
Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review




Abstract
Contents
- Introduction
- Characteristic features of mitochondrial carriers (MCs)
- Diseases caused by mutations in MCs
- 3.1.
- General information
- 3.2.
- SLC25A1 (citrate carrier, CIC) deficiency
- 3.3.
- SLC25A3 (phosphate carrier, PiC) deficiency
- 3.4.
- SLC25A4 (ADP/ATP carrier 1, AAC1) deficiency
- 3.5.
- SLC25A10 (dicarboxylate carrier, DIC) deficiency
- 3.6.
- SLC25A12 (aspartate-glutamate carrier 1, AGC1, aralar) deficiency
- 3.7.
- SLC25A13 (aspartate-glutamate carrier 2, AGC2, citrin) deficiency
- 3.8.
- SLC25A15 (ornithine carrier 1, ORC1) deficiency
- 3.9.
- SLC25A16 deficiency
- 3.10.
- SLC25A19 (thiamine pyrophosphate carrier, TPC) deficiency
- 3.11.
- SLC25A20 (carnitine-acylcarnitine carrier, CAC) deficiency
- 3.12.
- SLC25A21 (oxodicarboxylate carrier, ODC) deficiency
- 3.13.
- SLC25A22 (glutamate carrier 1, GC1) deficiency
- 3.14.
- SLC25A24 (ATP-Mg2+/phosphate carrier 1, APC1) deficiency
- 3.15.
- SLC25A26 (S-adenosylmethionine carrier, SAMC) deficiency
- 3.16.
- SLC25A32 deficiency
- 3.17.
- SLC25A38 (glycine carrier, GlyC) deficiency
- 3.18.
- SLC25A42 (CoA and PAP carrier) deficiency
- 3.19.
- SLC25A46 deficiency
- Multigenic diseases with mutations in MCs
- Concluding remarks
1. Introduction
2. Characteristic Features of Mitochondrial Carriers
3. Diseases Associated with Mutations in MCs
3.1. General Information
3.2. SLC25A1 (Citrate Carrier, CIC) Deficiency
3.3. SLC25A3 (Phosphate Carrier, PiC) Deficiency
3.4. SLC25A4 (ADP/ATP Carrier 1, AAC1) Deficiency
3.5. SLC25A10 (Dicarboxylate Carrier, DIC) Deficiency
3.6. SLC25A12 (Aspartate-Glutamate Carrier 1, AGC1, Aralar) Deficiency
3.7. SLC25A13 (Aspartate-Glutamate Carrier 2, AGC2, Citrin) Deficiency
3.8. SLC25A15 (Ornithine Carrier 1, ORC1) Deficiency
3.9. SLC25A16 Deficiency
3.10. SLC25A19 (Thiamine Pyrophosphate Carrier, TPC) Deficiency
3.11. SLC25A20 (Carnitine-Acylcarnitine Carrier, CAC) Deficiency
3.12. SLC25A21 (Oxodicarboxylate Carrier, ODC) Deficiency
3.13. SLC25A22 (Glutamate Carrier 1, GC1) Deficiency
3.14. SLC25A24 (ATP-Mg2+/Phosphate Carrier 1, APC1) Deficiency
3.15. SLC25A26 (S-Adenosylmethionine Carrier, SAMC) Deficiency
3.16. SLC25A32 Deficiency
3.17. SLC25A38 (Glycine Carrier, GlyC) Deficiency
3.18. SLC25A42 (CoA and PAP Carrier) Deficiency
3.19. SLC25A46 Deficiency
4. Multigenic Diseases with Mutations in MCs
5. Concluding Remarks
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update: Recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr. Opin. Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef]
- Davis, R.L.; Liang, C.; Sue, C.M. Mitochondrial diseases. Handb. Clin. Neurol. 2018, 147, 125–141. [Google Scholar] [PubMed]
- Dard, L.; Blanchard, W.; Hubert, C.; Lacombe, D.; Rossignol, R. Mitochondrial functions and rare diseases. Mol. Aspects Med. 2020, 71, 100842. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. Strategies for fighting mitochondrial diseases. J. Intern. Med. 2020. (In press) [CrossRef]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Palmieri, F.; Agrimi, G.; Blanco, E.; Castegna, A.; Di Noia, M.A.; Iacobazzi, V.; Lasorsa, F.M.; Marobbio, C.M.T.; Palmieri, L.; Scarcia, P.; et al. Identification of mitochondrial carriers in Saccharomyces cerevisiae by transport assay of reconstituted recombinant proteins. Biochim. Biophys. Acta 2006, 1757, 1249–1262. [Google Scholar] [CrossRef]
- Picault, N.; Hodges, M.; Palmieri, L.; Palmieri, F. The growing family of mitochondrial carriers in Arabidopsis. Trends Plant Sci. 2004, 9, 138–146. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Saraste, M.; Walker, J.E. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Lett. 1982, 144, 250–254. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial carrier proteins. FEBS Lett. 1994, 346, 48–54. [Google Scholar] [CrossRef]
- Fiermonte, G.; Walker, J.E.; Palmieri, F. Abundant bacterial expression and reconstitution of an intrinsic membrane-transport protein from bovine mitochondria. Biochem. J. 1993, 294, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Indiveri, C.; Bisaccia, F.; Iacobazzi, V. Mitochondrial metabolite carrier proteins: Purification, reconstitution, and transport studies. Methods Enzymol. 1995, 260, 349–369. [Google Scholar] [PubMed]
- Palmieri, F.; Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Monné, M.; Palmieri, F. Antiporters of the mitochondrial carrier family. Curr. Top. Membr. 2014, 73, 289–320. [Google Scholar]
- Palmieri, F.; Pierri, C.L. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar]
- Palmieri, F.; Pierri, C.L.; De Grassi, A.; Nunes-Nesi, A.; Fernie, A.R. Evolution, structure and function of mitochondrial carriers: A review with new insights. Plant J. 2011, 66, 161–181. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrústegui, J.; et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef]
- Fiermonte, G.; De Leonardis, F.; Todisco, S.; Palmieri, L.; Lasorsa, F.M.; Palmieri, F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J. Biol. Chem. 2004, 279, 30722–30730. [Google Scholar] [CrossRef]
- Del Arco, A.; Satrústegui, J. Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J. Biol. Chem. 2004, 279, 24701–24713. [Google Scholar] [CrossRef]
- Zara, V.; Palmieri, F.; Mahlke, K.; Pfanner, N. The cleavable presequence is not essential for import and assembly of the phosphate carrier of mammalian mitochondria but enhances the specificity and efficiency of import. J. Biol. Chem. 1992, 267, 12077–12081. [Google Scholar] [PubMed]
- Palmieri, F.; Bisaccia, F.; Capobianco, L.; Dolce, V.; Fiermonte, G.; Iacobazzi, V.; Zara, V. Transmembrane topology, genes, and biogenesis of the mitochondrial phosphate and oxoglutarate carriers. J. Bioenerg. Biomembr. 1993, 25, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Zara, V.; Ferramosca, A.; Palmisano, I.; Palmieri, F.; Rassow, J. Biogenesis of rat mitochondrial citrate carrier (CIC): The N-terminal presequence facilitates the solubility of the preprotein but does not act as a targeting signal. J. Mol. Biol. 2003, 325, 399–408. [Google Scholar] [CrossRef]
- Zara, V.; Ferramosca, A.; Robitaille-Foucher, P.; Palmieri, F.; Young, J.C. Mitochondrial carrier protein biogenesis: Role of the chaperones Hsc70 and Hsp90. Biochem. J. 2009, 419, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R.S. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef]
- Klingenberg, M. The ADP, ATP shuttle of the mitochondrion. Trends Biochem. Sci. 1979, 4, 249–252. [Google Scholar] [CrossRef]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef]
- Robinson, A.J.; Kunji, E.R.S. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 2617–2622. [Google Scholar] [CrossRef]
- Monné, M.; Miniero, D.V.; Daddabbo, L.; Robinson, A.J.; Kunji, E.R.S.; Palmieri, F. Substrate specificity of the two mitochondrial ornithine carriers can be swapped by single mutation in substrate binding site. J. Biol. Chem. 2012, 287, 7925–7934. [Google Scholar] [CrossRef] [PubMed]
- Monné, M.; Palmieri, F.; Kunji, E.R.S. The substrate specificity of mitochondrial carriers: Mutagenesis revisited. Mol. Membr. Biol. 2013, 30, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Marobbio, C.M.T.; Giannuzzi, G.; Paradies, E.; Pierri, C.L.; Palmieri, F. α-Isopropylmalate, a leucine biosynthesis intermediate in yeast, is transported by the mitochondrial oxalacetate carrier. J. Biol. Chem. 2008, 283, 28445–28553. [Google Scholar] [CrossRef] [PubMed]
- Tonazzi, A.; Console, L.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Identification by site-directed mutagenesis of a hydrophobic binding site of the mitochondrial carnitine/acylcarnitine carrier involved in the interaction with acyl groups. Biochim. Biophys. Acta 2012, 1817, 697–704. [Google Scholar] [CrossRef]
- Huizing, M.; Iacobazzi, V.; Ijlst, L.; Savelkoul, P.; Ruitenbeek, W.; van den Heuvel, L.; Indiveri, C.; Smeitink, J.; Trijbels, F.; Wanders, R.; et al. Cloning of the human carnitine-acylcarnitine carrier cDNA and identification of the molecular defect in a patient. Am. J. Hum. Genet. 1997, 61, 1239–1245. [Google Scholar] [CrossRef]
- Palmieri, F. Diseases caused by defects of mitochondrial carriers: A review. Biochim. Biophys. Acta 2008, 1777, 564–578. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef]
- Rusecka, J.; Kaliszewska, M.; Bartnik, E.; Tońska, K. Nuclear genes involved in mitochondrial diseases caused by instability of mitochondrial DNA. J. Appl. Genet. 2018, 59, 43–57. [Google Scholar] [CrossRef]
- Edvardson, S.; Porcelli, V.; Jalas, C.; Soiferman, D.; Kellner, Y.; Shaag, A.; Korman, S.H.; Pierri, C.L.; Scarcia, P.; Fraenkel, N.D.; et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J. Med. Genet. 2013, 50, 240–245. [Google Scholar] [CrossRef]
- Nota, B.; Struys, E.A.; Pop, A.; Jansen, E.E.; Fernandez Ojeda, M.R.; Kanhai, W.A.; Kranendijk, M.; van Dooren, S.J.M.; Bevova, M.R.; Sistermans, E.A.; et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2013, 92, 627–631. [Google Scholar] [CrossRef]
- Chaouch, A.; Porcelli, V.; Cox, D.; Edvardson, S.; Scarcia, P.; De Grassi, A.; Pierri, C.L.; Cossins, J.; Laval, S.H.; Griffin, H.; et al. Mutations in the Mitochondrial Citrate Carrier SLC25A1 are Associated with Impaired Neuromuscular Transmission. J. Neuromuscul. Dis. 2014, 1, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Prasun, P.; Young, S.; Salomons, G.; Werneke, A.; Jiang, Y.-H.; Struys, E.; Paige, M.; Avantaggiati, M.L.; McDonald, M. Expanding the Clinical Spectrum of Mitochondrial Citrate Carrier (SLC25A1) Deficiency: Facial Dysmorphism in Siblings with Epileptic Encephalopathy and Combined D,L-2-Hydroxyglutaric Aciduria. JIMD Rep. 2015, 19, 111–115. [Google Scholar] [PubMed]
- Smith, A.; McBride, S.; Marcadier, J.L.; Michaud, J.; Al-Dirbashi, O.Y.; Schwartzentruber, J.; Beaulieu, C.L.; Katz, S.L.; FORGE Canada Consortium; Majewski, J.; et al. Severe Neonatal Presentation of Mitochondrial Citrate Carrier (SLC25A1) Deficiency. JIMD Rep. 2016, 30, 73–79. [Google Scholar]
- Cohen, I.; Staretz-Chacham, O.; Wormser, O.; Perez, Y.; Saada, A.; Kadir, R.; Birk, O.S. A novel homozygous SLC25A1 mutation with impaired mitochondrial complex V: Possible phenotypic expansion. Am. J. Med. Genet. A. 2018, 176, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, M.; Ozaki, E.; Yamauchi, T.; Ohta, M.; Higaki, T.; Masuda, K.; Imoto, I.; Ishii, E.; Eguchi-Ishimae, M. Manifestation of recessive combined D-2-, L-2-hydroxyglutaric aciduria in combination with 22q11.2 deletion syndrome. Am. J. Med. Genet. A. 2018, 176, 351–358. [Google Scholar] [CrossRef]
- Palmieri, F.; Stipani, I.; Quagliariello, E.; Klingenberg, M. Kinetic study of the tricarboxylate carrier in rat liver mitochondria. Eur. J. Biochem. 1972, 26, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Bisaccia, F.; De Palma, A.; Palmieri, F. Identification and purification of the tricarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta 1989, 977, 171–176. [Google Scholar] [CrossRef]
- Bisaccia, F.; De Palma, A.; Dierks, T.; Krämer, R.; Palmieri, F. Reaction mechanism of the reconstituted tricarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta 1993, 1142, 139–145. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflüg. Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Huizing, M.; Ruitenbeek, W.; van den Heuvel, L.P.; Dolce, V.; Iacobazzi, V.; Smeitink, J.A.; Palmieri, F.; Trijbels, J.M. Human mitochondrial transmembrane metabolite carriers: Tissue distribution and its implication for mitochondrial disorders. J. Bioenerg. Biomembr. 1998, 30, 277–284. [Google Scholar] [CrossRef]
- Pop, A.; Williams, M.; Struys, E.A.; Monné, M.; Jansen, E.E.W.; De Grassi, A.; Kanhai, W.A.; Scarcia, P.; Ojeda, M.R.F.; Porcelli, V.; et al. An overview of combined D-2- and L-2-hydroxyglutaric aciduria: Functional analysis of CIC variants. J. Inherit. Metab. Dis. 2018, 41, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Majd, H.; King, M.S.; Smith, A.C.; Kunji, E.R.S. Pathogenic mutations of the human mitochondrial citrate carrier SLC25A1 lead to impaired citrate export required for lipid, dolichol, ubiquinone and sterol synthesis. Biochim. Biophys. Acta 2018, 1859, 1–7. [Google Scholar] [CrossRef]
- Pierri, C.L.; Palmieri, F.; De Grassi, A. Single-nucleotide evolution quantifies the importance of each site along the structure of mitochondrial carriers. Cell. Mol. Life Sci. 2013, 71, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [PubMed]
- Kranendijk, M.; Struys, E.A.; Salomons, G.S.; Van der Knaap, M.S.; Jakobs, C. Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 2012, 35, 571–587. [Google Scholar] [CrossRef]
- Dolce, V.; Iacobazzi, V.; Palmieri, F.; Walker, J.E. The sequences of human and bovine genes of the phosphate carrier from mitochondria contain evidence of alternatively spliced forms. J. Biol. Chem. 1994, 269, 10451–10460. [Google Scholar]
- Dolce, V.; Fiermonte, G.; Palmieri, F. Tissue-specific expression of the two isoforms of the mitochondrial phosphate carrier in bovine tissues. FEBS Lett. 1996, 399, 95–98. [Google Scholar] [CrossRef]
- Fiermonte, G.; Dolce, V.; Palmieri, F. Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J. Biol. Chem. 1998, 273, 22782–22787. [Google Scholar] [CrossRef]
- Palmieri, F.; Quagliariello, E.; Klingenberg, M. Quantitative correlation between the distribution of anions and the pH difference across the mitochondrial membrane. Eur. J. Biochem. 1970, 17, 230–238. [Google Scholar] [CrossRef]
- Mayr, J.A.; Merkel, O.; Kohlwein, S.D.; Gebhardt, B.R.; Böhles, H.; Fötschl, U.; Koch, J.; Jaksch, M.; Lochmüller, H.; Horváth, R.; et al. Mitochondrial phosphate-carrier deficiency: A novel disorder of oxidative phosphorylation. Am. J. Hum. Genet. 2007, 80, 478–484. [Google Scholar] [CrossRef]
- Mayr, J.A.; Zimmermann, F.A.; Horváth, R.; Schneider, H.-C.; Schoser, B.; Holinski-Feder, E.; Czermin, B.; Freisinger, P.; Sperl, W. Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul. Disord. 2011, 21, 803–808. [Google Scholar] [CrossRef]
- Bhoj, E.J.; Li, M.; Ahrens-Nicklas, R.; Pyle, L.C.; Wang, J.; Zhang, V.W.; Clarke, C.; Wong, L.J.; Sondheimer, N.; Ficicioglu, C.; et al. Pathologic Variants of the Mitochondrial Phosphate Carrier SLC25A3: Two New Patients and Expansion of the Cardiomyopathy/Skeletal Myopathy Phenotype With and Without Lactic Acidosis. JIMD Rep. 2015, 19, 59–66. [Google Scholar] [PubMed]
- Kwong, J.Q.; Davis, J.; Baines, C.P.; Sargent, M.A.; Karch, J.; Wang, X.; Huang, T.; Molkentin, J.D. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Prezioso, G.; Quagliariello, E.; Klingenberg, M. Kinetic study of the dicarboxylate carrier in rat liver mitochondria. Eur. J. Biochem. 1971, 22, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Bisaccia, F.; Indiveri, C.; Palmieri, F. Purification and reconstitution of two anion carriers from rat liver mitochondria: The dicarboxylate and the 2-oxoglutarate carrier. Biochim. Biophys. Acta 1988, 933, 229–240. [Google Scholar] [CrossRef]
- Indiveri, C.; Capobianco, L.; Krämer, R.; Palmieri, F. Kinetics of the reconstituted dicarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta 1989, 977, 187–193. [Google Scholar] [CrossRef]
- Indiveri, C.; Dierks, T.; Krämer, R.; Palmieri, F. Kinetic discrimination of two substrate binding sites of the reconstituted dicarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta 1989, 977, 194–199. [Google Scholar] [CrossRef]
- Fiermonte, G.; Palmieri, L.; Dolce, V.; Lasorsa, F.M.; Palmieri, F.; Runswick, M.J.; Walker, J.E. The sequence, bacterial expression, and functional reconstitution of the rat mitochondrial dicarboxylate transporter cloned via distant homologs in yeast and Caenorhabditis elegans. J. Biol. Chem. 1998, 273, 24754–24759. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef]
- Gorgoglione, R.; Porcelli, V.; Santoro, A.; Daddabbo, L.; Vozza, A.; Monné, M.; Di Noia, M.A.; Palmieri, L.; Fiermonte, G.; Palmieri, F. The human uncoupling proteins 5 and 6 (UCP5/SLC25A14 and UCP6/SLC25A30) transport sulfur oxyanions, phosphate and dicarboxylates. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 724–733. [Google Scholar] [CrossRef]
- Dolce, V.; Scarcia, P.; Iacopetta, D.; Palmieri, F. A fourth ADP/ATP carrier isoform in man: Identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 2005, 579, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.P.; Keränen, S.; Peltonen, L.; Suomalainen, A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000, 289, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Alberio, S.; Pisano, I.; Lodi, T.; Meznaric-Petrusa, M.; Zidar, J.; Santoro, A.; Scarcia, P.; Fontanesi, F.; Lamantea, E.; et al. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum. Mol. Genet. 2005, 14, 3079–3088. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Chassagne, M.; Ceresuela, J.; Rouvet, I.; Padet, S.; Acquaviva, C.; Nataf, S.; Vinzio, S.; Bozon, D.; Mousson de Camaret, B. Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J. Med. Genet. 2012, 49, 146–150. [Google Scholar] [CrossRef]
- Körver-Keularts, I.M.L.W.; de Visser, M.; Bakker, H.D.; Wanders, R.J.A.; Vansenne, F.; Scholte, H.R.; Dorland, L.; Nicolaes, G.A.F.; Spaapen, L.M.J.; Smeets, H.J.M.; et al. Two Novel Mutations in the SLC25A4 Gene in a Patient with Mitochondrial Myopathy. JIMD Rep. 2015, 22, 39–45. [Google Scholar]
- Tosserams, A.; Papadopoulos, C.; Jardel, C.; Lemière, I.; Romero, N.B.; De Lonlay, P.; Wahbi, K.; Voermans, N.; Hogrel, J.-Y.; Laforêt, P. Two new cases of mitochondrial myopathy with exercise intolerance, hyperlactatemia and cardiomyopathy, caused by recessive SLC25A4 mutations. Mitochondrion 2018, 39, 26–29. [Google Scholar] [CrossRef]
- Thompson, K.; Majd, H.; Dallabona, C.; Reinson, K.; King, M.S.; Alston, C.L.; He, L.; Lodi, T.; Jones, S.A.; Fattal-Valevski, A.; et al. Recurrent De Novo Dominant Mutations in SLC25A4 Cause Severe Early-Onset Mitochondrial Disease and Loss of Mitochondrial DNA Copy Number. Am. J. Hum. Genet. 2016, 99, 860–876. [Google Scholar] [CrossRef]
- Von Renesse, A.; Morales-Gonzalez, S.; Gill, E.; Salomons, G.S.; Stenzel, W.; Schuelke, M. Muscle Weakness, Cardiomyopathy, and L-2-Hydroxyglutaric Aciduria Associated with a Novel Recessive SLC25A4 Mutation. JIMD Rep. 2019, 43, 27–35. [Google Scholar]
- King, M.S.; Thompson, K.; Hopton, S.; He, L.; Kunji, E.R.S.; Taylor, R.W.; Ortiz-Gonzalez, X.R. Expanding the phenotype of de novo SLC25A4-linked mitochondrial disease to include mild myopathy. Neurol. Genet. 2018, 4, e256. [Google Scholar] [CrossRef] [PubMed]
- Punzi, G.; Porcelli, V.; Ruggiu, M.; Hossain, M.F.; Menga, A.; Scarcia, P.; Castegna, A.; Gorgoglione, R.; Pierri, C.L.; Laera, L.; et al. SLC25A10 biallelic mutations in intractable epileptic encephalopathy with complex I deficiency. Hum. Mol. Genet. 2018, 27, 499–504. [Google Scholar] [CrossRef] [PubMed]
- McGivan, J.D.; Klingenberg, M. Correlation between H+ and anion movement in mitochondria and the key role of the phosphate carrier. Eur. J. Biochem. 1971, 20, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Palmieri, F.; Capano, M.; Quagliariello, E. The transport of sulphate and sulphite in rat liver mitochondria. Biochem. J. 1974, 142, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Palmieri, F.; Capano, M.; Quagliariello, E. The transport of thiosulphate in rat liver mitochondria. FEBS Lett. 1974, 46, 247–250. [Google Scholar] [CrossRef]
- Crompton, M.; Palmieri, F.; Capano, M.; Quagliariello, E. A kinetic study of sulphate transport in rat liver mitochondria. Biochem. J. 1975, 146, 667–673. [Google Scholar] [CrossRef]
- Bisaccia, F.; Palmieri, F. Specific elution from hydroxylapatite of the mitochondrial phosphate carrier by cardiolipin. Biochim. Biophys. Acta 1984, 766, 386–394. [Google Scholar] [CrossRef]
- Palmieri, F.; Quagliariello, E.; Klingenberger, M. Kinetics and specificity of the oxoglutarate carrier in rat-liver mitochondria. Eur. J. Biochem. 1972, 29, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Bisaccia, F.; Indiveri, C.; Palmieri, F. Purification of reconstitutively active alpha-oxoglutarate carrier from pig heart mitochondria. Biochim. Biophys. Acta 1985, 810, 362–369. [Google Scholar] [CrossRef]
- Indiveri, C.; Palmieri, F.; Bisaccia, F.; Krämer, R. Kinetics of the reconstituted 2-oxoglutarate carrier from bovine heart mitochondria. Biochim. Biophys. Acta 1987, 890, 310–318. [Google Scholar] [CrossRef]
- Runswick, M.J.; Walker, J.E.; Bisaccia, F.; Iacobazzi, V.; Palmieri, F. Sequence of the bovine 2-oxoglutarate/malate carrier protein: Structural relationship to other mitochondrial transport proteins. Biochemistry 1990, 29, 11033–11040. [Google Scholar] [CrossRef]
- Wibom, R.; Lasorsa, F.M.; Töhönen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 deficiency associated with global cerebral hypomyelination. N. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N.; et al. AGC1 Deficiency Causes Infantile Epilepsy, Abnormal Myelination, and Reduced N-Acetylaspartate. JIMD Rep. 2014, 14, 77–85. [Google Scholar]
- Lasorsa, F.M.; Pinton, P.; Palmieri, L.; Fiermonte, G.; Rizzuto, R.; Palmieri, F. Recombinant expression of the Ca(2+)-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated Chinese hamster ovary cells. J. Biol. Chem. 2003, 278, 38686–38692. [Google Scholar] [CrossRef] [PubMed]
- Thangaratnarajah, C.; Ruprecht, J.J.; Kunji, E.R.S. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat. Commun. 2014, 5, 5491. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Krämer, R.; Palmieri, F. Reconstitution of the malate/aspartate shuttle from mitochondria. J. Biol. Chem. 1987, 262, 15979–15983. [Google Scholar]
- Pfeiffer, B.; Sen, K.; Kaur, S.; Pappas, K. Expanding Phenotypic Spectrum of Cerebral Aspartate-Glutamate Carrier Isoform 1 (AGC1) Deficiency. Neuropediatrics 2020, 51, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Jalil, M.A.; Begum, L.; Contreras, L.; Pardo, B.; Iijima, M.; Li, M.X.; Ramos, M.; Marmol, P.; Horiuchi, M.; Shimotsu, K.; et al. Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J. Biol. Chem. 2005, 280, 31333–31339. [Google Scholar] [CrossRef]
- Profilo, E.; Peña-Altamira, L.E.; Corricelli, M.; Castegna, A.; Danese, A.; Agrimi, G.; Petralla, S.; Giannuzzi, G.; Porcelli, V.; Sbano, L.; et al. Down-regulation of the mitochondrial aspartate-glutamate carrier isoform 1 AGC1 inhibits proliferation and N-acetylaspartate synthesis in Neuro2A cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1422–1435. [Google Scholar] [CrossRef]
- Petralla, S.; Peña-Altamira, L.E.; Poeta, E.; Massenzio, F.; Virgili, M.; Barile, S.N.; Sbano, L.; Profilo, E.; Corricelli, M.; Danese, A.; et al. Deficiency of Mitochondrial Aspartate-Glutamate Carrier 1 Leads to Oligodendrocyte Precursor Cell Proliferation Defects Both In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 4486. [Google Scholar] [CrossRef]
- Napolioni, V.; Persico, A.M.; Porcelli, V.; Palmieri, L. The mitochondrial aspartate/glutamate carrier AGC1 and calcium homeostasis: Physiological links and abnormalities in autism. Mol. Neurobiol. 2011, 44, 83–92. [Google Scholar] [CrossRef]
- Shelton, G.D.; Minor, K.M.; Li, K.; Naviaux, J.C.; Monk, J.; Wang, L.; Guzik, E.; Guo, L.T.; Porcelli, V.; Gorgoglione, R.; et al. A Mutation in the Mitochondrial Aspartate/Glutamate Carrier Leads to a More Oxidizing Intramitochondrial Environment and an Inflammatory Myopathy in Dutch Shepherd Dogs. J. Neuromuscul. Dis. 2019, 6, 485–501. [Google Scholar] [CrossRef]
- Dahlin, M.; Martin, D.A.; Hedlund, Z.; Jonsson, M.; von Döbeln, U.; Wedell, A. The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 2015, 56, e176–e181. [Google Scholar] [CrossRef] [PubMed]
- Monné, M.; Vozza, A.; Lasorsa, F.M.; Porcelli, V.; Palmieri, F. Mitochondrial Carriers for Aspartate, Glutamate and Other Amino Acids: A Review. Int. J. Mol. Sci. 2019, 20, 4456. [Google Scholar] [CrossRef]
- Kobayashi, K.; Sinasac, D.S.; Iijima, M.; Boright, A.P.; Begum, L.; Lee, J.R.; Yasuda, T.; Ikeda, S.; Hirano, R.; Terazono, H.; et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat. Genet. 1999, 22, 159–163. [Google Scholar] [CrossRef]
- Saheki, T.; Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 2002, 47, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Saheki, T.; Song, Y. Citrin Deficiency. In GeneReviews®-NCBI Bookshelf; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Tabata, A.; Sheng, J.-S.; Ushikai, M.; Song, Y.-Z.; Gao, H.-Z.; Lu, Y.-B.; Okumura, F.; Iijima, M.; Mutoh, K.; Kishida, S.; et al. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J. Hum. Genet. 2008, 53, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Hayasaka, K.; Numakura, C.; Yamakawa, M.; Mitsui, T.; Watanabe, H.; Haga, H.; Yazaki, M.; Ohira, H.; Ochiai, Y.; Tahara, T.; et al. Medium-chain triglycerides supplement therapy with a low-carbohydrate formula can supply energy and enhance ammonia detoxification in the hepatocytes of patients with adult-onset type II citrullinemia. J. Inherit. Metab. Dis. 2018, 41, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Saheki, T.; Moriyama, M.; Kuroda, E.; Funahashi, A.; Yasuda, I.; Setogawa, Y.; Gao, Q.; Ushikai, M.; Furuie, S.; Yamamura, K.-I.; et al. Pivotal role of inter-organ aspartate metabolism for treatment of mitochondrial aspartate-glutamate carrier 2 (citrin) deficiency, based on the mouse model. Sci. Rep. 2019, 9, 4179. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Identification and purification of the ornithine/citrulline carrier from rat liver mitochondria. Eur. J. Biochem. 1992, 207, 449–454. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Stipani, I.; Palmieri, F. The purified and reconstituted ornithine/citrulline carrier from rat liver mitochondria: Electrical nature and coupling of the exchange reaction with H+ translocation. Biochem. J. 1997, 327, 349–355. [Google Scholar] [CrossRef]
- Fiermonte, G.; Dolce, V.; David, L.; Santorelli, F.M.; Dionisi-Vici, C.; Palmieri, F.; Walker, J.E. The mitochondrial ornithine transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 2003, 278, 32778–32783. [Google Scholar] [CrossRef]
- Camacho, J.A.; Obie, C.; Biery, B.; Goodman, B.K.; Hu, C.-A.; Almashanu, S.; Steel, G.; Casey, R.; Lambert, M.; Mitchell, G.A.; et al. Hyperornithinaemia- syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat. Genet. 1999, 22, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Diodato, D.; Ponzi, E.; Monné, M.; Boenzi, S.; Bertini, E.; Fiermonte, G.; Dionisi-Vici, C. The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Orphanet J. Rare Dis. 2015, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Häberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders-update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Camacho, J.A.; Mardach, R.; Rioseco-Camacho, N.; Ruiz-Pesini, E.; Derbeneva, O.; Andrade, D.; Zaldivar, F.; Qu, Y.; Cederbaum, S.D. Clinical and functional characterization of a human ORNT1 mutation (T32R) in the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr. Res. 2006, 60, 423–429. [Google Scholar] [CrossRef]
- Tessa, A.; Fiermonte, G.; Dionisi-Vici, C.; Paradies, E.; Baumgartner, M.R.; Chien, Y.-H.; Loguercio, C.; de Baulny, H.O.; Nassogne, M.-C.; Schiff, M.; et al. Identification of novel mutations in the SLC25A15 gene in hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome: A clinical, molecular, and functional study. Hum. Mutat. 2009, 30, 741–748. [Google Scholar] [CrossRef]
- Ersoy Tunali, N.; Marobbio, C.M.T.; Tiryakioğlu, N.O.; Punzi, G.; Saygılı, S.K.; Onal, H.; Palmieri, F. A novel mutation in the SLC25A15 gene in a Turkish patient with HHH syndrome: Functional analysis of the mutant protein. Mol. Genet. Metab. 2014, 112, 25–29. [Google Scholar] [CrossRef]
- Zarrilli, R.; Oates, E.L.; McBride, O.W.; Lerman, M.I.; Chan, J.Y.; Santisteban, P.; Ursini, M.V.; Notkins, A.L.; Kohn, L.D. Sequence and chromosomal assignment of a novel cDNA identified by immunoscreening of a thyroid expression library: Similarity to a family of mitochondrial solute carrier proteins. Mol. Endocrinol. 1989, 3, 1498–1505. [Google Scholar] [CrossRef]
- Fiermonte, G.; Runswick, M.J.; Walker, J.E.; Palmieri, F. Sequence and pattern of expression of a bovine homologue of a human mitochondrial transport protein associated with Grave’s disease. DNA Seq. 1992, 3, 71–78. [Google Scholar] [CrossRef]
- Prohl, C.; Pelzer, W.; Diekert, K.; Kmita, H.; Bedekovics, T.; Kispal, G.; Lill, R. The yeast mitochondrial carrier Leu5p and its human homologue Graves’ disease protein are required for accumulation of coenzyme A in the matrix. Mol. Cell Biol. 2001, 21, 1089–1097. [Google Scholar] [CrossRef]
- Fiermonte, G.; Paradies, E.; Todisco, S.; Marobbio, C.M.T.; Palmieri, F. A novel member of solute carrier family 25 (SLC25A42) is a transporter of coenzyme A and adenosine 3′,5′-diphosphate in human mitochondria. J. Biol. Chem. 2009, 284, 18152–18159. [Google Scholar] [CrossRef]
- Khan, S.; Ansar, M.; Khan, A.K.; Shah, K.; Muhammad, N.; Shahzad, S.; Nickerson, D.A.; Bamshad, M.J.; Santos-Cortez, R.L.P.; Leal, S.M.; et al. A homozygous missense mutation in SLC25A16 associated with autosomal recessive isolated fingernail dysplasia in a Pakistani family. Br. J. Dermatol. 2018, 178, 556–558. [Google Scholar] [CrossRef]
- Rosenberg, M.J.; Agarwala, R.; Bouffard, G.; Davis, J.; Fiermonte, G.; Hilliard, M.S.; Koch, T.; Kalikin, L.M.; Makalowska, I.; Morton, D.H.; et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat. Genet. 2002, 32, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Shaag, A.; Edvardson, S.; Mandel, H.; Stepensky, P.; Shalev, S.A.; Horovitz, Y.; Pines, O.; Elpeleg, O. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann. Neurol. 2009, 66, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Ortigoza-Escobar, J.D.; Alfadhel, M.; Molero-Luis, M.; Darin, N.; Spiegel, R.; de Coo, I.F.; Gerards, M.; Taylor, R.W.; Artuch, R.; Nashabat, M.; et al. Thiamine deficiency in childhood with attention to genetic causes: Survival and outcome predictors. Ann. Neurol. 2017, 82, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Bottega, R.; Perrone, M.D.; Vecchiato, K.; Taddio, A.; Sabui, S.; Pecile, V.; Said, H.M.; Faletra, F. Functional analysis of the third identified SLC25A19 mutation causative for the thiamine metabolism dysfunction syndrome 4. J. Hum. Genet. 2019, 64, 1075–1081. [Google Scholar] [CrossRef]
- Gowda, V.K.; Srinivasan, V.M.; Jehta, K.; Bhat, M.D. Bilateral Striatal Necrosis with Polyneuropathy with a Novel SLC25A19 (Mitochondrial Thiamine Pyrophosphate Carrier OMIMI*606521) Mutation: Treatable Thiamine Metabolic Disorder-A Report of Two Indian Cases. Neuropediatrics 2019, 50, 313–317. [Google Scholar] [CrossRef]
- Dolce, V.; Fiermonte, G.; Runswick, M.J.; Palmieri, F.; Walker, J.E. The human mitochondrial deoxynucleotide carrier and its role in the toxicity of nucleoside antivirals. Proc. Natl. Acad. Sci. USA 2001, 98, 2284–2288. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Fiermonte, G.; Song, S.; Struys, E.; De Leonardis, F.; Schwartzberg, P.L.; Chen, A.; Castegna, A.; Verhoeven, N.; Mathews, C.K.; et al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc. Natl. Acad. Sci. USA 2006, 103, 15927–15932. [Google Scholar] [CrossRef]
- Marobbio, C.M.T.; Vozza, A.; Harding, M.; Bisaccia, F.; Palmieri, F.; Walker, J.E. Identification and reconstitution of the yeast mitochondrial transporter for thiamine pyrophosphate. EMBO J. 2002, 21, 5653–5661. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Mol. Aspects Med. 2011, 32, 223–233. [Google Scholar] [CrossRef]
- Stanley, C.A.; Palmieri, F.; Bennett, M.J. Disorders of the Mitochondrial Carnitine Shuttle. In Online Molecular and Metabolic Basis of Inherited Disease; Valle, D., Vogelstein, B., Kinzler, K., Antonarakis, S., Ballagio, A., Gibson, K., Mitchell, G., Eds.; McGraw Hill: New York, NY, USA, 2013. [Google Scholar]
- Stanley, C.A.; Hale, D.E.; Berry, G.T.; Deleeuw, S.; Boxer, J.; Bonnefont, J.P. Brief report: A deficiency of carnitine-acylcarnitine translocase in the inner mitochondrial membrane. N. Engl. J. Med. 1992, 327, 19–23. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Giangregorio, N.; Palmieri, F. The mitochondrial carnitine carrier protein: CDNA cloning, primary structure and comparison with other mitochondrial transport proteins. Biochem. J. 1997, 321, 713–719. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Invernizzi, F.; Baratta, S.; Pons, R.; Chung, W.; Garavaglia, B.; Dionisi-Vici, C.; Ribes, A.; Parini, R.; Huertas, M.D.; et al. Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum. Mutat. 2004, 24, 312–320. [Google Scholar] [CrossRef]
- De Lucas, J.R.; Indiveri, C.; Tonazzi, A.; Perez, P.; Giangregorio, N.; Iacobazzi, V.; Palmieri, F. Functional characterization of residues within the carnitine/acylcarnitine translocase RX2PANAAXF distinct motif. Mol. Membr. Biol. 2008, 25, 152–163. [Google Scholar] [CrossRef]
- IJlst, L.; van Roermund, C.W.; Iacobazzi, V.; Oostheim, W.; Ruiter, J.P.; Williams, J.C.; Palmieri, F.; Wanders, R.J. Functional analysis of mutant human carnitine acylcarnitine translocases in yeast. Biochem. Biophys. Res. Commun. 2001, 280, 700–706. [Google Scholar] [CrossRef]
- Pérez, P.; Martínez, O.; Romero, B.; Olivas, I.; Pedregosa, A.M.; Palmieri, F.; Laborda, F.; Ramón De Lucas, J. Functional analysis of mutations in the human carnitine/acylcarnitine translocase in Aspergillus nidulans. Fungal Genet. Biol. 2003, 39, 211–220. [Google Scholar] [CrossRef]
- Boczonadi, V.; King, M.S.; Smith, A.C.; Olahova, M.; Bansagi, B.; Roos, A.; Eyassu, F.; Borchers, C.; Ramesh, V.; Lochmüller, H.; et al. Mitochondrial oxodicarboxylate carrier deficiency is associated with mitochondrial DNA depletion and spinal muscular atrophy-like disease. Genet. Med. 2018, 20, 1224–1235. [Google Scholar] [CrossRef]
- Fiermonte, G.; Dolce, V.; Palmieri, L.; Ventura, M.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the human mitochondrial oxodicarboxylate carrier. Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J. Biol. Chem. 2001, 276, 8225–8230. [Google Scholar] [CrossRef]
- Palmieri, L.; Agrimi, G.; Runswick, M.J.; Fearnley, I.M.; Palmieri, F.; Walker, J.E. Identification in Saccharomyces cerevisiae of two isoforms of a novel mitochondrial transporter for 2-oxoadipate and 2-oxoglutarate. J. Biol. Chem. 2001, 276, 1916–1922. [Google Scholar] [CrossRef]
- Scarcia, P.; Palmieri, L.; Agrimi, G.; Palmieri, F.; Rottensteiner, H. Three mitochondrial transporters of Saccharomyces cerevisiae are essential for ammonium fixation and lysine biosynthesis in synthetic minimal medium. Mol. Genet. Metab. 2017, 122, 54–60. [Google Scholar] [CrossRef]
- Molinari, F.; Raas-Rothschild, A.; Rio, M.; Fiermonte, G.; Encha-Razavi, F.; Palmieri, L.; Palmieri, F.; Ben-Neriah, Z.; Kadhom, N.; Vekemans, M.; et al. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am. J. Hum. Genet. 2005, 76, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Kaminska, A.; Fiermonte, G.; Boddaert, N.; Raas-Rothschild, A.; Plouin, P.; Palmieri, L.; Brunelle, F.; Palmieri, F.; Dulac, O.; et al. Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin. Genet. 2009, 76, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Lemattre, C.; Imbert-Bouteille, M.; Gatinois, V.; Benit, P.; Sanchez, E.; Guignard, T.; Tran Mau-Them, F.; Haquet, E.; Rivier, F.; Carme, E.; et al. Report on three additional patients and genotype-phenotype correlation in SLC25A22-related disorders group. Eur. J. Hum. Genet. 2019, 27, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Poduri, A.; Heinzen, E.; Chitsazzadeh, V.; Lasorsa, F.; LaCoursiere, C.; Martin, E.; Yusakaitis, C.; Hill, R.; Elhosary, P.; Atabay, K.; et al. SLC25A22 is a novel gene for migrating partial seizures in infancy. Ann. Neurol. 2013, 76, 873–882. [Google Scholar] [CrossRef]
- Fiermonte, G.; Palmieri, L.; Todisco, S.; Agrimi, G.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 2002, 277, 19289–19294. [Google Scholar] [CrossRef]
- Goubert, E.; Mircheva, Y.; Lasorsa, F.M.; Melon, C.; Profilo, E.; Sutera, J.; Becq, H.; Palmieri, F.; Palmieri, L.; Aniksztejn, L.; et al. Inhibition of the Mitochondrial Glutamate Carrier SLC25A22 in Astrocytes Leads to Intracellular Glutamate Accumulation. Front. Cell. Neurosci. 2017, 11, 149. [Google Scholar] [CrossRef]
- Trabelsi, Y.; Amri, M.; Becq, H.; Molinari, F.; Aniksztejn, L. The conversion of glutamate by glutamine synthase in neocortical astrocytes from juvenile rat is important to limit glutamate spillover and peri/extrasynaptic activation of NMDA receptors. Glia 2017, 65, 401–415. [Google Scholar] [CrossRef]
- Reid, E.S.; Williams, H.; Anderson, G.; Benatti, M.; Chong, K.; James, C.; Ocaka, L.; GOSgene; Hemingway, C.; Little, D.; et al. Mutations in SLC25A22: Hyperprolinaemia, vacuolated fibroblasts and presentation with developmental delay. J. Inherit. Metab. Dis. 2017, 40, 385–394. [Google Scholar] [CrossRef]
- Ryu, J.; Ko, J.M.; Shin, C.-H. A 9-year-old Korean girl with Fontaine progeroid syndrome: A case report with further phenotypical delineation and description of clinical course during long-term follow-up. BMC Med. Genet. 2019, 20, 188. [Google Scholar] [CrossRef]
- Ehmke, N.; Graul-Neumann, L.; Smorag, L.; Koenig, R.; Segebrecht, L.; Magoulas, P.; Scaglia, F.; Kilic, E.; Hennig, A.F.; Adolphs, N.; et al. De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction. Am. J. Hum. Genet. 2017, 101, 833–843. [Google Scholar] [CrossRef]
- Writzl, K.; Maver, A.; Kovačič, L.; Martinez-Valero, P.; Contreras, L.; Satrustegui, J.; Castori, M.; Faivre, L.; Lapunzina, P.; van Kuilenburg, A.B.P.; et al. De Novo Mutations in SLC25A24 Cause a Disorder Characterized by Early Aging, Bone Dysplasia, Characteristic Face, and Early Demise. Am. J. Hum. Genet. 2017, 101, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Harborne, S.P.D.; Ruprecht, J.J.; Kunji, E.R.S. Calcium-induced conformational changes in the regulatory domain of the human mitochondrial ATP-Mg/Pi carrier. Biochim. Biophys. Acta 2015, 1847, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Monné, M.; Daddabbo, L.; Giannossa, L.C.; Nicolardi, M.C.; Palmieri, L.; Miniero, D.V.; Mangone, A.; Palmieri, F. Mitochondrial ATP-Mg/phosphate carriers transport divalent inorganic cations in complex with ATP. J. Bioenerg. Biomembr. 2017, 49, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Agrimi, G.; Di Noia, M.A.; Marobbio, C.M.T.; Fiermonte, G.; Lasorsa, F.M.; Palmieri, F. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 2004, 379, 183–190. [Google Scholar] [CrossRef]
- Marobbio, C.M.T.; Agrimi, G.; Lasorsa, F.M.; Palmieri, F. Identification and functional reconstitution of yeast mitochondrial carrier for S-adenosylmethionine. EMBO J. 2003, 22, 5975–5982. [Google Scholar] [CrossRef]
- Palmieri, L.; Arrigoni, R.; Blanco, E.; Carrari, F.; Zanor, M.I.; Studart-Guimaraes, C.; Fernie, A.R.; Palmieri, F. Molecular identification of an Arabidopsis S-adenosylmethionine transporter. Analysis of organ distribution, bacterial expression, reconstitution into liposomes, and functional characterization. Plant Physiol. 2006, 142, 855–865. [Google Scholar] [CrossRef]
- Kishita, Y.; Pajak, A.; Bolar, N.A.; Marobbio, C.M.T.; Maffezzini, C.; Miniero, D.V.; Monné, M.; Kohda, M.; Stranneheim, H.; Murayama, K.; et al. Intra-mitochondrial Methylation Deficiency Due to Mutations in SLC25A26. Am. J. Med. Genet. 2015, 97, 761–768. [Google Scholar] [CrossRef]
- Schiff, M.; Veauville-Merllié, A.; Su, C.H.; Tzagoloff, A.; Rak, M.; Ogier de Baulny, H.; Boutron, A.; Smedts-Walters, H.; Romero, N.B.; Rigal, O.; et al. SLC25A32 Mutations and Riboflavin-Responsive Exercise Intolerance. N. Engl. J. Med. 2016, 374, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.E.I.; Sallevelt, S.C.E.H.; Theunissen, T.E.J.; Hendrickx, A.T.M.; Gottschalk, R.W.; Hoeijmakers, J.G.J.; Habets, D.D.; Bierau, J.; Schoonderwoerd, K.G.; Smeets, H.J.M. Novel SLC25A32 mutation in a patient with a severe neuromuscular phenotype. Eur. J. Hum. Genet. 2017, 25, 886–888. [Google Scholar] [CrossRef]
- Tzagoloff, A.; Jang, J.; Glerum, D.M.; Wu, M. FLX1 codes for a carrier protein involved in maintaining a proper balance of flavin nucleotides in yeast mitochondria. J. Biol. Chem. 1996, 271, 7392–7397. [Google Scholar] [CrossRef]
- Titus, S.A.; Moran, R.G. Retrovirally mediated complementation of the glyB phenotype. Cloning of a human gene encoding the carrier for entry of folates into mitochondria. J. Biol. Chem. 2000, 275, 36811–36817. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; Ijlst, L.; van Roermund, C.W.T.; Wijburg, F.A.; Wanders, R.J.A.; Waterham, H.R. Identification of the human mitochondrial FAD transporter and its potential role in multiple acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2005, 86, 441–447. [Google Scholar] [CrossRef]
- Todisco, S.; Agrimi, G.; Castegna, A.; Palmieri, F. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2006, 281, 1524–1531. [Google Scholar] [CrossRef]
- Kim, J.; Lei, Y.; Guo, J.; Kim, S.-E.; Wlodarczyk, B.J.; Cabrera, R.M.; Lin, Y.L.; Nilsson, T.K.; Zhang, T.; Ren, A.; et al. Formate rescues neural tube defects caused by mutations in Slc25a32. Proc. Natl. Acad. Sci. USA 2018, 115, 4690–4695. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef]
- Le Rouzic, M.-A.; Fouquet, C.; Leblanc, T.; Touati, M.; Fouyssac, F.; Vermylen, C.; Jäkel, N.; Guichard, J.-F.; Maloum, K.; Toutain, F.; et al. Non syndromic childhood onset congenital sideroblastic anemia: A report of 13 patients identified with an ALAS2 or SLC25A38 mutation. Blood Cells. Mol. Dis. 2017, 66, 11–18. [Google Scholar] [CrossRef]
- Lunetti, P.; Damiano, F.; De Benedetto, G.; Siculella, L.; Pennetta, A.; Muto, L.; Paradies, E.; Marobbio, C.M.T.; Dolce, V.; Capobianco, L. Characterization of Human and Yeast Mitochondrial Glycine Carriers with Implications for Heme Biosynthesis and Anemia. J. Biol. Chem. 2016, 291, 19746–19759. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Smith, L.L.; Kentab, A.; Alkhalidi, H.; Summers, B.; Alsedairy, H.; Xiong, Y.; Gupta, V.A.; Alkuraya, F.S. Mutation of the mitochondrial carrier SLC25A42 causes a novel form of mitochondrial myopathy in humans. Hum. Genet. 2016, 135, 21–30. [Google Scholar] [CrossRef]
- Almannai, M.; Alasmari, A.; Alqasmi, A.; Faqeih, E.; Al Mutairi, F.; Alotaibi, M.; Samman, M.M.; Eyaid, W.; Aljadhai, Y.I.; Shamseldin, H.E.; et al. Expanding the phenotype of SLC25A42-associated mitochondrial encephalomyopathy. Clin. Genet. 2018, 93, 1097–1102. [Google Scholar] [CrossRef]
- Iuso, A.; Alhaddad, B.; Weigel, C.; Kotzaeridou, U.; Mastantuono, E.; Schwarzmayr, T.; Graf, E.; Terrile, C.; Prokisch, H.; Strom, T.M.; et al. A Homozygous Splice Site Mutation in SLC25A42, Encoding the Mitochondrial Transporter of Coenzyme A, Causes Metabolic Crises and Epileptic Encephalopathy. JIMD Rep. 2019, 44, 1–7. [Google Scholar]
- Vozza, A.; De Leonardis, F.; Paradies, E.; De Grassi, A.; Pierri, C.L.; Parisi, G.; Marobbio, C.M.T.; Lasorsa, F.M.; Muto, L.; Capobianco, L.; et al. Biochemical characterization of a new mitochondrial transporter of dephosphocoenzyme A in Drosophila melanogaster. Biochim. Biophys. Acta 2017, 1858, 137–146. [Google Scholar] [CrossRef]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef]
- Wan, J.; Steffen, J.; Yourshaw, M.; Mamsa, H.; Andersen, E.; Rudnik-Schöneborn, S.; Pope, K.; Howell, K.B.; McLean, C.A.; Kornberg, A.J.; et al. Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain J. Neurol. 2016, 139, 2877–2890. [Google Scholar] [CrossRef]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Des Rosiers, C.; Forest, A.; Lin, Z.-Y.; Gingras, A.-C.; et al. SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef]
- Hammer, M.B.; Ding, J.; Mochel, F.; Eleuch-Fayache, G.; Charles, P.; Coutelier, M.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Hernandez, D.G.; et al. SLC25A46 Mutations Associated with Autosomal Recessive Cerebellar Ataxia in North African Families. Neurodegener. Dis. 2017, 17, 208–212. [Google Scholar] [CrossRef]
- Nguyen, M.; Boesten, I.; Hellebrekers, D.M.E.I.; Mulder-den Hartog, N.M.; de Coo, I.F.M.; Smeets, H.J.M.; Gerards, M. Novel pathogenic SLC25A46 splice-site mutation causes an optic atrophy spectrum disorder. Clin. Genet. 2017, 91, 121–125. [Google Scholar] [CrossRef]
- Sulaiman, R.A.; Patel, N.; Alsharif, H.; Arold, S.T.; Alkuraya, F.S. A novel mutation in SLC25A46 causes optic atrophy and progressive limb spasticity, with no cerebellar atrophy or axonal neuropathy. Clin. Genet. 2017, 92, 230–231. [Google Scholar] [CrossRef]
- Braunisch, M.C.; Gallwitz, H.; Abicht, A.; Diebold, I.; Holinski-Feder, E.; Van Maldergem, L.; Lammens, M.; Kovács-Nagy, R.; Alhaddad, B.; Strom, T.M.; et al. Extension of the phenotype of biallelic loss-of-function mutations in SLC25A46 to the severe form of pontocerebellar hypoplasia type I. Clin. Genet. 2018, 93, 255–265. [Google Scholar] [CrossRef]
- Abrams, A.J.; Fontanesi, F.; Tan, N.B.L.; Buglo, E.; Campeanu, I.J.; Rebelo, A.P.; Kornberg, A.J.; Phelan, D.G.; Stark, Z.; Zuchner, S. Insights into the genotype-phenotype correlation and molecular function of SLC25A46. Hum. Mutat. 2018, 39, 1995–2007. [Google Scholar] [CrossRef]
- Walder, K.; Norman, R.A.; Hanson, R.L.; Schrauwen, P.; Neverova, M.; Jenkinson, C.P.; Easlick, J.; Warden, C.H.; Pecqueur, C.; Raimbault, S.; et al. Association between uncoupling protein polymorphisms (UCP2-UCP3) and energy metabolism/obesity in Pima indians. Hum. Mol. Genet. 1998, 7, 1431–1435. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schneitler, C.; Oberkofler, H.; Ebenbichler, C.; Paulweber, B.; Sandhofer, F.; Ladurner, G.; Hell, E.; Strosberg, A.D.; Patsch, J.R.; et al. A common polymorphism in the promoter of UCP2 is associated with decreased risk of obesity in middle-aged humans. Nat. Genet. 2001, 28, 178–183. [Google Scholar] [CrossRef]
- Ferrara, C.T.; Boodhansingh, K.E.; Paradies, E.; Fiermonte, G.; Steinkrauss, L.J.; Topor, L.S.; Quintos, J.B.; Ganguly, A.; De Leon, D.D.; Palmieri, F.; et al. Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J. Clin. Endocrinol. Metab. 2017, 102, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, E.A.; Ranchalis, J.; Crosslin, D.R.; Burt, A.; Brunzell, J.D.; Motulsky, A.G.; Nickerson, D.A.; NHLBI GO Exome Sequencing Project; Wijsman, E.M.; Jarvik, G.P. Joint linkage and association analysis with exome sequence data implicates SLC25A40 in hypertriglyceridemia. Am. J. Hum. Genet. 2013, 93, 1035–1045. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Buffet, A.; Morin, A.; Castro-Vega, L.-J.; Habarou, F.; Lussey-Lepoutre, C.; Letouzé, E.; Lefebvre, H.; Guilhem, I.; Haissaguerre, M.; Raingeard, I.; et al. Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res. 2018, 78, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mo, W.; Zhang, Z.; Yu, H.; Yang, A.; Qu, F.; Hu, P.; Liu, Z.; Wang, S. Single Nucleotide Polymorphisms in SLC19A1 and SLC25A9 Are Associated with Childhood Autism Spectrum Disorder in the Chinese Han Population. J. Mol. Neurosci. 2017, 62, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Köse, M.D.; Kagnici, M.; Özdemir, T.R.; Erdur, C.B.; Erdemir, G.; Karakoyun, M.; Guzin, Y.; Ceylaner, S.; Genel, F. Clinical findings in five Turkish patients with citrin deficiency and identification of a novel mutation on SLC25A13. J. Pediatr. Endocrinol. Metab. 2020, 33, 157–163. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
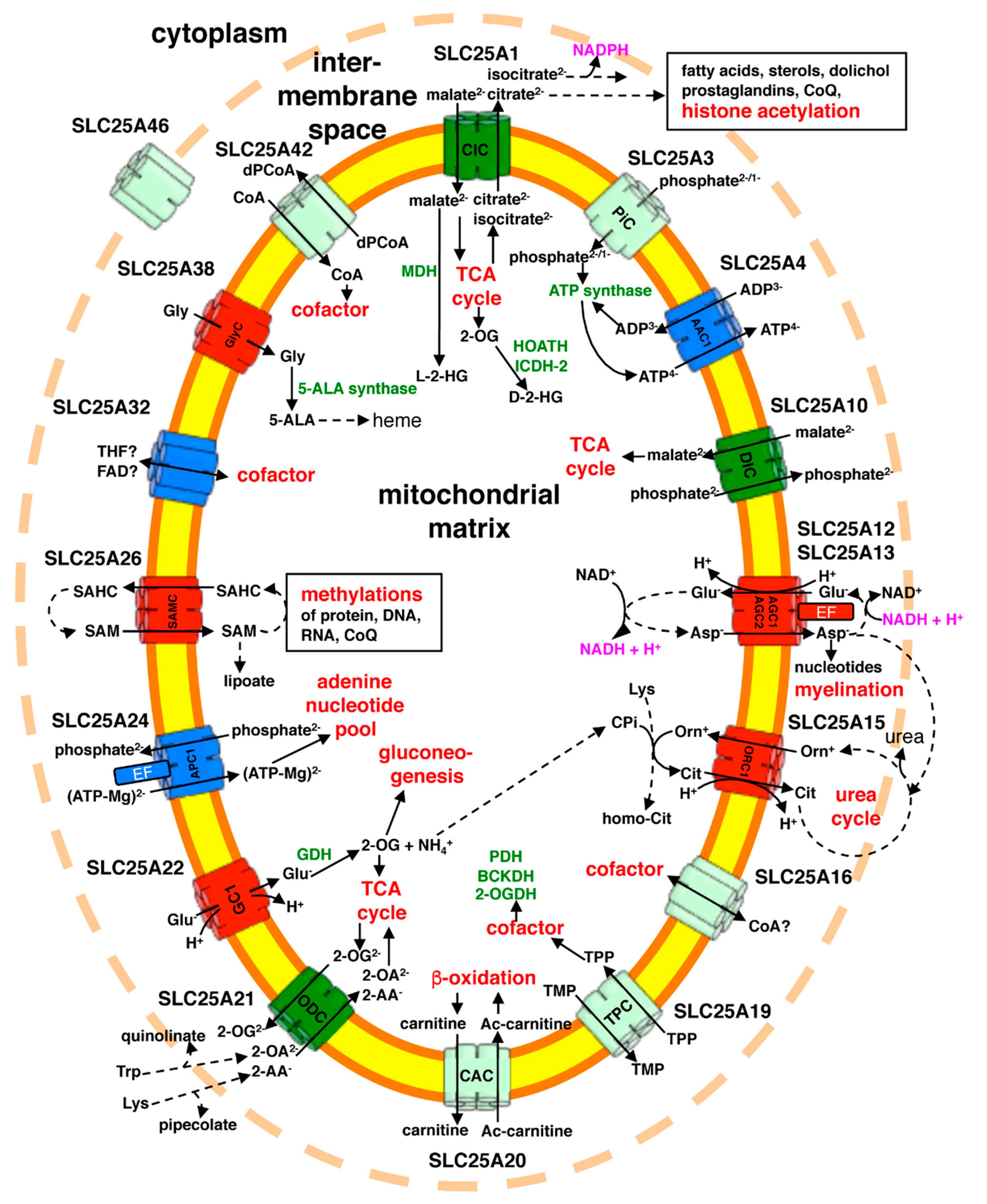{kind=link}
| Affected MC | Phenotype | OMIM/Inheritance | Mutations/Patients | References of First Reported Case |
|---|---|---|---|---|
| SLC25A1, citrate carrier (CIC) | Combined D-2- and L-2-hydroxyglutaric aciduria Congenital myasthenic syndrome 23 | 615182/AR 618197/AR | 24/40 | (Edvardson et al., 2013) (Nota et al., 2013) (Chaouch et al., 2014) |
| SLC25A3, phosphate carrier (PiC) | Mitochondrial phosphate carrier deficiency | 610773 | 4/7 | (Mayr et al., 2007) |
| SLC25A4, ADP/ATP carrier 1 (AAC1) | Autosomal dominant progressive external ophthalmoplegia with mitochondrial DNA deletions 2 (AdPEO2) Mitochondrial DNA depletion syndrome (MTDPS) 12B (cardiomyopathic type) Mitochondrial DNA depletion syndrome (MTDPS) 12A (cardiomyopathic type) | 609283/AD 615418/AR 617184/AD | 5/9? 6/7 3/5 | (Kaukonen et al., 2000) (Palmieri et al., 2005) (Thompson et al., 2016) |
| SLC25A10, dicarboxylate carrier (DIC) | Intractable epileptic encephalopathy with complex I deficiency | AR | 3/1 | (Punzi et al., 2018) |
| SLC25A12, aspartate/glutamate carrier 1 (AGC1) | Early infantile epileptic encephalopathy 39 (AGC1 deficiency) | 612949/AR | 3/4 | (Wibom et al., 2009) |
| SLC25A13, aspartate/glutamate carrier 2 (AGC2) | Adult-onset citrullinemia type II (CTLN2) Neonatal-onset citrullinemia type II (NICCD) | 603471/AR 605814/AR | 117/>600 | (Saheki and Kobayashi, 2002) (Kobayashi et al., 1999) |
| SLC25A15, ornithine carrier 1 (ORC1) | Hyperornithinemia-hyperammonemia-homocitrullinemia (HHH) syndrome | 238970/AR | 38/91 | (Camacho et al., 1999) |
| SLC25A16 | Fingernail dysplasia | AR | 1/9 | (Khan et al., 2018) |
| SLC25A19, thiamine pyrophosphate carrier (TPC) | Amish microcephaly Thiamine metabolism dysfunction syndrome 4 (progressive polyneuropathy type) | 607196/AR 613710/AR | 1/? 5/8 | (Rosenberg et al., 2002) (Spiegel et al., 2009) |
| SLC25A20, carnitine/acylcarnitine carrier (CAC) | Carnitine-acylcarnitine translocase deficiency (CAC deficiency) | 212138/AR | 38/43? | (Huizing et al., 1997) |
| SLC25A21, oxodicarboxylate carrier (ODC) | Mitochondrial DNA depletion and spinal muscular atrophy–like disease | 618811 | 1/1 | (Boczonadi et al., 2018) |
| SLC25A22, glutamate carrier 1 (GC1) | Early infantile epileptic encephalopathy 3 (EIEE3) | 609304/AR | 11/17 | (Molinari et al., 2005) |
| SLC25A24, ATP-Mg/phosphate carrier 1 (APC1) | Fontaine progeroid syndrome | 612289/AD | 2/11 | (Ehmke et al., 2017) (Writzl et al., 2017) |
| SLC25A26, S-adenosylmethionine carrier (SAMC) | Combined oxidative phosphorylation deficiency 28 | 616794/AR | 4/3 | (Kishita et al., 2015) |
| SLC25A32 | Riboflavin-responsive exercise intolerance | 616839/AR | 3/2 | (Schiff et al., 2016) |
| SLC25A38, glycine carrier (GlyC) | Congenital sideroblastic anemia 2 (pyrodoxine-refractory) | 205950/AR | 25/39 | (Guernsey et al., 2009) |
| SLC25A42, CoA and PAP carrier | Recurrent metabolic crises with variable encephalomyopathic features and neurologic regression | 618416/AR | 2/15 | (Shamseldin et al., 2015) |
| SLC25A46 | Hereditary motor and sensory neuropathy type VIB | 616505/AR | 16/25 | (Abrams et al., 2015) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmieri, F.; Scarcia, P.; Monné, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. https://doi.org/10.3390/biom10040655
Palmieri F, Scarcia P, Monné M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules. 2020; 10(4):655. https://doi.org/10.3390/biom10040655
Chicago/Turabian StylePalmieri, Ferdinando, Pasquale Scarcia, and Magnus Monné. 2020. "Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review" Biomolecules 10, no. 4: 655. https://doi.org/10.3390/biom10040655
APA StylePalmieri, F., Scarcia, P., & Monné, M. (2020). Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules, 10(4), 655. https://doi.org/10.3390/biom10040655