In Silico Analysis of a Highly Mutated Gene in Cancer Provides Insight into Abnormal mRNA Splicing: Splicing Factor 3B Subunit 1K700E Mutant

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. SF3B1 Gene Has the Highest Number of Mutations in the Pre-mRNA Splicing Network
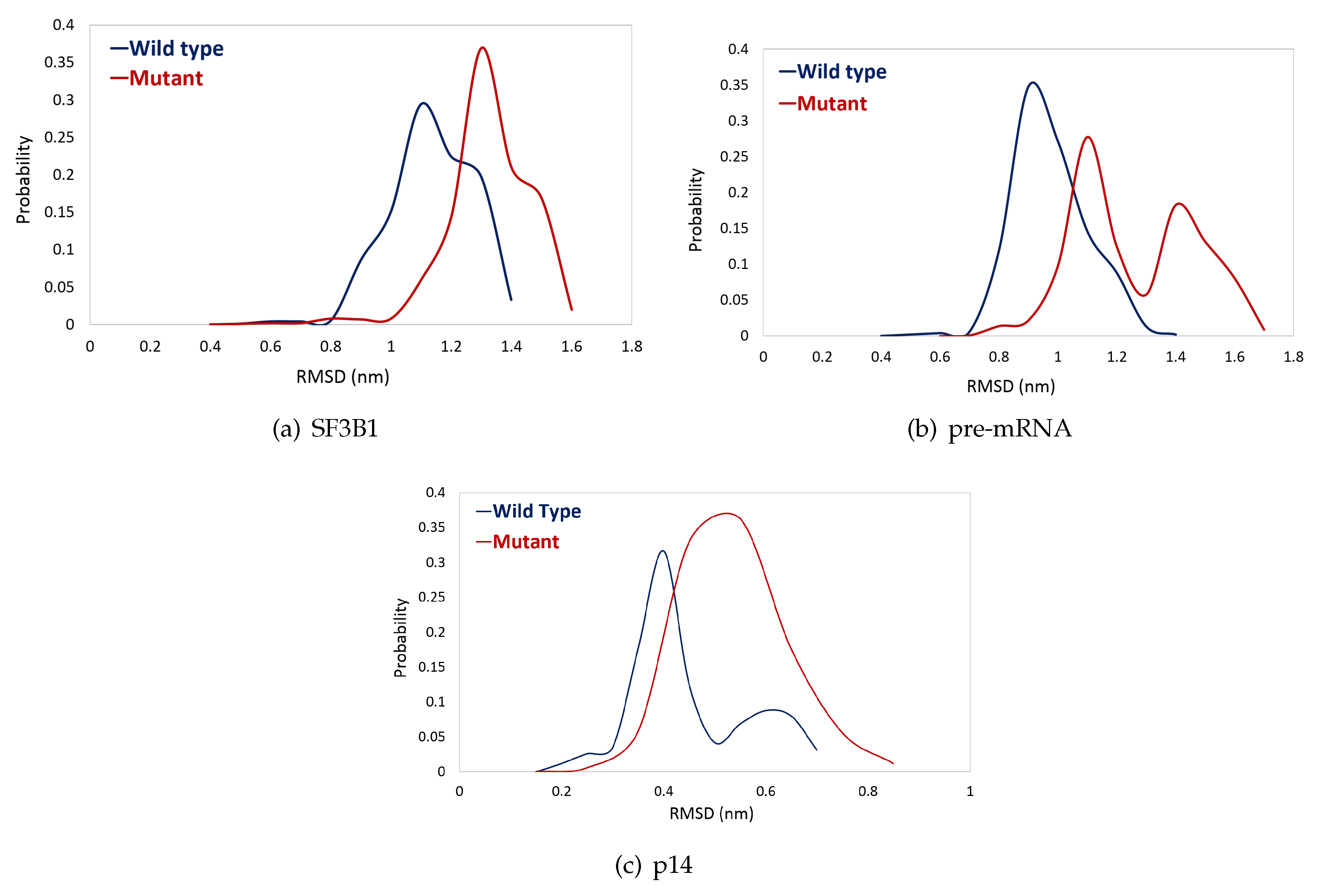
2.2. K700E Mutation Affects the Stability of SF3B1, Pre-mRNA, and p14
2.3. K700E Mutation Weakens Interactions Formed with Pre-mRNA
2.4. K700E Mutation Impacts the Global Dynamics of SF3B1
2.5. K700E Mutation Changes the Correlation between Residues in SF3B1 Protein
3. Discussion
4. Materials and Methods
4.1. Identification of Genes Participating in Pre-mRNA Splicing and Constructing the Related Gene Network
- For practical reasons, we chose breast cancer, one of the most common cancer type, with large number of samples in COSMIC.
- Only “Substitution-Missense” somatic mutations are included. According to COSMIC statistics, of breast carcinoma patients have somatic mutations of type “Substitution-Missense”.
- Only somatic mutations that are predicted strictly as pathogenic mutations are included. Functional Analysis through Hidden Markov Models (FATHMM) is a predictive tool for the mutation’s impact and classifies any mutation as one of the following classes; pathogenic, neutral or not specified. In addition, FATHMM scores are given in the form of p-value that ranges from 0 to 1. By default, the pathogenic mutations have scores greater than . For our purposes, we further increased the threshold for the pathogenic mutations to to avoid false positive predictions.
4.2. Assessment of the Impact of the Mutation by Means of Molecular Dynamics Simulations
4.2.1. System Setup
4.2.2. Simulation Protocol
4.2.3. Simulation Analysis Tools
Root Mean Square Deviation (RMSD)
Calculation of Number of Contacts
Principal Component Analysis (PCA)
Dynamic Cross Correlation Map (DCCM)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hortobagyi, G.N.; de la Garza Salazar, J.; Pritchard, K.; Amadori, D.; Haidinger, R.; Hudis, C.A.; Khaled, H.; Liu, M.C.; Martin, M.; Namer, M.; et al. The global breast cancer burden: Variations in epidemiology and survival. Clin. Breast Cancer 2005, 6, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-associated SF3B1 hotspot mutations induce cryptic 3’ splice site selection through use of a different branch point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome sequencing reveals potential mechanism of cryptic 3’ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol. 2015, 11, e1004105. [Google Scholar] [CrossRef]
- Venables, J.P. Unbalanced alternative splicing and its significance in cancer. Bioessays 2006, 28, 378–386. [Google Scholar] [CrossRef]
- Ghigna, C.; Valacca, C.; Biamonti, G. Alternative splicing and tumor progression. Curr. Genom. 2008, 9, 556–570. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5’ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Gozani, O.; Feld, R.; Reed, R. Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes Dev. 1996, 10, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Schneider, C.; MacMillan, A.M.; Katopodis, N.F.; Neubauer, G.; Wilm, M.; Lührmann, R.; Query, C.C. A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J. 2001, 20, 4536–4546. [Google Scholar] [CrossRef] [PubMed]
- Query, C.C.; McCaw, P.S.; Sharp, P.A. A minimal spliceosomal complex A recognizes the branch site and polypyrimidine tract. Mol. Cell. Biol. 1997, 17, 2944–2953. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gozani, O.; Potashkin, J.; Reed, R. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol. Cell. Biol. 1998, 18, 4752–4760. [Google Scholar] [CrossRef]
- Cretu, C.; Schmitzová, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Lührmann, R.; Pena, V. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [Google Scholar] [CrossRef]
- Cass, D.M.; Berglund, J.A. The SF3b155 N-terminal domain is a scaffold important for splicing. Biochemistry 2006, 45, 10092–10101. [Google Scholar] [CrossRef]
- Wang, C.; Chua, K.; Seghezzi, W.; Lees, E.; Gozani, O.; Reed, R. Phosphorylation of spliceosomal protein SAP 155 coupled with splicing catalysis. Genes Dev. 1998, 12, 1409–1414. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, C.; Zhan, X.; Li, L.; Lei, J.; Shi, Y. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018, 28, 307. [Google Scholar] [CrossRef]
- Schellenberg, M.J.; Edwards, R.A.; Ritchie, D.B.; Kent, O.A.; Golas, M.M.; Stark, H.; Lührmann, R.; Glover, J.M.; MacMillan, A.M. Crystal structure of a core spliceosomal protein interface. Proc. Natl. Acad. Sci. USA 2006, 103, 1266–1271. [Google Scholar] [CrossRef]
- Spadaccini, R.; Reidt, U.; Dybkov, O.; Will, C.; Frank, R.; Stier, G.; Corsini, L.; Wahl, M.C.; Lührmann, R.; Sattler, M. Biochemical and NMR analyses of an SF3b155–p14–U2AF-RNA interaction network involved in branch point definition during pre-mRNA splicing. RNA 2006, 12, 410–425. [Google Scholar] [CrossRef]
- Andrade, M.A.; Perez-Iratxeta, C.; Ponting, C.P. Protein repeats: Structures, functions, and evolution. J. Struct. Biol. 2001, 134, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Avishai, N.; Mahfouz, R.; Tabarroki, A.; Cowen, J.; Sharghi-Moshtaghin, R.; Hitomi, M.; Rogers, H.; Hasrouni, E.; Phillips, J.; et al. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia 2015, 29, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchiò, C.; Ng, C.K.; Sapino, A.; Salomon, A.V.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Fackenthal, J.D.; Godley, L.A. Aberrant RNA splicing and its functional consequences in cancer Cells. Dis. Model. Mech. 2008, 1, 37–42. [Google Scholar] [CrossRef]
- Dolatshad, H.; Pellagatti, A.; Liberante, F.G.; Llorian, M.; Repapi, E.; Steeples, V.; Roy, S.; Scifo, L.; Armstrong, R.N.; Shaw, J.; et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 2016, 30, 2322. [Google Scholar] [CrossRef]
- Borišek, J.; Saltalamacchia, A.; Gallì, A.; Palermo, G.; Molteni, E.; Malcovati, L.; Magistrato, A. Disclosing the Impact of Carcinogenic SF3b Mutations on Pre-mRNA Recognition Via All-Atom Simulations. Biomolecules 2019, 9, 633. [Google Scholar] [CrossRef]
- Borišek, J.; Saltalamacchia, A.; Spinello, A.; Magistrato, A. Exploiting Cryo-EM Structural Information and All-Atom Simulations to Decrypt the Molecular Mechanism of Splicing Modulators. J. Chem. Inf. Model. 2019. [Google Scholar] [CrossRef]
- Read, A.; Natrajan, R. Splicing dysregulation as a driver of breast cancer. Endocr.-Relat. Cancer 2018, 25, R467–R478. [Google Scholar] [CrossRef]
- MacMillan, A.M.; Query, C.C.; Allerson, C.R.; Chen, S.; Verdine, G.L.; Sharp, P.A. Dynamic association of proteins with the pre-mRNA branch region. Genes Dev. 1994, 8, 3008–3020. [Google Scholar] [CrossRef]
- Arenas, J.E.; Abelson, J.N. Prp43: An RNA helicase-like factor involved in spliceosome disassembly. Proc. Natl. Acad. Sci. USA 1997, 94, 11798–11802. [Google Scholar] [CrossRef]
- Fourmann, J.B.; Schmitzová, J.; Christian, H.; Urlaub, H.; Ficner, R.; Boon, K.L.; Fabrizio, P.; Lührmann, R. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev. 2013, 27, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.T.; Fu, R.H.; Yeh, F.L.; Tseng, C.K.; Lin, Y.C.; Huang, Y.H.; Cheng, S.C. Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev. 2005, 19, 2991–3003. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.I.; Voelker, R.B.; Henscheid, K.L.; Warf, M.B.; Berglund, J.A. Identification of motifs that function in the splicing of non-canonical introns. Genome Biol. 2008, 9, R97. [Google Scholar] [CrossRef] [PubMed]
- Ohi, M.D.; Vander Kooi, C.W.; Rosenberg, J.A.; Ren, L.; Hirsch, J.P.; Chazin, W.J.; Walz, T.; Gould, K.L. Structural and functional analysis of essential pre-mRNA splicing factor Prp19p. Mol. Cell. Biol. 2005, 25, 451–460. [Google Scholar] [CrossRef]
- Hogg, R.; McGrail, J.C.; O’Keefe, R.T. The function of the NineTeen Complex NTC in regulating spliceosome conformations and fidelity during pre-mRNA splicing. Biochem. Soc. Trans. 2010, 38, 1110–1115. [Google Scholar] [CrossRef]
- Das, S.; Krainer, A.R. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. 2014, 12, 1195–1204. [Google Scholar] [CrossRef]
- Huang, Y.; Gattoni, R.; Stévenin, J.; Steitz, J.A. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol. Cell 2003, 11, 837–843. [Google Scholar] [CrossRef]
- Chin, K.; Sharp, K.A.; Honig, B.; Pyle, A.M. Calculating the electrostatic properties of RNA provides new insights into molecular interactions and function. Nat. Struct. Mol. Biol. 1999, 6, 1055. [Google Scholar]
- Query, C.C.; Strobel, S.A.; Sharp, P.A. Three recognition events at the branch-site adenine. EMBO J. 1996, 15, 1392–1402. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD–Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kesarwani, A.K.; Ramirez, O.; Gupta, A.K.; Yang, X.; Murthy, T.; Minella, A.C.; Pillai, M.M. Cancer-associated SF3B1 mutants recognize otherwise inaccessible cryptic 3’ splice sites within RNA secondary structures. Oncogene 2017, 36, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Carrocci, T.J.; Zoerner, D.M.; Paulson, J.C.; Hoskins, A.A. SF3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res. 2017, 45, 4837–4852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ali, A.M.; Lieu, Y.K.; Liu, Z.; Gao, J.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-causing mutations in SF3B1 alter splicing by disrupting interaction with SUGP1. Mol. Cell 2019, 76, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Boudrez, A.; Beullens, M.; Waelkens, E.; Stalmans, W.; Bollen, M. Phosphorylation-dependent interaction between the splicing factors SAP155 and NIPP1. J. Biol. Chem. 2002, 277, 31834–31841. [Google Scholar] [CrossRef] [PubMed]
- Liberante, F.G.; Lappin, K.; Barros, E.M.; Vohhodina, J.; Grebien, F.; Savage, K.I.; Mills, K.I. Altered splicing and cytoplasmic levels of tRNA synthetases in SF3B1-mutant myelodysplastic syndromes as a therapeutic vulnerability. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497. [Google Scholar] [CrossRef]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef]
- Di, C.; Zhang, Q.; Chen, Y.; Wang, Y.; Zhang, X.; Liu, Y.; Sun, C.; Zhang, H.; Hoheisel, J.D. Function, clinical application, and strategies of Pre-mRNA splicing in cancer. Cell Death Differ. 2019, 26, 1181–1194. [Google Scholar] [CrossRef]
- Desmedt, C.; Haibe-Kains, B.; Wirapati, P.; Buyse, M.; Larsimont, D.; Bontempi, G.; Delorenzi, M.; Piccart, M.; Sotiriou, C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin. Cancer Res. 2008, 14, 5158–5165. [Google Scholar] [CrossRef]
- Bonifaci, N.; Berenguer, A.; Díez, J.; Reina, O.; Medina, I.; Dopazo, J.; Moreno, V.; Pujana, M.A. Biological processes, properties and molecular wiring diagrams of candidate low-penetrance breast cancer susceptibility genes. BMC Med. Genom. 2008, 1, 62. [Google Scholar] [CrossRef]
- Wang, B.D.; Lee, N.H. Aberrant RNA splicing in cancer and drug resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2013, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2014, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2007, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef]
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol. Ther. 2010, 10, 955–960. [Google Scholar] [CrossRef]
- Makki, J. Diversity of breast carcinoma: Histological subtypes and clinical relevance. Clin. Med. Insights Pathol. 2015, 8, S31563. [Google Scholar] [CrossRef]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Caesar-Johnson, S.J.; Demchok, J.A.; Felau, I.; et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018, 23, 282–296. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2015, 12, 405–413. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Wang, L.; Yuan, Y.; Chen, X.; Chen, J.; Guo, Y.; Li, M.; Li, C.; Pu, X. Probing the cooperative mechanism of the μ–δ opioid receptor heterodimer by multiscale simulation. Phys. Chem. Chem. Phys. 2018, 20, 29969–29982. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Filipek, S.; Palczewski, K.; Vogel, H. Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nat. Commun. 2014, 5, 4733. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. In Homology Modeling; Springer: Berlin, Germany, 2011; pp. 231–257. [Google Scholar]
- Aier, I.; Varadwaj, P.K.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef]
- Yang, L.; Song, G.; Carriquiry, A.; Jernigan, R.L. Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure 2008, 16, 321–330. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Mishra, S.K.; Jernigan, R.L. Protein dynamic communities from elastic network models align closely to the communities defined by molecular dynamics. PLoS ONE 2018, 13, e0199225. [Google Scholar] [CrossRef]
- Kasahara, K.; Fukuda, I.; Nakamura, H. A novel approach of dynamic cross correlation analysis on molecular dynamics simulations and its application to Ets1 dimer–DNA complex. PLoS ONE 2014, 9, e112419. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J. Protein dynamics. Rep. Prog. Phys. 1984, 47, 1. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samy, A.; Suzek, B.E.; Ozdemir, M.K.; Sensoy, O. In Silico Analysis of a Highly Mutated Gene in Cancer Provides Insight into Abnormal mRNA Splicing: Splicing Factor 3B Subunit 1K700E Mutant. Biomolecules 2020, 10, 680. https://doi.org/10.3390/biom10050680
Samy A, Suzek BE, Ozdemir MK, Sensoy O. In Silico Analysis of a Highly Mutated Gene in Cancer Provides Insight into Abnormal mRNA Splicing: Splicing Factor 3B Subunit 1K700E Mutant. Biomolecules. 2020; 10(5):680. https://doi.org/10.3390/biom10050680
Chicago/Turabian StyleSamy, Asmaa, Baris Ethem Suzek, Mehmet Kemal Ozdemir, and Ozge Sensoy. 2020. "In Silico Analysis of a Highly Mutated Gene in Cancer Provides Insight into Abnormal mRNA Splicing: Splicing Factor 3B Subunit 1K700E Mutant" Biomolecules 10, no. 5: 680. https://doi.org/10.3390/biom10050680
APA StyleSamy, A., Suzek, B. E., Ozdemir, M. K., & Sensoy, O. (2020). In Silico Analysis of a Highly Mutated Gene in Cancer Provides Insight into Abnormal mRNA Splicing: Splicing Factor 3B Subunit 1K700E Mutant. Biomolecules, 10(5), 680. https://doi.org/10.3390/biom10050680