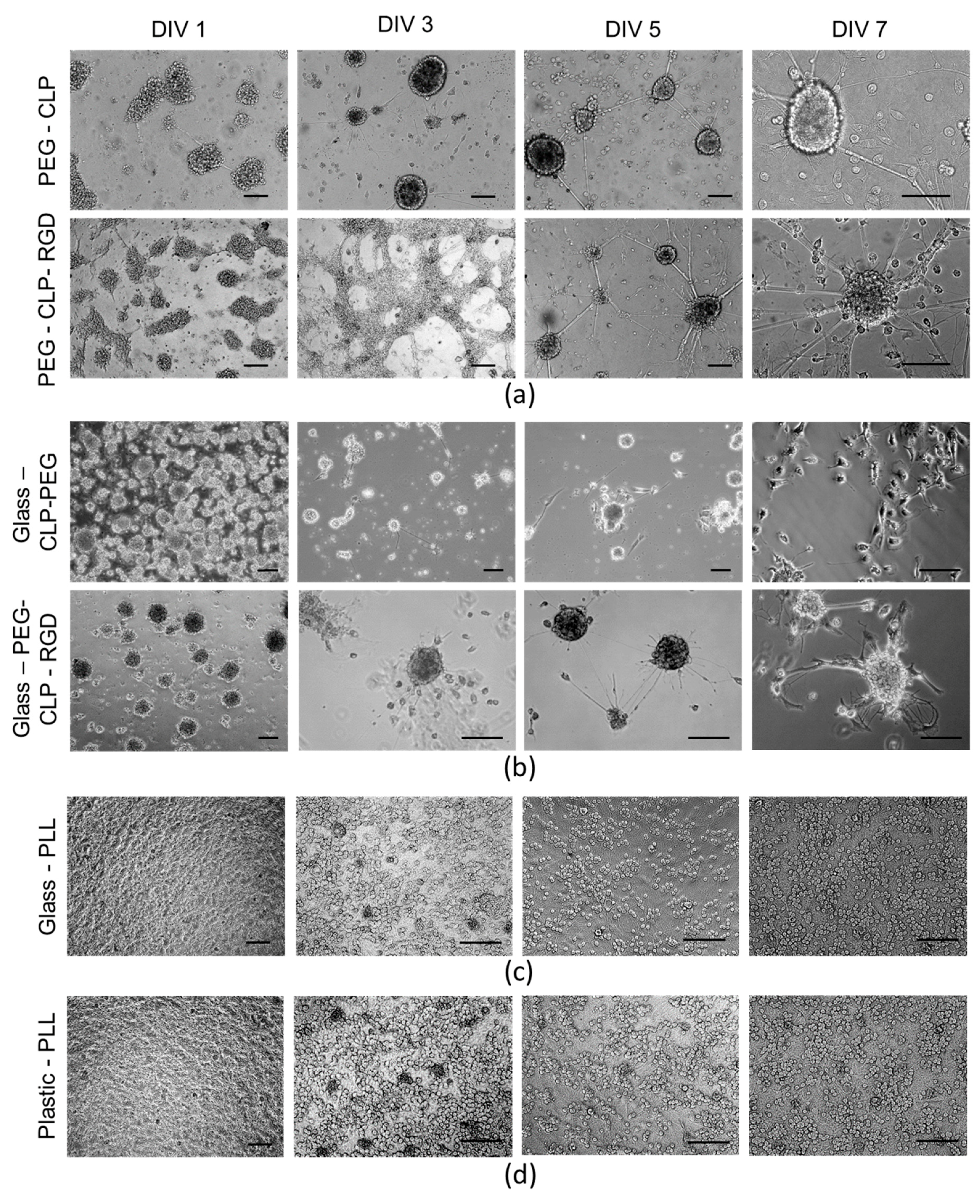
Interestingly, upon seeding the cells on the PEG-CLP-RGD hydrogel, they initially spread, and after several days, they started clustering. Whereas on the PEG-CLP hydrogel, the cells already formed compact spherical clusters during the first day after seeding. These cellular assemblies remained present during the whole monitoring period. Both collagen and fibronectin are known to have distinct roles in the formation of cerebellar shaping during development by cell differentiation and migration control [
14,
15,
30]. Thus, we mimicked this effect by employing the chimeric CLP-RGD peptide rather than formulating hydrogels from individual motifs. The introduction of RGD made the cultures more “relaxed,” not so tightly packed, and potentially more prone to migration, as one might guess from the observed more scattered glial cell distribution.
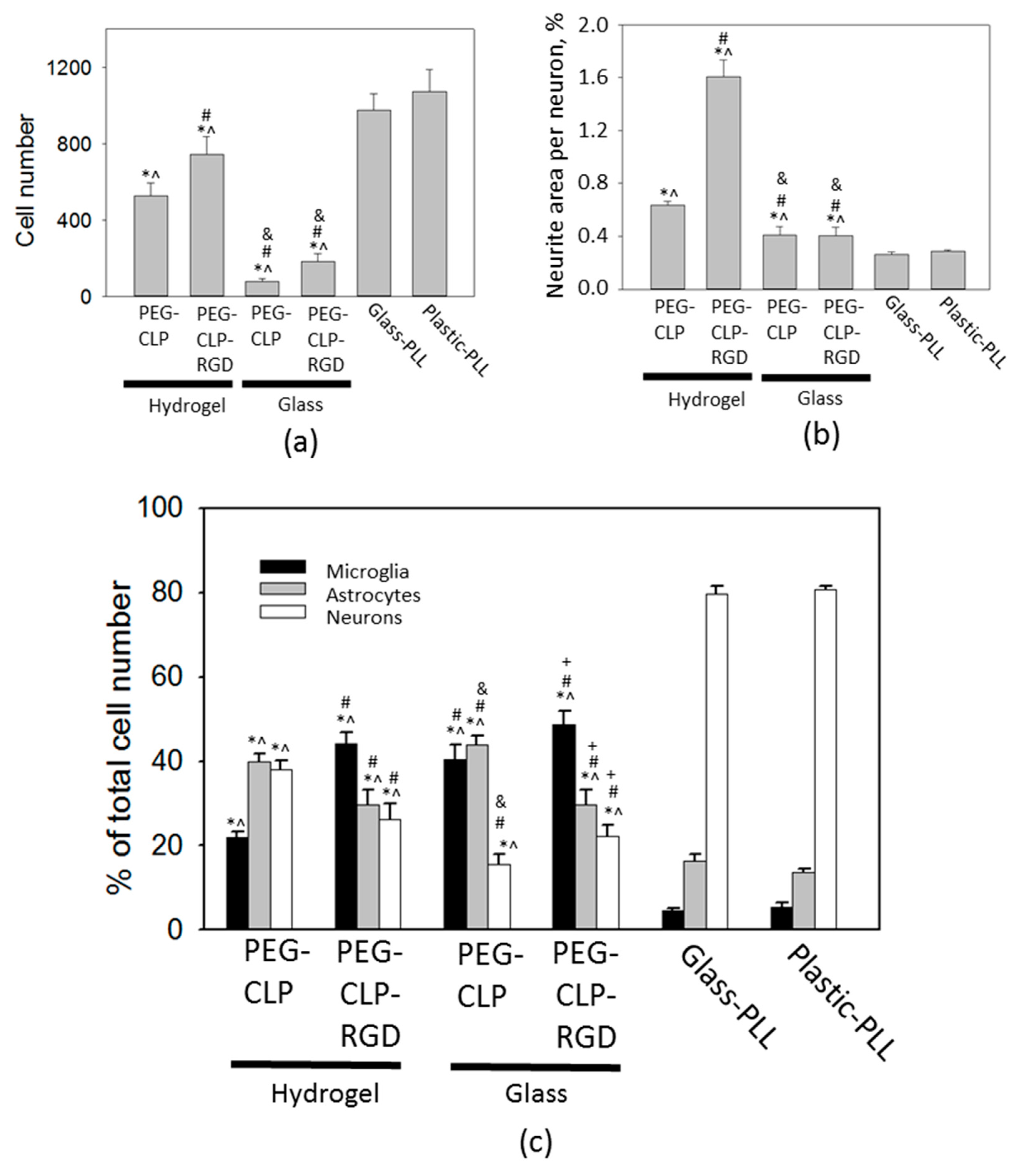
The highest number of cells was on the poly-L-lysine-coated plastic and glass surfaces. Moreover, the neuronal cells highly predominated in these cultures. Not surprisingly, these coatings are very common for neural cell cultures in the laboratories. Giving such a high yield of firmly attached neuronal cells, poly-L-lysine made the neural cell cultures easy manageable. On the other hand, the cells in such cultures were immobilized and did not show any indication for organization or communication. Contrary to this behavior, on the hydrogel membranes, the cerebellar granule cells were gathered to compact clusters resembling the cerebellar granule layer [
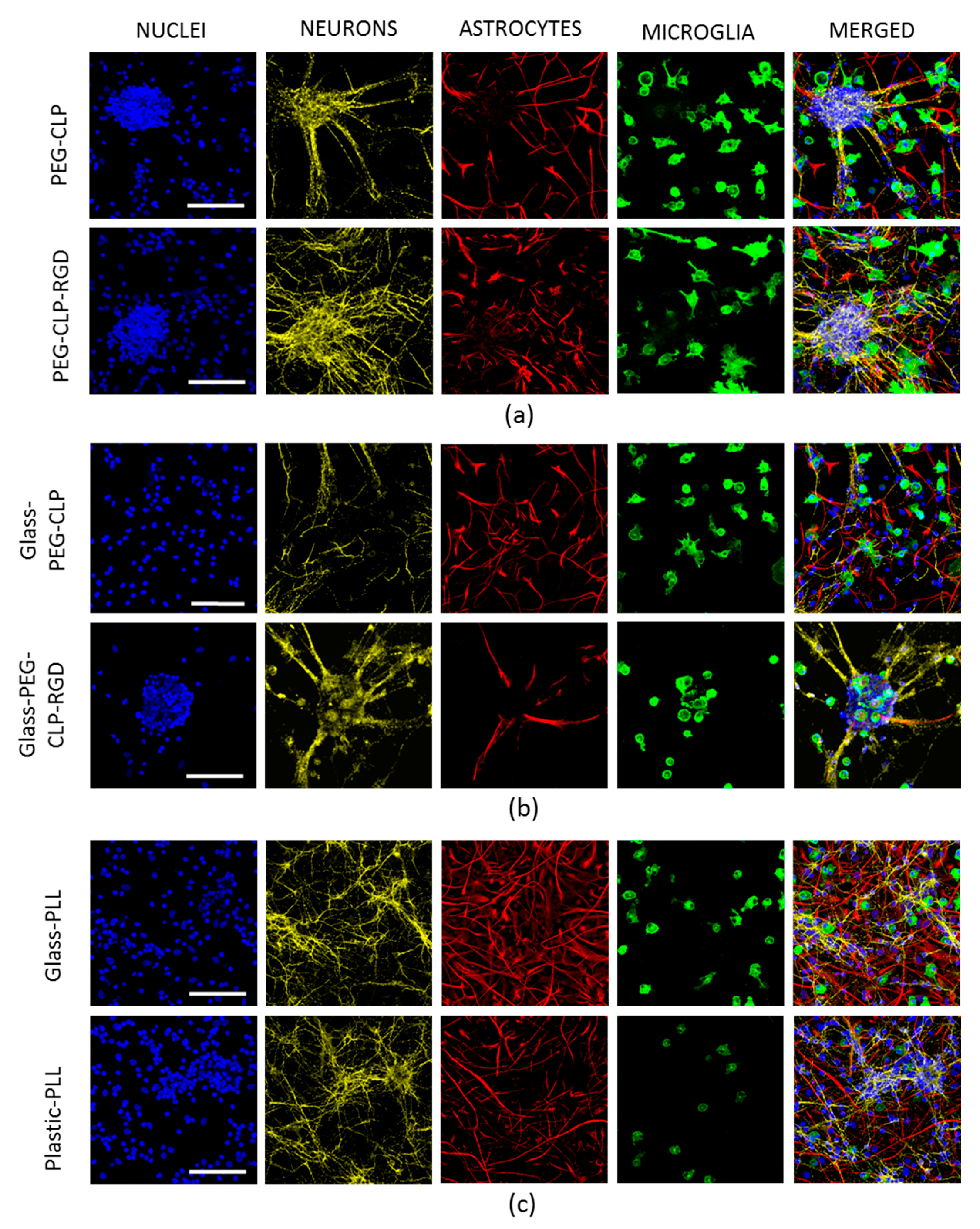
15]. Additionally, the astrocytes were distributed alongside the neurite fibers that connected the clusters into a distinct network. Such a cellular organization suggests there was ongoing functional communication between the neuronal and glial cells in these complex cultures.
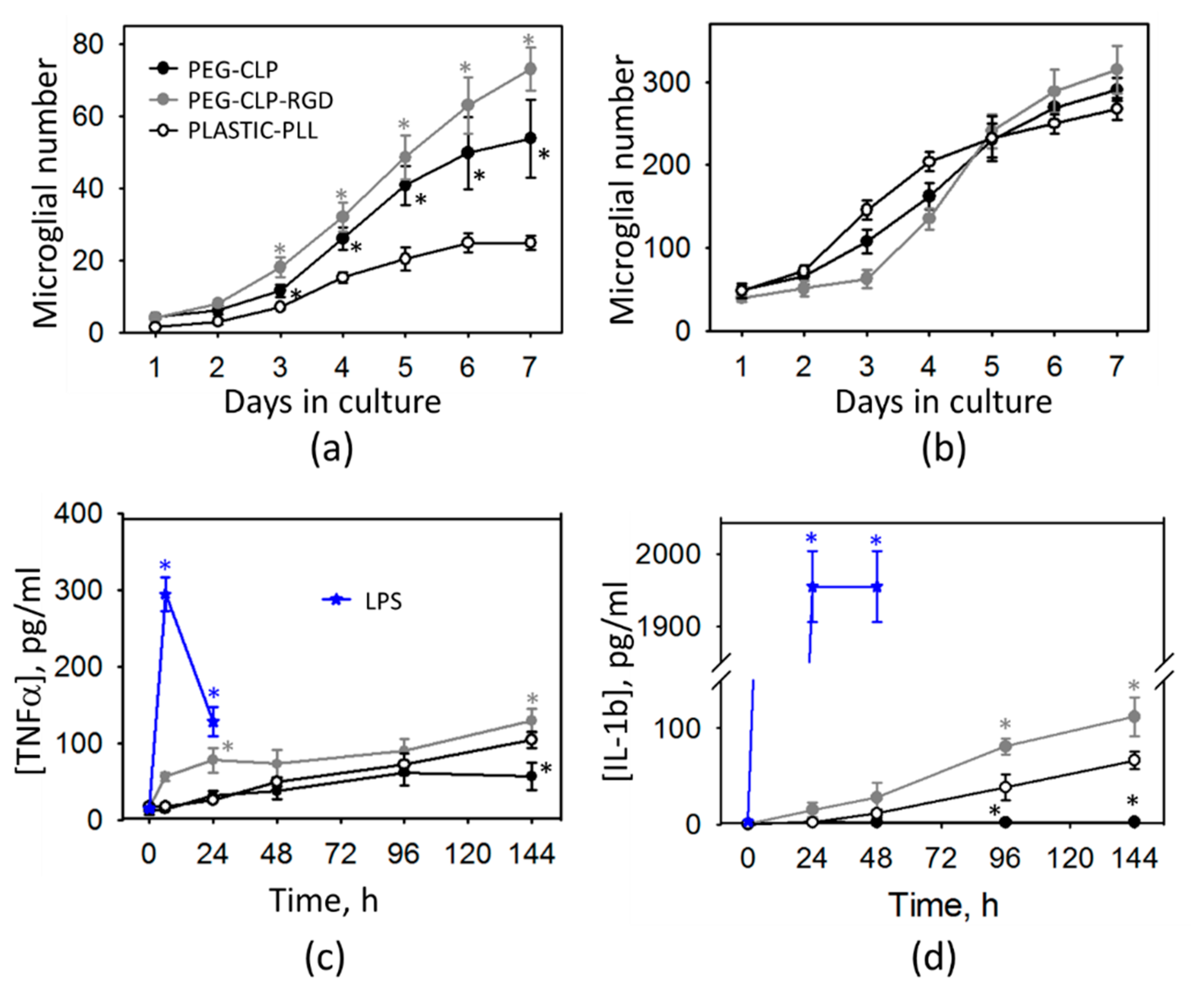
The cerebellar cells grown on hydrogels and PEG-peptide-coated surfaces had higher glial to neuron ratios and elevated numbers of microglia. Perez-Pouchoulen and colleagues found that microglial percentages in developing rat cerebellum vary from about 20 to 60 during the postnatal 21 day period [
44]. Microglial numbers on both the PEG-CLP and PEG-CLP-RGD hydrogels and glass surfaces were within this range, indicating the materials support in vivo-relevant microglial/non-microglial cell ratios. On the other hand, the total cell numbers on PEG-peptide-coated glass surfaces were more than five times lower than on hydrogels and did not support functional cell assemblies. On poly-L-lysine-coated plastic or glass, microglial percentages were far below those found in real cerebellar tissue. In rat cerebellum, microglial numbers increase slightly more than twice, going from postnatal days 5–10 [
44]. In our study, cell culture was prepared from 5–7-day-old rat cerebella and monitored for 7 days in culture. Initially after seeding, the cell behavior might have been influenced by the isolation stress. From DIV2 to DIV7, microglial cell numbers in mixed neuronal-glial cultures increased about two times on the poly-L-lysine-coated plastic, about five times on the PEG-CLP hydrogel, and about seven times on the PEG-CLP-RGD hydrogel. Thus, the microglial proliferation rate on hydrogels was much higher than that observed in cerebellar tissue. However, it has to be taken into account that the cultures were grown with a fetal bovine serum supplement in the culture medium, which is known as microglia activating and proliferation stimulating factor [
45]. Thus, microglial proliferation in all samples examined in this study might have been shifted to higher rates due to the effect of serum supplement.
The cytokine assessment revealed that such microglial proliferation was not associated with any acute inflammatory response; however, there was a slight increase in both TNFα and IL-1β levels of the PEG-CLP-RGD containing hydrogel samples. The increased numbers of glia in the developing cerebellum is a typical sign of intensive brain remodeling processes because the microglial cells are the primary responsible for the neuronal network formation by phagocytosing unnecessary neurons and pruning inefficient synapses [
16,
36]. Fetal microglia secrete pro-inflammatory cytokines including, IL-1, IL-6, and TNFα, which are important regulators of glial proliferation, developmental apoptosis, and synaptogenesis [
46,
47]. Moreover, both in development and during the whole life span, TNFα and IL-1β have been implicated in regulating synaptic transmission and functional plasticity in normal healthy brains [
48]. The nanomolar concentrations of these cytokines are cytoprotective, but micromolar levels are neurotoxic and indicate neuroinflammation [
49]. In our study, the increase of TNFα and IL-1β in the medium of cells grown on PEG-CLP and PEG-CLP-RGD hydrogels was even lower than the nanomolar level; thus, it was unlikely to be a sign of acute neuroinflammation. If the cultures were not in the inflammatory state, the next possible explanation of such microglial proliferation is that the cells isolated from the developing cerebella continued to actively participate in functional neuronal network formation. Altogether, these features allowed us to assume that the PEG-CLP and PEG-CLP-RGD substrates promoted a glial-neuronal communication characteristic for the brain developmental processes.
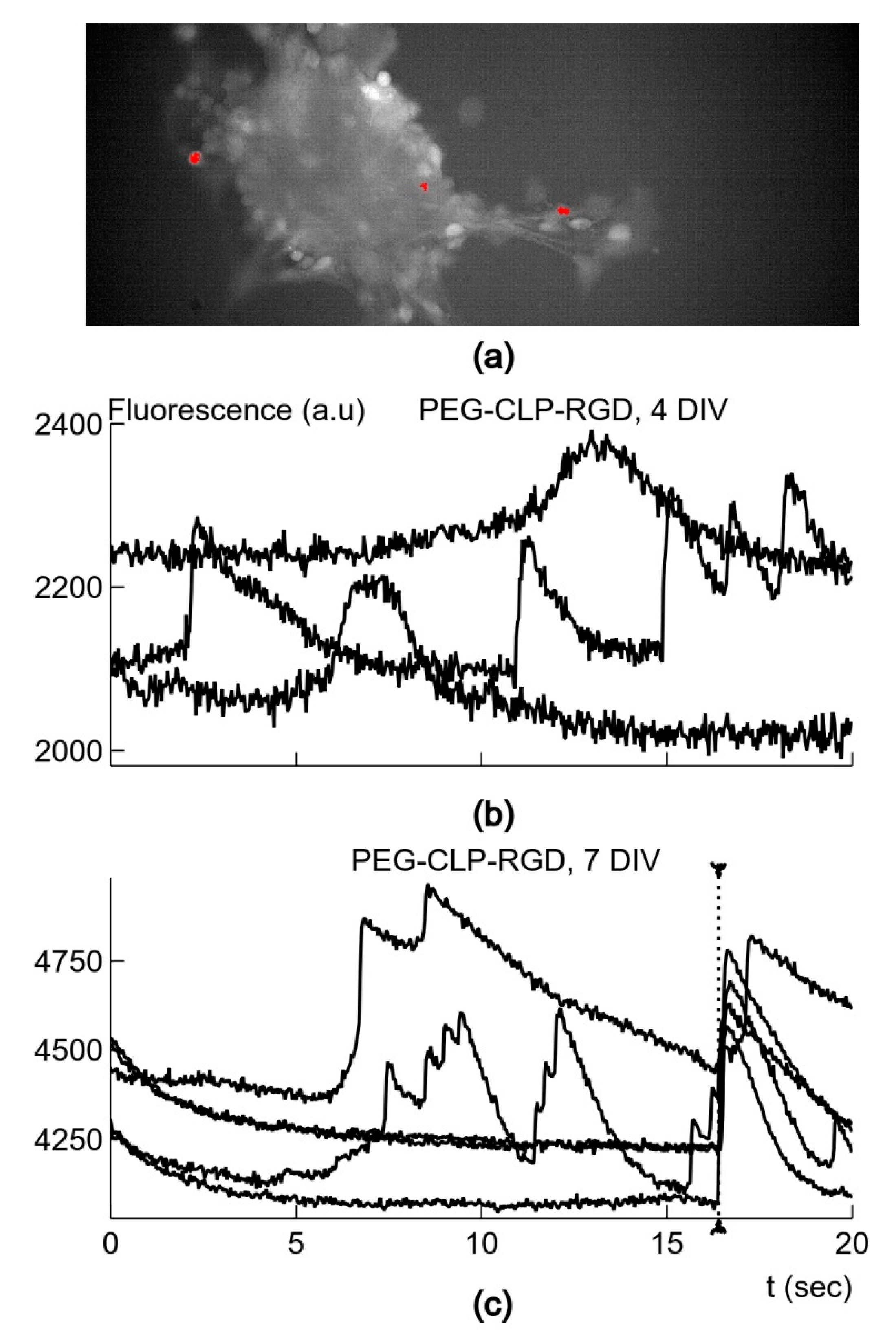
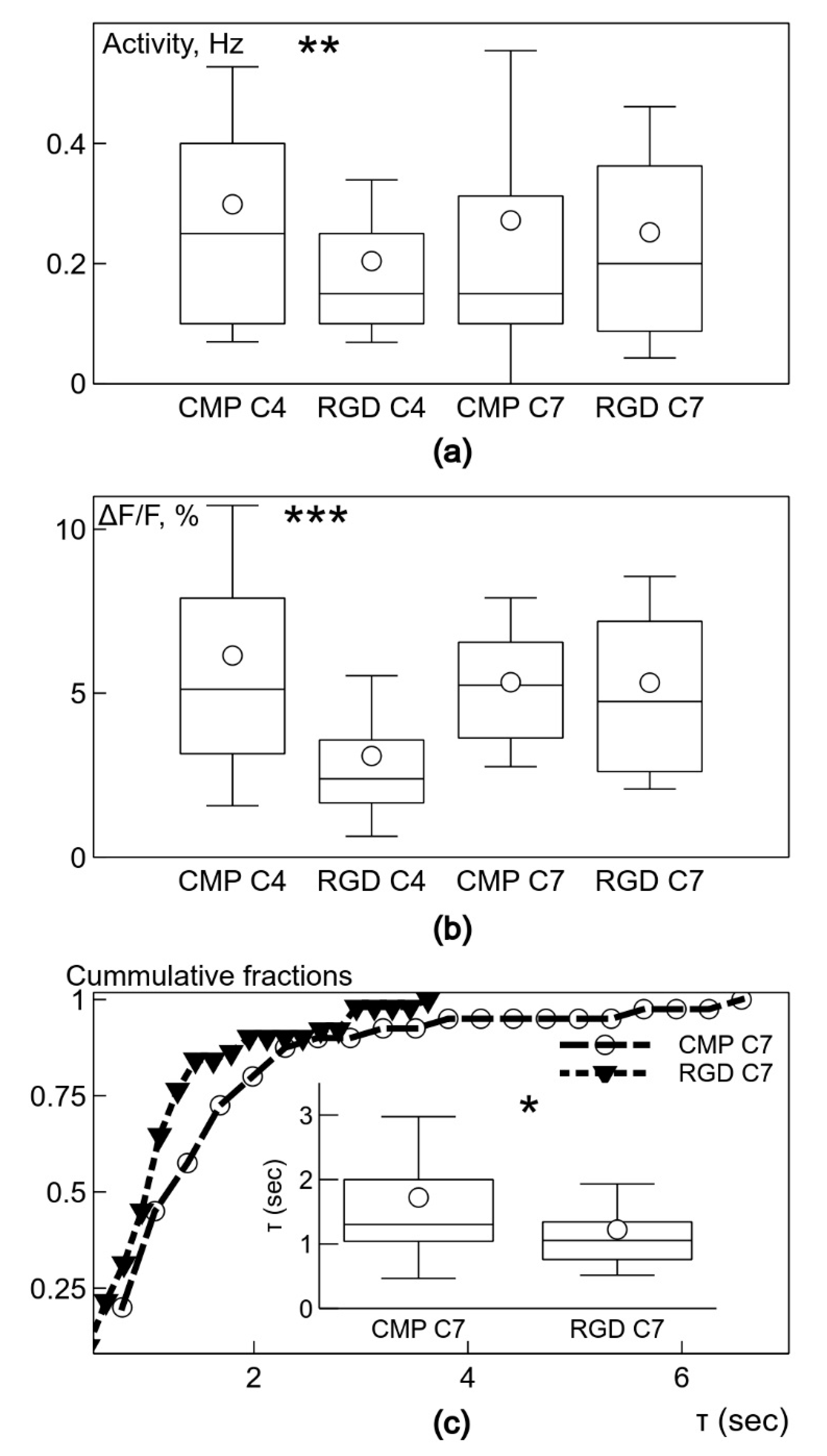
The important finding of the study is that the neuronal clusters on the PEG-CLP and PEG-CLP-RGD hydrogel membranes revealed the signs of spontaneous neuronal activity, as reflected in fast-rising Ca2+ signals on day 4 in culture. Though PEG-CLP induced faster neuronal maturation with a higher firing frequency and amplitude, the incorporation of the RGD motif ensured more qualitative synaptic signaling occurring later in time, characterized by a faster decay of intracellular Ca2+. Furthermore, the initial qualities of the PEG-CLP hydrogel-supported cultures become not significant later in time when the cultures reached DIV7. The pro-functional stimulation of the RGD peptide was also supported by its neurite outgrowth promoting activity that was so clearly visible in the neuritogenesis evaluation experiments. Such a fast and qualitative maturation of the functional cerebellar organoids suggests PEG-CLP and, especially, PEG-CLP-RGD as promising matrices for brain pathology in vitro modelling, as well as brain development, neuronal-glial communication, and other studies. Note that despite so many neurons in the cultures studied, there were no signs of functional activity on both glass and plastic poly-L-lysine-covered surfaces up to DIV7 in culture. Thus, to ensure functional cerebellar organoids, it is necessary to design matrices that not only ensure neuronal cell differentiation and viability but also promote enough glial cells for the neuronal maintenance and enough migratory capacity that would enable neurons to cluster and glial cells to respond to the neuronal signaling.
Regarding the current state of hydrogel use for brain in vitro cell research, the most popular hydrogels, according to the number of publications, are the cell culture-derived matrices Geltrex
TM and Matrigel
®. They are used to develop human brain organoids from iPSCs and even applied for neural tissue engineering in animal studies [
50,
51,
52]. The hydrogels well-support (although not so fast) neural cell differentiation and functional maturation [
53,
54,
55]. Though providing a nice in vivo-like environment for neural cells due to the rich composition of basal lamina, such matrices of biological origin are not defined, are expensive, and slightly vary from batch to batch. Thus, such scaffolds are not very favorable for highly standardized automated high throughput screening applications and tissue regeneration. RADA peptide-based synthetic hydrogel PuraMatrix
TM functionalized with laminin also supports neural cell survival; however, to our knowledge, there are no functional brain cell culture models described on this scaffold. All the above mentioned and other commercially available hydrogel systems provide scientists with the opportunity to self-tune hydrogel composition and stiffness, opening a lot of options for experimentation. On the other hand, this raises the issue of the experimental standardization and comparison of the results produced by different researchers. The chemically crosslinked PEG-peptide hydrogel membranes examined in this study have robust and well-standardized structure and stiffness. Therefore, they might be well-suited for such neural cell culture applications that require a high level of reproducibility, such as drug screening or tissue engineering.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}