Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment



Abstract
1. Introduction
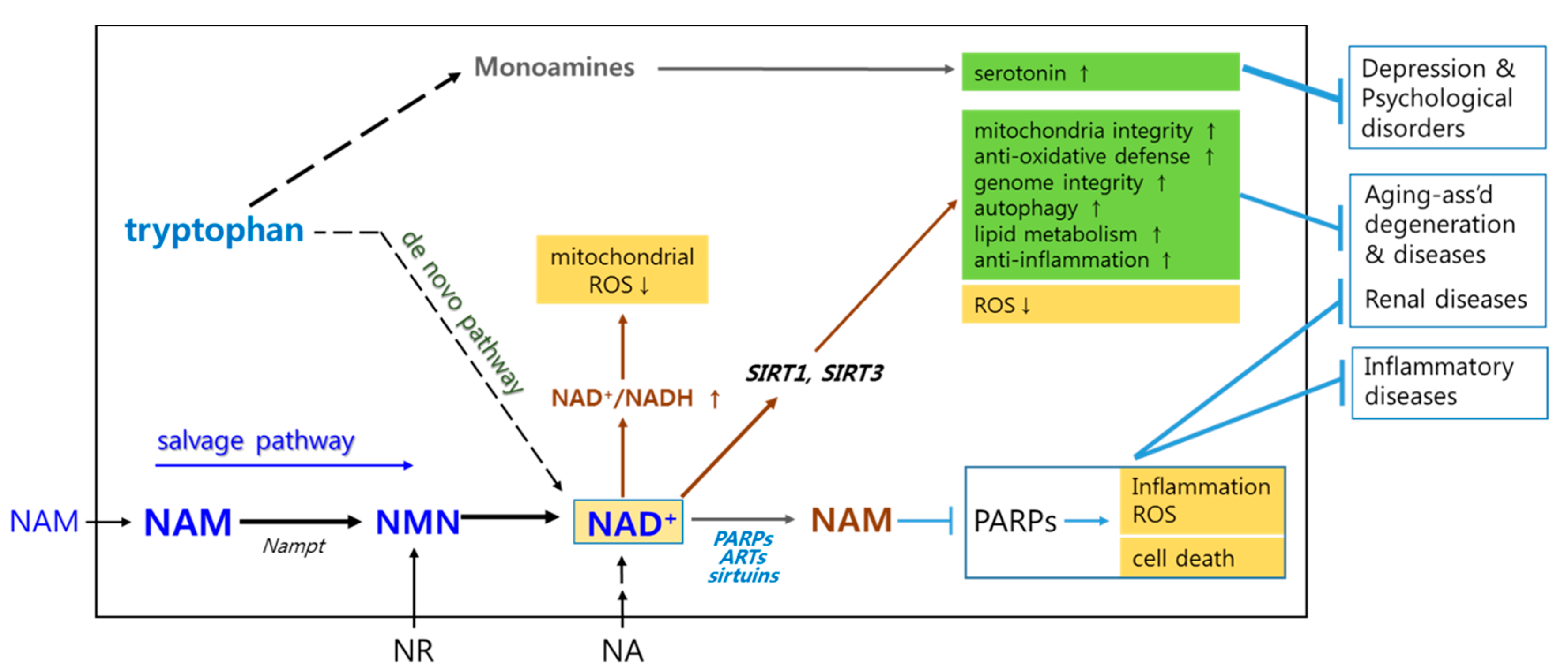
2. Briefs on Biochemistry Associated with Mechanisms Underlying NAM’s Positive Effects
3. Potential Toxicity and Adverse Effects of High Doses of NAM
3.1. Possible Genotoxicity and Carcinogenicity: Inconclusive Effects of NAM
3.2. Inhibition of Sirtuin Activity: An Effect that May Not Be Important In Vivo
3.3. High NAD+/NADH Ratio: Concerns Regarding Energy Metabolism
3.4. High-Level NAD+: Effect on Protein Translation
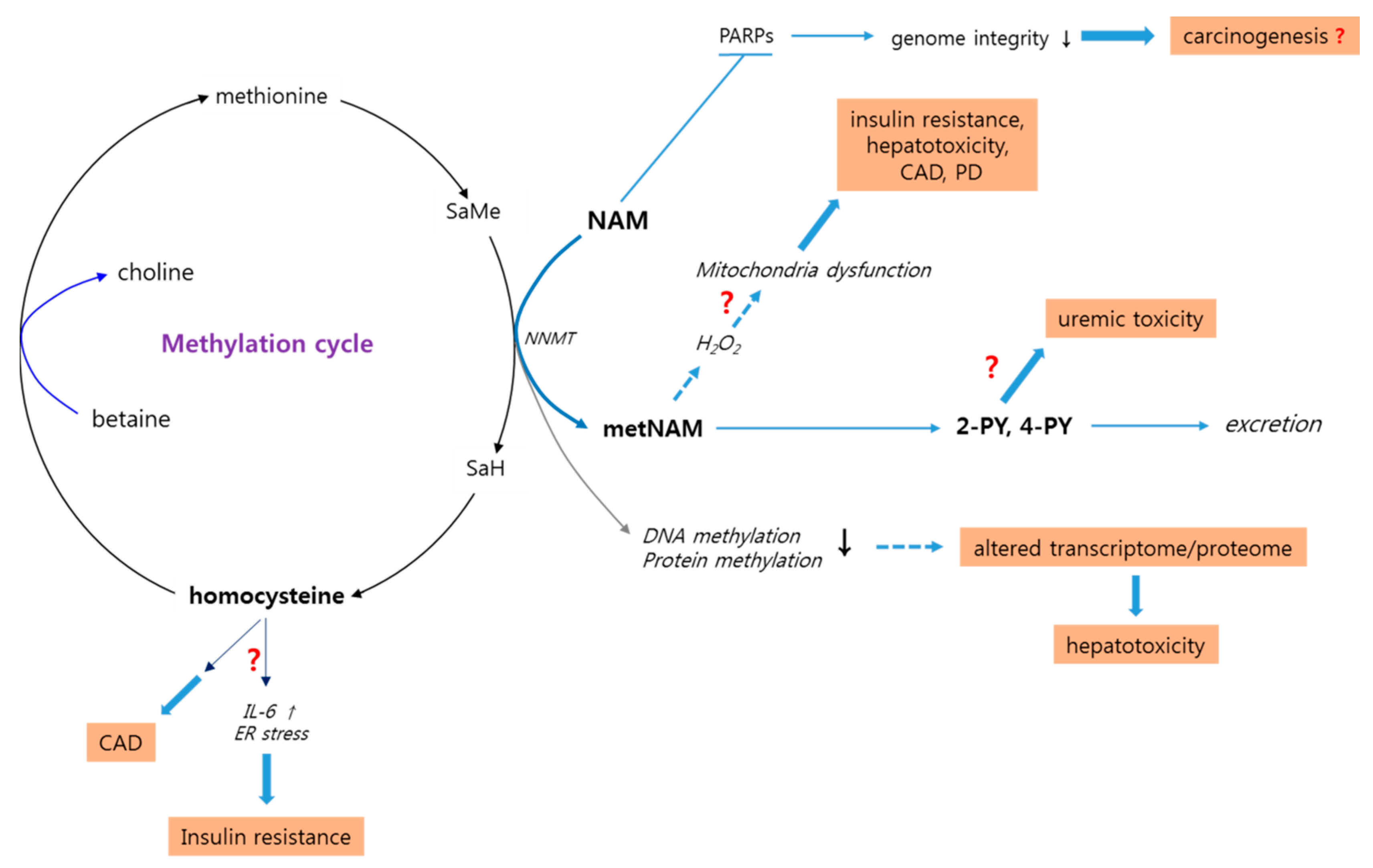
3.5. High-Level NAM Methylation: Potential Effects of Altered Methyl Pool
3.6. High-Level NAM Methylation: Potential Adverse Effects of Altered Methyl Pool
3.6.1. Insulin Resistance and Metabolic Syndrome
3.6.2. Parkinson’s Disease
3.6.3. Cardiac Diseases
3.6.4. Liver Toxicity
3.7. Potential Positive Effects of metNAM: Contradiction to the Proposed Adverse Effects
3.8. N-Methyl-2-Pyridone-5-Carboxamide: A Potential Uremic Toxin
4. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Elvehjem, C.A.; Madden, R.J.; Strong, F.M.; Wolley, D.W. The isolation and identification of the anti-black tongue factor. J. Biol. Chem. 1938, 123, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Klaidman, L.K.; Nalbandian, A.; Oliver, J.; Chang, M.L.; Chan, P.H.; Adams, J.D., Jr. The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in wistar rats. Neurosci. Lett. 2002, 333, 91–94. [Google Scholar] [CrossRef]
- Anderson, D.W.; Bradbury, K.A.; Schneider, J.S. Broad neuroprotective profile of nicotinamide in different mouse models of mptp-induced parkinsonism. Eur. J. Neurosci. 2008, 28, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Mokudai, T.; Ayoub, I.A.; Sakakibara, Y.; Lee, E.J.; Ogilvy, C.S.; Maynard, K.I. Delayed treatment with nicotinamide (vitamin b(3)) improves neurological outcome and reduces infarct volume after transient focal cerebral ischemia in wistar rats. Stroke 2000, 31, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Miao, C.Y. Nampt as a therapeutic target against stroke. Trends Pharmacol. Sci. 2015, 36, 891–905. [Google Scholar] [CrossRef]
- Sheline, C.T.; Cai, A.L.; Zhu, J.; Shi, C. Serum or target deprivation-induced neuronal death causes oxidative neuronal accumulation of zn2+ and loss of nad+. Eur. J. Neurosci. 2010, 32, 894–904. [Google Scholar] [CrossRef]
- Cai, A.L.; Zipfel, G.J.; Sheline, C.T. Zinc neurotoxicity is dependent on intracellular nad levels and the sirtuin pathway. Eur. J. Neurosci. 2006, 24, 2169–2176. [Google Scholar] [CrossRef]
- Hathorn, T.; Snyder-Keller, A.; Messer, A. Nicotinamide improves motor deficits and upregulates pgc-1alpha and bdnf gene expression in a mouse model of huntington’s disease. Neurobiol. Dis. 2011, 41, 43–50. [Google Scholar] [CrossRef]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laco, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in huntington’s disease models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of thr231-phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef]
- Bold, J.M.; Gardner, C.R.; Walker, R.J. Central effects of nicotinamide and inosine which are not mediated through benzodiazepine receptors. Br. J. Pharmacol. 1985, 84, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Hiromatsu, Y.; Sato, M.; Yamada, K.; Nonaka, K. Inhibitory effects of nicotinamide on recombinant human interferon-gamma-induced intercellular adhesion molecule-1 (icam-1) and hla-dr antigen expression on cultured human endothelial cells. Immunol. Lett. 1992, 31, 35–39. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Matsui, T.C.; Godin, A.M.; Gomides, L.F.; Pereira-Silva, P.E.; Duarte, I.D.; Menezes, G.B.; Coelho, M.M.; Klein, A. Neutrophil recruitment is inhibited by nicotinamide in experimental pleurisy in mice. Eur. J. Pharmacol. 2012, 685, 198–204. [Google Scholar] [CrossRef]
- Miesel, R.; Kurpisz, M.; Kroger, H. Modulation of inflammatory arthritis by inhibition of poly(adp ribose) polymerase. Inflammation 1995, 19, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Snaidr, V.A.; Damian, D.L.; Halliday, G.M. Nicotinamide for photoprotection and skin cancer chemoprevention: A review of efficacy and safety. Exp. Dermatol. 2019, 28 (Suppl. 1), 15–22. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.F. Nicotinamide: An oral antimicrobial agent with activity against both mycobacterium tuberculosis and human immunodeficiency virus. Clin. Infect. Dis. 2003, 36, 453–460. [Google Scholar] [CrossRef]
- Murray, M.F.; Srinivasan, A. Nicotinamide inhibits hiv-1 in both acute and chronic in vitro infection. Biochem. Biophys. Res. Commun. 1995, 210, 954–959. [Google Scholar] [CrossRef]
- Crino, A.; Schiaffini, R.; Manfrini, S.; Mesturino, C.; Visalli, N.; Beretta Anguissola, G.; Suraci, C.; Pitocco, D.; Spera, S.; Corbi, S.; et al. A randomized trial of nicotinamide and vitamin e in children with recent onset type 1 diabetes (imdiab ix). Eur. J. Endocrinol. 2004, 150, 719–724. [Google Scholar] [CrossRef]
- Elliott, R.B.; Pilcher, C.C.; Fergusson, D.M.; Stewart, A.W. A population based strategy to prevent insulin-dependent diabetes using nicotinamide. J. Pediatric Endocrinol. Metab. 1996, 9, 501–509. [Google Scholar] [CrossRef]
- Gale, E.A.; Bingley, P.J.; Emmett, C.L.; Collier, T.; European Nicotinamide Diabetes Intervention Trial Group. European nicotinamide diabetes intervention trial (endit): A randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 2004, 363, 925–931. [Google Scholar] [CrossRef]
- Staeva-Vieira, T.; Peakman, M.; von Herrath, M. Translational mini-review series on type 1 diabetes: Immune-based therapeutic approaches for type 1 diabetes. Clin. Exp. Immunol. 2007, 148, 17–31. [Google Scholar] [CrossRef]
- Maes, M.; Galecki, P.; Chang, Y.S.; Berk, M. A review on the oxidative and nitrosative stress (o&ns) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 676–692. [Google Scholar]
- Hollis, F.; van der Kooij, M.A.; Zanoletti, O.; Lozano, L.; Canto, C.; Sandi, C. Mitochondrial function in the brain links anxiety with social subordination. Proc. Natl. Acad. Sci. USA 2015, 112, 15486–15491. [Google Scholar] [CrossRef]
- Dedee, F.; Murrell, M.R.-Q. Management and Prognosis of Bullous Pemphigoid. 2019. Available online: https://www.uptodate.com/contents/management-and-prognosis-of-bullous-pemphigoid (accessed on 23 April 2020).
- Starr, P. Oral nicotinamide prevents common skin cancers in high-risk patients, reduces costs. Am. Health Drug. Benefits 2015, 8, 13–14. [Google Scholar] [PubMed]
- Damian, D.L.; Patterson, C.R.; Stapelberg, M.; Park, J.; Barnetson, R.S.; Halliday, G.M. Uv radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J. Investig. Dermatol. 2008, 128, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Park, J.S.; Chung, G.J.; Lee, I.H.; Hwang, E.S. Diverse therapeutic efficacies and more diverse mechanisms of nicotinamide. Metabolomics 2019, 15, 137. [Google Scholar] [CrossRef] [PubMed]
- Prousky, J. Vitamin b3 for depression: Case report and review of the literature. J. Orthomol. Med. 2010, 25, 137–147. [Google Scholar]
- Saini, J.S.; Corneo, B.; Miller, J.D.; Kiehl, T.R.; Wang, Q.; Boles, N.C.; Blenkinsop, T.A.; Stern, J.H.; Temple, S. Nicotinamide ameliorates disease phenotypes in a human ipsc model of age-related macular degeneration. Cell Stem Cell 2017, 20, 635-647.e7. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cardozo, B.H.; Cochran, K.E.; John, S.W.M. Nicotinamide and wld(s) act together to prevent neurodegeneration in glaucoma. Front. Neurosci. 2017, 11, 232. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; John, S.W.M. Glaucoma as a metabolic optic neuropathy: Making the case for nicotinamide treatment in glaucoma. J. Glaucoma 2017, 26, 1161–1168. [Google Scholar] [CrossRef]
- Nakajima, H.; Yamada, K.; Hanafusa, T.; Fujino-Kurihara, H.; Miyagawa, J.; Miyazaki, A.; Saitoh, R.; Minami, Y.; Kono, N.; Nonaka, K.; et al. Elevated antibody-dependent cell-mediated cytotoxicity and its inhibition by nicotinamide in the diabetic nod mouse. Immunol. Lett. 1986, 12, 91–94. [Google Scholar] [CrossRef]
- Monfrecola, G.; Gaudiello, F.; Cirillo, T.; Fabbrocini, G.; Balato, A.; Lembo, S. Nicotinamide downregulates gene expression of interleukin-6, interleukin-10, monocyte chemoattractant protein-1, and tumour necrosis factor-alpha gene expression in hacat keratinocytes after ultraviolet b irradiation. Clin. Exp. Dermatol. 2013, 38, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Nagai, A.; Matsumiya, H.; Hayashi, M.; Yasui, S.; Okamoto, H.; Konno, K. Effects of nicotinamide and niacin on bleomycin-induced acute injury and subsequent fibrosis in hamster lungs. Exp. Lung Res. 1994, 20, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Gurujeyalakshmi, G.; Iyer, S.N.; Hollinger, M.A.; Giri, S.N. Procollagen gene expression is down-regulated by taurine and niacin at the transcriptional level in the bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1996, 277, 1152–1157. [Google Scholar]
- Kim, S.K.; Yun, S.J.; Kim, J.; Lee, O.J.; Bae, S.C.; Kim, W.J. Identification of gene expression signature modulated by nicotinamide in a mouse bladder cancer model. PLoS ONE 2011, 6, e26131. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Lee, J.W.; Quan, C.; Youn, H.J.; Kim, H.M.; Bae, S.C. Nicotinamide inhibits growth of carcinogen induced mouse bladder tumor and human bladder tumor xenograft through up-regulation of runx3 and p300. J. Urol. 2011, 185, 2366–2375. [Google Scholar] [CrossRef]
- Omidian, M.; Khazanee, A.; Yaghoobi, R.; Ghorbani, A.R.; Pazyar, N.; Beladimousavi, S.S.; Ghadimi, M.; Mohebbipour, A.; Feily, A. Therapeutic effect of oral nicotinamide on refractory uremic pruritus: A randomized, double-blind study. Saudi J. Kidney Dis. Transplant. 2013, 24, 995–999. [Google Scholar]
- Pozzilli, P.; Visalli, N.; Buzzetti, R.; Cavallo, M.G.; Marietti, G.; Hawa, M.; Leslie, R.D. Metabolic and immune parameters at clinical onset of insulin-dependent diabetes: A population-based study. Imdiab study group. Immunotherapy diabetes. Metabolism 1998, 47, 1205–1210. [Google Scholar] [CrossRef]
- Kalckar, H.M.; Maxwell, E.S.; Strominger, J.L. Some properties of uridine diphosphoglucose dehydrogenase. Arch. Biochem. Biophys. 1956, 65, 2–10. [Google Scholar]
- Muller, D.; Mehling, H.; Otto, B.; Bergmann-Lips, R.; Luft, F.; Jordan, J.; Kettritz, R. Niacin lowers serum phosphate and increases hdl cholesterol in dialysis patients. Clin. J. Am. Soc. Nephrol. 2007, 2, 1249–1254. [Google Scholar] [CrossRef]
- Surjana, D.; Halliday, G.M.; Martin, A.J.; Moloney, F.J.; Damian, D.L. Oral nicotinamide reduces actinic keratoses in phase ii double-blinded randomized controlled trials. J. Investig. Dermatol. 2012, 132, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Hoyer, M.; Honess, D.J.; Dennis, I.F.; Overgaard, J. Nicotinamide pharmacokinetics in humans and mice: A comparative assessment and the implications for radiotherapy. Radiother. Oncol. 1993, 27, 131–139. [Google Scholar] [CrossRef]
- Hoffer, A. Biochemistry of nicotinic acid and nicotinamide. Psychosomatics 1967, 8, 95–100. [Google Scholar] [CrossRef]
- Greenbaum, C.J.; Kahn, S.E.; Palmer, J.P. Nicotinamide’s effects on glucose metabolism in subjects at risk for iddm. Diabetes 1996, 45, 1631–1634. [Google Scholar] [CrossRef]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of sirt1 in vitro, but can be a stimulator in cells. Cell. Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.L.; Boyer, J.L. Hepatic toxicity from large doses of vitamin b3 (nicotinamide). N. Engl. J. Med. 1973, 289, 1180–1182. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Bodeau, S.; Louvet, L.; Mary, A.; Boullier, A.; Lemaire-Hurtel, A.S.; Jonet, A.; Sonnet, P.; Kamel, S.; et al. N-methyl-2-pyridone-5-carboxamide (2py)-major metabolite of nicotinamide: An update on an old uremic toxin. Toxins (Basel) 2016, 8, 339. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Esper, N.E.; Brisset, S.; Mansour, J.; Lemaire-Hurtel, A.S.; Mary, A.; Brazier, M.; Kamel, S.; Mentaverri, R.; et al. Efficacy and safety of nicotinamide in haemodialysis patients: The nicoren study. Nephrol. Dial. Transplant. 2017, 32, 1597. [Google Scholar] [CrossRef]
- Jiao, X.; Doamekpor, S.K.; Bird, J.G.; Nickels, B.E.; Tong, L.; Hart, R.P.; Kiledjian, M. 5’ end nicotinamide adenine dinucleotide cap in human cells promotes rna decay through dxo-mediated denadding. Cell 2017, 168, 1015-1027.e10. [Google Scholar] [CrossRef]
- Zhou, S.S.; Li, D.; Zhou, Y.M.; Sun, W.P.; Liu, Q.G. B-vitamin consumption and the prevalence of diabetes and obesity among the us adults: Population based ecological study. BMC Public Health 2010, 10, 746. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of nad(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, M.; Schemainda, I. Fk866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Ghosh, D.; LeVault, K.R.; Barnett, A.J.; Brewer, G.J. A reversible early oxidized redox state that precedes macromolecular ros damage in aging nontransgenic and 3xtg-ad mouse neurons. J. Neurosci. 2012, 32, 5821–5832. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Jang, S.Y.; Kang, H.T.; Wei, B.; Jeoun, U.W.; Yoon, G.S.; Hwang, E.S. Modulation of mitochondrial membrane potential and ros generation by nicotinamide in a manner independent of sirt1 and mitophagy. Mol. Cells 2017, 40, 503–514. [Google Scholar]
- Aman, Y.; Qiu, Y.; Tao, J.; Fang, E.F. Therapeutic potential of boosting nad+ in aging and age-related diseases. Transl. Med. Aging 2018, 2, 30–37. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ros signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenet. 2016, 8, 61. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of foxo transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by sirt3-mediated sod2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3 Cell Metab-mediated deacetylation of evolutionarily conserved lysine 122 regulates mnsod activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mptp by sirt3-mediated deacetylation of cypd at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010, 2, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. Nicotinamide modulates mitochondrial membrane potential and cysteine protease activity during cerebral vascular endothelial cell injury. J. Vasc. Res. 2002, 39, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Chong, Z.Z.; Maiese, K. Nicotinamide: A nutritional supplement that provides protection against neuronal and vascular injury. J. Med. Food 2001, 4, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Lee, H.I.; Hwang, E.S. Nicotinamide extends replicative lifespan of human cells. Aging Cell 2006, 5, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high nad+/nadh ratio and sirt1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Hwang, E.S. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell 2009, 8, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Ok, J.S.; Song, S.B.; Hwang, E.S. Enhancement of replication and differentiation potential of human bone marrow stem cells by nicotinamide treatment. Int. J. Stem Cells 2018, 11, 13–25. [Google Scholar] [CrossRef]
- Berger, N.A. Poly(adp-ribose) in the cellular response to DNA damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef]
- Zhang, J.; Dawson, V.L.; Dawson, T.M.; Snyder, S.H. Nitric oxide activation of poly(adp-ribose) synthetase in neurotoxicity. Science 1994, 263, 687–689. [Google Scholar] [CrossRef]
- Ying, W.; Garnier, P.; Swanson, R.A. Nad+ repletion prevents parp-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem. Biophys. Res. Commun. 2003, 308, 809–813. [Google Scholar] [CrossRef]
- Zong, W.X.; Ditsworth, D.; Bauer, D.E.; Wang, Z.Q.; Thompson, C.B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004, 18, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, S.; Pioli, C. Multifaceted role of parp-1 in DNA repair and inflammation: Pathological and therapeutic implications in cancer and non-cancer diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, A. Treatment of arthritis by nicotinic acid and nicotinamide. Can. Med. Assoc. J. 1959, 81, 235–238. [Google Scholar] [PubMed]
- William, K. The Common form of Joint Dysfunction: Its Incidence and Treatment; E. L. Hildreth & Company: Brattleboro, VT, USA, 1949. [Google Scholar]
- Namazi, M.R. Nicotinamide: A potential addition to the anti-psoriatic weaponry. FASEB J. 2003, 17, 1377–1379. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.; Janjua, M.Z. The optimum dose of nicotinamide for protection of pancreatic beta-cells against the cytotoxic effect of streptozotocin in albino rat. J. Ayub Med. Coll. Abbottabad 2001, 13, 26–30. [Google Scholar]
- Kolb, H.; Burkart, V. Nicotinamide in type 1 diabetes. Mechanism of action revisited. Diabetes Care 1999, 22 (Suppl. 2), B16–B20. [Google Scholar]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of sirt1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of pgc-1alpha and sirt1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Ahn, J.; Cho, I.; Kim, S.; Kwon, D.; Ha, T. Dietary resveratrol alters lipid metabolism-related gene expression of mice on an atherogenic diet. J. Hepatol. 2008, 49, 1019–1028. [Google Scholar] [CrossRef]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.H.; Min, B.H.; Lee, K.M.; Cho, M.H.; Park, G.H.; Lee, K.H. Sirt1 promotes DNA repair activity and deacetylation of ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. Sirt1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of foxos and p53 by sirt1 modulators under oxidative stress. PLoS ONE 2013, 8, e73875. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.J.; Liu, N.; Xiao, Z.; Sun, T.; Wu, S.H.; Sun, W.X.; Xu, Z.G.; Yuan, H. Renal protective effect of sirtuin 1. J. Diabetes Res. 2014, 2014, 843786. [Google Scholar] [CrossRef]
- Delmez, J.A.; Slatopolsky, E. Hyperphosphatemia: Its consequences and treatment in patients with chronic renal disease. Am. J. Kidney Dis. 1992, 19, 303–317. [Google Scholar] [CrossRef]
- Thomas, P.; Dousa, S.A.K. Role of nicotinamide adenine dinucleotide (nad) in control of proximal renal tubular phosphate transport. In Urolithiasis; Springer: Boston, MA, USA, 1981; pp. 741–745. [Google Scholar]
- Eto, N.; Miyata, Y.; Ohno, H.; Yamashita, T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol. Dial. Transplant. 2005, 20, 1378–1384. [Google Scholar] [CrossRef]
- Rennie, G.; Chen, A.C.; Dhillon, H.; Vardy, J.; Damian, D.L. Nicotinamide and neurocognitive function. Nutr. Neurosci. 2015, 18, 193–200. [Google Scholar] [CrossRef]
- Slominski, A.; Semak, I.; Pisarchik, A.; Sweatman, T.; Szczesniewski, A.; Wortsman, J. Conversion of l-tryptophan to serotonin and melatonin in human melanoma cells. FEBS Lett. 2002, 511, 102–106. [Google Scholar] [CrossRef]
- McCarty, M.F. High-dose pyridoxine as an ’anti-stress’ strategy. Med. Hypotheses 2000, 54, 803–807. [Google Scholar] [CrossRef]
- Unilever. Niacinamide: Safety Assessment (Document Number d97/059), Section 5; Safety Assessment of Topically Applied Niacinamide; CTFA (Personal Care Products Council): Washington, DC, USA, 1998. [Google Scholar]
- Li, D.; Tian, Y.J.; Guo, J.; Sun, W.P.; Lun, Y.Z.; Guo, M.; Luo, N.; Cao, Y.; Cao, J.M.; Gong, X.J.; et al. Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats. Br. J. Nutr. 2013, 110, 2156–2164. [Google Scholar] [CrossRef]
- Griffin, S.M.; Pickard, M.R.; Orme, R.P.; Hawkins, C.P.; Fricker, R.A. Nicotinamide promotes neuronal differentiation of mouse embryonic stem cells in vitro. Neuroreport 2013, 24, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Pubchem. 13 Toxicity. National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nicotinamide (accessed on 23 April 2020).
- Lewis, C.M.; Canafax, D.M.; Sprafka, J.M.; Barbosa, J.J. Double-blind randomized trial of nicotinamide on early-onset diabetes. Diabetes Care 1992, 15, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Lopez, E.; Saleh-Gohari, N.; Helleday, T. Poly(adp-ribose) polymerase (parp-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003, 31, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Cefle, K.; Ucur, A.; Guney, N.; Ozturk, S.; Palanduz, S.; Tas, F.; Asoglu, O.; Bayrak, A.; Muslumanoglu, M.; Aydiner, A. Increased sister chromatid exchange frequency in young women with breast cancer and in their first-degree relatives. Cancer Genet. Cytogenet. 2006, 171, 65–67. [Google Scholar] [CrossRef]
- Utakoji, T.; Hosoda, K.; Umezawa, K.; Sawamura, M.; Matsushima, T.; Miwa, M.; Sugimura, T. Induction of sister chromatid exchanges by nicotinamide in chinese hamster lung fibroblasts and human lymphoblastoid cells. Biochem. Biophys. Res. Commun. 1979, 90, 1147–1152. [Google Scholar] [CrossRef]
- Oikawa, A.; Tohda, H.; Kanai, M.; Miwa, M.; Sugimura, T. Inhibitors of poly(adenosine diphosphate ribose) polymerase induce sister chromatid exchanges. Biochem. Biophys. Res. Commun. 1980, 97, 1311–1316. [Google Scholar] [CrossRef]
- Ishidate, M., Jr.; Harnois, M.C.; Sofuni, T. A comparative analysis of data on the clastogenicity of 951 chemical substances tested in mammalian cell cultures. Mutat. Res. 1988, 195, 151–213. [Google Scholar] [CrossRef]
- Lindahl-Kiessling, K.; Shall, S. Nicotinamide deficiency and benzamide-induced sister chromatid exchanges. Carcinogenesis 1987, 8, 1185–1188. [Google Scholar] [CrossRef]
- Ishidate, M., Jr.; Sofuni, T.; Yoshikawa, K.; Hayashi, M.; Nohmi, T.; Sawada, M.; Matsuoka, A. Primary mutagenicity screening of food additives currently used in japan. Food Chem. Toxicol. 1984, 22, 623–636. [Google Scholar] [CrossRef]
- Riklis, E.; Kol, R.; Marko, R. Trends and developments in radioprotection: The effect of nicotinamide on DNA repair. Int. J. Radiat. Biol. 1990, 57, 699–708. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, Y.; Li, L.; Wang, H.H.; Ma, X.S.; Qian, W.P.; Shen, W.; Schatten, H.; Sun, Q.Y. Sirt1, 2, 3 protect mouse oocytes from postovulatory aging. Aging (Albany NY) 2016, 8, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Rakieten, N.; Gordon, B.S.; Beaty, A.; Cooney, D.A.; Davis, R.D.; Schein, P.S. Pancreatic islet cell tumors produced by the combined action of streptozotocin and nicotinamide. Proc. Soc. Exp. Biol. Med. 1971, 137, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Schoental, R. The role of nicotinamide and of certain other modifying factors in diethylnitrosamine carcinogenesis: Fusaria mycotoxins and "spontaneous" tumors in animals and man. Cancer 1977, 40, 1833–1840. [Google Scholar] [CrossRef]
- Handler, P.; Dann, W.J. The inhibition of rat growth by nicotinamide. J. Biol. Chem. 1942, 146, 357–368. [Google Scholar]
- Handler, P. The effect of excessive nicotinamide feeding on rabbits and guinea pigs. J. Biol. Chem. 1944, 154, 203–206. [Google Scholar]
- Kang-Lee, Y.A.; McKee, R.W.; Wright, S.M.; Swendseid, M.E.; Jenden, D.J.; Jope, R.S. Metabolic effects of nicotinamide administration in rats. J. Nutr. 1983, 113, 215–221. [Google Scholar] [CrossRef]
- Mahmoud, Y.I.; Mahmoud, A.A. Role of nicotinamide (vitamin b3) in acetaminophen-induced changes in rat liver: Nicotinamide effect in acetaminophen-damged liver. Exp. Toxicol. Pathol. 2016, 68, 345–354. [Google Scholar] [CrossRef]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. Nnmt activation can contribute to the development of fatty liver disease by modulating the nad (+) metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef]
- Harrison, I.F.; Powell, N.M.; Dexter, D.T. The histone deacetylase inhibitor nicotinamide exacerbates neurodegeneration in the lactacystin rat model of parkinson’s disease. J. Neurochem. 2019, 148, 136–156. [Google Scholar] [CrossRef]
- Rakieten, N.; Gordon, B.S.; Beaty, A.; Cooney, D.A.; Schein, P.S.; Dixon, R.L. Modification of renal tumorigenic effect of streptozotocin by nicotinamide: Spontaneous reversibility of streptozotocin diabetes. Proc. Soc. Exp. Biol. Med. 1976, 151, 356–361. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, W.; Miner, J.; Fromm, M. Cross regulation of sirtuin 1, ampk, and ppargamma in conjugated linoleic acid treated adipocytes. PLoS ONE 2012, 7, e48874. [Google Scholar] [CrossRef]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/pbef/visfatin regulates insulin secretion in beta cells as a systemic nad biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Both, G.W.; Furuichi, Y.; Shatkin, A.J. 5’-terminal 7-methylguanosine in eukaryotic mrna is required for translation. Nature 1975, 255, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Rupprecht, K.M.; Hecht, S.M.; Shatkin, A.J. Eukaryotic mrna cap binding protein: Purification by affinity chromatography on sepharose-coupled m7gdp. Proc. Natl. Acad. Sci. USA 1979, 76, 4345–4349. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Douek, I.F.; Moore, W.P.; Gillmor, H.A.; McLean, A.E.; Bingley, P.J.; Gale, E.A.; European Nicotinamide Diabetes Intervention Trial Group. Safety of high-dose nicotinamide: A review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Felsted, R.L.; Chaykin, S. N1-methylnicotinamide oxidation in a number of mammals. J. Biol. Chem. 1967, 242, 1274–1279. [Google Scholar] [PubMed]
- Mrochek, J.E.; Jolley, R.L.; Young, D.S.; Turner, W.J. Metabolic response of humans to ingestion of nicotinic acid and nicotinamide. Clin. Chem. 1976, 22, 1821–1827. [Google Scholar] [CrossRef]
- Attwood, J.T.; Yung, R.L.; Richardson, B.C. DNA methylation and the regulation of gene transcription. Cell. Mol. Life Sci. 2002, 59, 241–257. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Drong, A.W.; Lindgren, C.M.; McCarthy, M.I. The genetic and epigenetic basis of type 2 diabetes and obesity. Clin. Pharmacol. Ther. 2012, 92, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Teyssier, C.; Strahl, B.D.; Stallcup, M.R. Role of protein methylation in regulation of transcription. Endocr. Rev. 2005, 26, 147–170. [Google Scholar] [CrossRef]
- Finkelstein, J.D.; Martin, J.J. Homocysteine. Int. J. Biochem. Cell Biol. 2000, 32, 385–389. [Google Scholar] [CrossRef]
- Cosmetic Ingredient Review Expert Panel. Final report of the safety assessment of niacinamide and niacin. Int. J. Toxicol. 2005, 24 (Suppl. 5), 1–31. [Google Scholar]
- Pissios, P. Nicotinamide n-methyltransferase: More than a vitamin b3 clearance enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.J.; Li, D.; Ma, Q.; Gu, X.Y.; Guo, M.; Lun, Y.Z.; Sun, W.P.; Wang, X.Y.; Cao, Y.; Zhou, S.S. Excess nicotinamide increases plasma serotonin and histamine levels. Sheng Li Xue Bao 2013, 65, 33–38. [Google Scholar] [PubMed]
- Burgos, E.S.; Schramm, V.L. Weak coupling of atp hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry 2008, 47, 11086–11096. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Tsuchiya, M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS ONE 2011, 6, e22781. [Google Scholar] [CrossRef]
- Zhou, S.S.; Li, D.; Sun, W.P.; Guo, M.; Lun, Y.Z.; Zhou, Y.M.; Xiao, F.C.; Jing, L.X.; Sun, S.X.; Zhang, L.B.; et al. Nicotinamide overload may play a role in the development of type 2 diabetes. World J. Gastroenterol. 2009, 15, 5674–5684. [Google Scholar] [CrossRef]
- Kannt, A.; Rajagopal, S.; Kadnur, S.V.; Suresh, J.; Bhamidipati, R.K.; Swaminathan, S.; Hallur, M.S.; Kristam, R.; Elvert, R.; Czech, J.; et al. A small molecule inhibitor of nicotinamide n-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 2018, 8, 3660. [Google Scholar] [CrossRef]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Riederer, M.; Erwa, W.; Zimmermann, R.; Frank, S.; Zechner, R. Adipose tissue as a source of nicotinamide n-methyltransferase and homocysteine. Atherosclerosis 2009, 204, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Hemati, T.; Moghadami-Tabrizi, N.; Davari-Tanha, F.; Salmanian, B.; Javadian, P. High plasma homocysteine and insulin resistance in patients with polycystic ovarian syndrome. Iran. J. Reprod. Med. 2011, 9, 223–228. [Google Scholar]
- Yang, N.; Yao, Z.; Miao, L.; Liu, J.; Gao, X.; Fan, H.; Hu, Y.; Zhang, H.; Xu, Y.; Qu, A.; et al. Novel clinical evidence of an association between homocysteine and insulin resistance in patients with hypothyroidism or subclinical hypothyroidism. PLoS ONE 2015, 10, e0125922. [Google Scholar] [CrossRef]
- Akintunde, A.; Nondi, J.; Gogo, K.; Jones, E.S.W.; Rayner, B.L.; Hackam, D.G.; Spence, J.D. Physiological phenotyping for personalized therapy of uncontrolled hypertension in africa. Am. J. Hypertens. 2017, 30, 923–930. [Google Scholar] [CrossRef]
- Feng, X.; Xu, Y. Hyperhomocysteinemia as a metabolic risk factor for glucose intolerance among high-risk groups of Chinese adults. Med. Sci. Monit. 2017, 23, 2775–2781. [Google Scholar] [CrossRef]
- Cardellini, M.; Perego, L.; D’Adamo, M.; Marini, M.A.; Procopio, C.; Hribal, M.L.; Andreozzi, F.; Frontoni, S.; Giacomelli, M.; Paganelli, M.; et al. C-174g polymorphism in the promoter of the interleukin-6 gene is associated with insulin resistance. Diabetes Care 2005, 28, 2007–2012. [Google Scholar] [CrossRef][Green Version]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Mooney, R.A. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 2002, 51, 3391–3399. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Jiang, C.; Xu, M.; Pang, Y.; Feng, J.; Xiang, X.; Kong, W.; Xu, G.; Li, Y.; et al. Hyperhomocysteinemia promotes insulin resistance by inducing endoplasmic reticulum stress in adipose tissue. J. Biol. Chem. 2013, 288, 9583–9592. [Google Scholar] [CrossRef]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Asp. Med. 2019, 66, 49–61. [Google Scholar] [CrossRef]
- Xu, J.; Xu, S.Q.; Liang, J.; Lu, Y.; Luo, J.H.; Jin, J.H. Protective effect of nicotinamide in a mouse parkinson’s disease model. Zhejiang Da Xue Xue Bao Yi Xue Ban 2012, 41, 146–152. [Google Scholar] [PubMed]
- Williams, A.C.; Ramsden, D.B. Autotoxicity, methylation and a road to the prevention of parkinson’s disease. J. Clin. Neurosci. 2005, 12, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Aoyama, K.; Suno, M.; Awaya, T. N-methylation underlying parkinson’s disease. Neurotoxicol. Teratol. 2002, 24, 593–598. [Google Scholar] [CrossRef]
- Parsons, R.B.; Smith, S.W.; Waring, R.H.; Williams, A.C.; Ramsden, D.B. High expression of nicotinamide n-methyltransferase in patients with idiopathic parkinson’s disease. Neurosci. Lett. 2003, 342, 13–16. [Google Scholar] [CrossRef]
- Fukushima, T. Niacin metabolism and parkinson’s disease. Environ. Health Prev. Med. 2005, 10, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Kaetsu, A.; Lim, H.; Moriyama, M. Possible role of 1-methylnicotinamide in the pathogenesis of parkinson’s disease. Exp. Toxicol. Pathol. 2002, 53, 469–473. [Google Scholar] [CrossRef]
- Liu, M.; Chu, J.; Gu, Y.; Shi, H.; Zhang, R.; Wang, L.; Chen, J.; Shen, L.; Yu, P.; Chen, X.; et al. Serum n1-methylnicotinamide is associated with coronary artery disease in chinese patients. J. Am. Heart Assoc. 2017, 6, e004328. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Shai, I.; Stampfer, M.J.; Ma, J.; Manson, J.E.; Hankinson, S.E.; Cannuscio, C.; Selhub, J.; Curhan, G.; Rimm, E.B. Homocysteine as a risk factor for coronary heart diseases and its association with inflammatory biomarkers, lipids and dietary factors. Atherosclerosis 2004, 177, 375–381. [Google Scholar] [CrossRef]
- Zhang, R.; Shen, Y.; Zhou, L.; Sangwung, P.; Fujioka, H.; Zhang, L.; Liao, X. Short-term administration of nicotinamide mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell. Cardiol. 2017, 112, 64–73. [Google Scholar] [CrossRef]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide n-methyltransferase. Cdna cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [PubMed]
- Parsons, W.B., Jr. Studies of nicotinic acid use in hypercholesteremia. Changes in hepatic function, carbohydrate tolerance, and uric acid metabolism. Arch. Intern. Med. 1961, 107, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Volpi, E.; Lucidi, P.; Cruciani, G.; Monacchia, F.; Reboldi, G.; Brunetti, P.; Bolli, G.B.; De Feo, P. Nicotinamide counteracts alcohol-induced impairment of hepatic protein metabolism in humans. J. Nutr. 1997, 127, 2199–2204. [Google Scholar] [CrossRef]
- Strom, K.; Morales-Alamo, D.; Ottosson, F.; Edlund, A.; Hjort, L.; Jorgensen, S.W.; Almgren, P.; Zhou, Y.; Martin-Rincon, M.; Ekman, C.; et al. N(1)-methylnicotinamide is a signalling molecule produced in skeletal muscle coordinating energy metabolism. Sci. Rep. 2018, 8, 3016. [Google Scholar] [CrossRef]
- Kannt, A.; Pfenninger, A.; Teichert, L.; Tonjes, A.; Dietrich, A.; Schon, M.R.; Kloting, N.; Bluher, M. Association of nicotinamide-n-methyltransferase mrna expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance. Diabetologia 2015, 58, 799–808. [Google Scholar] [CrossRef]
- Hong, S.; Moreno-Navarrete, J.M.; Wei, X.; Kikukawa, Y.; Tzameli, I.; Prasad, D.; Lee, Y.; Asara, J.M.; Fernandez-Real, J.M.; Maratos-Flier, E.; et al. Nicotinamide n-methyltransferase regulates hepatic nutrient metabolism through sirt1 protein stabilization. Nat. Med. 2015, 21, 887–894. [Google Scholar] [CrossRef]
- Takeuchi, K.; Yokouchi, C.; Goto, H.; Umehara, K.; Yamada, H.; Ishii, Y. Alleviation of fatty liver in a rat model by enhancing n(1)-methylnicotinamide bioavailability through aldehyde oxidase inhibition. Biochem. Biophys. Res. Commun. 2018, 507, 203–210. [Google Scholar] [CrossRef]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-methylnicotinamide (mna), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef]
- Menon, R.M.; Adams, M.H.; Gonzalez, M.A.; Tolbert, D.S.; Leu, J.H.; Cefali, E.A. Plasma and urine pharmacokinetics of niacin and its metabolites from an extended-release niacin formulation. Int. J. Clin. Pharmacol. Ther. 2007, 45, 448–454. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-methyl-2-pyridone-5-carboxamide: A novel uremic toxin? Kidney Int. 2003, 63, S19–S21. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Martin, A.J.; Choy, B.; Fernandez-Penas, P.; Dalziell, R.A.; McKenzie, C.A.; Scolyer, R.A.; Dhillon, H.M.; Vardy, J.L.; Kricker, A.; et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N. Engl. J. Med. 2015, 373, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.; Silverberg, J.D.; Nguyen, T.T. Nicotinic acid-induced toxicity associated with cytopenia and decreased levels of thyroxine-binding globulin. Mayo Clin. Proc. 1992, 67, 465–468. [Google Scholar] [CrossRef]
- Tian, Y.J.; Luo, N.; Chen, N.N.; Lun, Y.Z.; Gu, X.Y.; Li, Z.; Ma, Q.; Zhou, S.S. Maternal nicotinamide supplementation causes global DNA hypomethylation, uracil hypo-incorporation and gene expression changes in fetal rats. Br. J. Nutr. 2014, 111, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, M.; Chan, X.Y.; Tan, S.Y.; Subramaniam, S.; Fan, Y.; Loh, E.; Chang, K.T.E.; Tan, T.C.; Chen, Q. Uncovering the mystery of opposite circadian rhythms between mouse and human leukocytes in humanized mice. Blood 2017, 130, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Otterness, D.M.; Weinshilboum, R.M. Human nicotinamide n-methyltransferase pharmacogenetics: Gene sequence analysis and promoter characterization. Pharmacogenetics 1999, 9, 307–316. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Effects | Examples of Effects | References |
|---|---|---|
| Protection against ATP depletion | [2] | |
| Decreased AD pathology and cognitive decline | [10] | |
| Improved sensory and motor neurological behavior | [3] | |
| Increased recovery from bilateral frontal brain injury | [4] | |
| Neuroprotection | Prevention/delay of ischemic stroke in stroke-prone hypertensive rats | [5] |
| Reduced lateral geniculate nucleus neuronal death | [6] | |
| Attenuated hippocampal neuronal death after global ischemia | [7] | |
| Improved motor deficits associated with Huntington’s disease phenotype | [8] | |
| Increased NAD+ level and mitochondrial function | [9] | |
| Amelioration of depression and psychological disorders | Amelioration of depression | [28] |
| Increased social interaction | [11] | |
| Anti-inflammation | Attenuated neutrophil recruitment in carrageenan-induced pleurisy or in lesions of autoimmune disease | [12,13] |
| Reduced arthritis activity | [14] | |
| Protection against vision and hearing loss | Attenuated retinal pigment cell death and age-related macular degeneration in animals | [29] |
| Reduced incidence of optic nerve degeneration and glaucoma | [30,31] | |
| Immune modulation | Improved mouse survival after lethal Staphylococcus enterotoxin B challenge | [32] |
| Skin protection/anti- skin disorders/cosmetic effects | Downregulation of the expression of inflammatory cytokines and protection against UV light | [33] |
| Anti-fibrosis | Attenuated development of pulmonary fibrosis | [34,35] |
| Anti-metastasis and adjuvant cancer therapy | Decreased growth and progression of bladder tumors | [36,37] |
| Photo-protection and reduced incidence of skin cancers | [15] | |
| Anti-HIV and -AIDS | Decreased provirus integration | [16] |
| Decreased viral RNA expression | [17] |
| Affected Organs and Conditions 2 | Observed Effects | Dose and Duration | References |
|---|---|---|---|
| Beneficial effects | |||
| Joints | Reduced itching in uremic patients | 550 mg twice a day (4 weeks) | [38] |
| pancreatic β-cell | β-cell function preserved and improved | 25 mg/kg daily intake (4 weeks) | [18,39] |
| Reduced the rate of diabetes incidence | 500 mg twice per day (2.5 years) | [19] | |
| No effect on the incidence of being diabetes-free | 1200 mg daily intake (5 years) | [20] | |
| Ineffective in prevention or delaying clinical onset of diabetes | 1.2 g daily intake (3 years) | [21] | |
| Skin | Reduced acne lesions and severity | 4% gel applied twice daily (8 weeks) | [26] |
| Attenuated immunosuppression with alterations in metabolism and apoptosis | 5% lotion applied before UV exposure | [40] | |
| Psychology | Improvements against depression | 0.5–1.5 g daily intake (3 weeks) | [22] |
| Relief from anxiety | A dose of 2 ug 3 h prior to test | [23] | |
| Kidney | Lowered serum concentrations of phosphorus, parathyroid hormone, and LDL, and increased serum HDL | 500 mg/day (with and increment every 2 weeks) (12 weeks) | [41] |
| Skin cancers non-melanoma | Reduced incidence of various types of skin cancers and actinic keratoses | 500 mg twice daily (4 months) | [42] |
| Adverse Effects | |||
| Minor effects | Frontal dull headaches, nausea, headache, dizziness | 1–18 g immediate | [43,44] |
| Pancreatic β-cell/plasma | Decreased insulin sensitivity, increased oxidative stress (H2O2) | 2 g daily (2 weeks) | [45,46] |
| Liver | Parenchymal-cell injury, portal fibrosis and cholestasis, liver injury | 3, 9 g daily (10 days) | [47] |
| Lymphocytes, platelets | Uremic toxicity-related cancer and thrombocytopenia | 1300, 1500 mg daily (24 weeks) | [48] |
| Kidney/platelets | Decreased serum phosphorus and thrombocytopenia | 0.52–2 g daily (3–6 months) | [49,50] |
| Subjects | Examples of Effects | Dose | Duration | Ref. |
|---|---|---|---|---|
| Death of mouse embryonic stem cells | 20 mM | 3–4 days | [95] | |
| Tumorigenicity. DNA damage, and sister chromatid exchanges | 1–10 mM 10 mM | 3 h 40 h | [100,101] | |
| Cells | 25 mM | 48 h | [102] | |
| Decreased SIRT1 activity. Increased intracellular ROS, spindle defects, and mitochondria dysfunction | 5 mM | 6, 12, 24 h | [106] | |
| Blocked mitochondria-related transcription. Worsened motor disturbance in Huntington’s disease model | 0.5, 1 mM | 96 h | [9] | |
| Mice and Rats | Oxidative DNA damage in hepatic and renal tissues. Impaired glucose tolerance and insulin sensitivity | 1 or 4 g/kg, d.w. | 8 weeks | [94] |
| Increased lethality | 4.5 g/kg, d.w., 2.5 g/kg, i.p. | 40 days | [44] | |
| Occurrence of pancreatic islet cell tumor | 350 mg/kg, i.p. | 226 days | [107] | |
| Increased incidence of kidney tumors | 350 mg/kg, i.p. | until die | [108] | |
| Decreased growth rate | 1, 2 %, d.w. 1, 2 %, d.w. | 24 days 20 days | [109,110] | |
| Growth inhibition, methyl deficiency, reduced tissue choline level, and increased hepatic lipids | 6, 20, 60 mg/100 g bw, i.p. | 2, 5 weeks | [111] | |
| Amelioration of acetaminophen-induced biochemical changes but occurrence of hepatotoxicity in healthy animals | 500 mg/kg, i.p. | 1.5 h | [112] | |
| Development of hepatic steatosis and fibrosis | 1%, d.w. | 6 weeks, 7 months | [113] | |
| Neurodegeneration of dopaminergic neurons Behavioral deficits and structural brain changes | 500 mg/kg, i.p. | 28 days | [114] | |
| Blocked mitochondrial-related transcription, worsened motor phenotype | 250mg/kg/day, s.c. | 28 days | [9] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, E.S.; Song, S.B. Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules 2020, 10, 687. https://doi.org/10.3390/biom10050687
Hwang ES, Song SB. Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules. 2020; 10(5):687. https://doi.org/10.3390/biom10050687
Chicago/Turabian StyleHwang, Eun Seong, and Seon Beom Song. 2020. "Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment" Biomolecules 10, no. 5: 687. https://doi.org/10.3390/biom10050687
APA StyleHwang, E. S., & Song, S. B. (2020). Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules, 10(5), 687. https://doi.org/10.3390/biom10050687