Antimicrobial and Antibiofilm Peptides




Abstract
1. Introduction
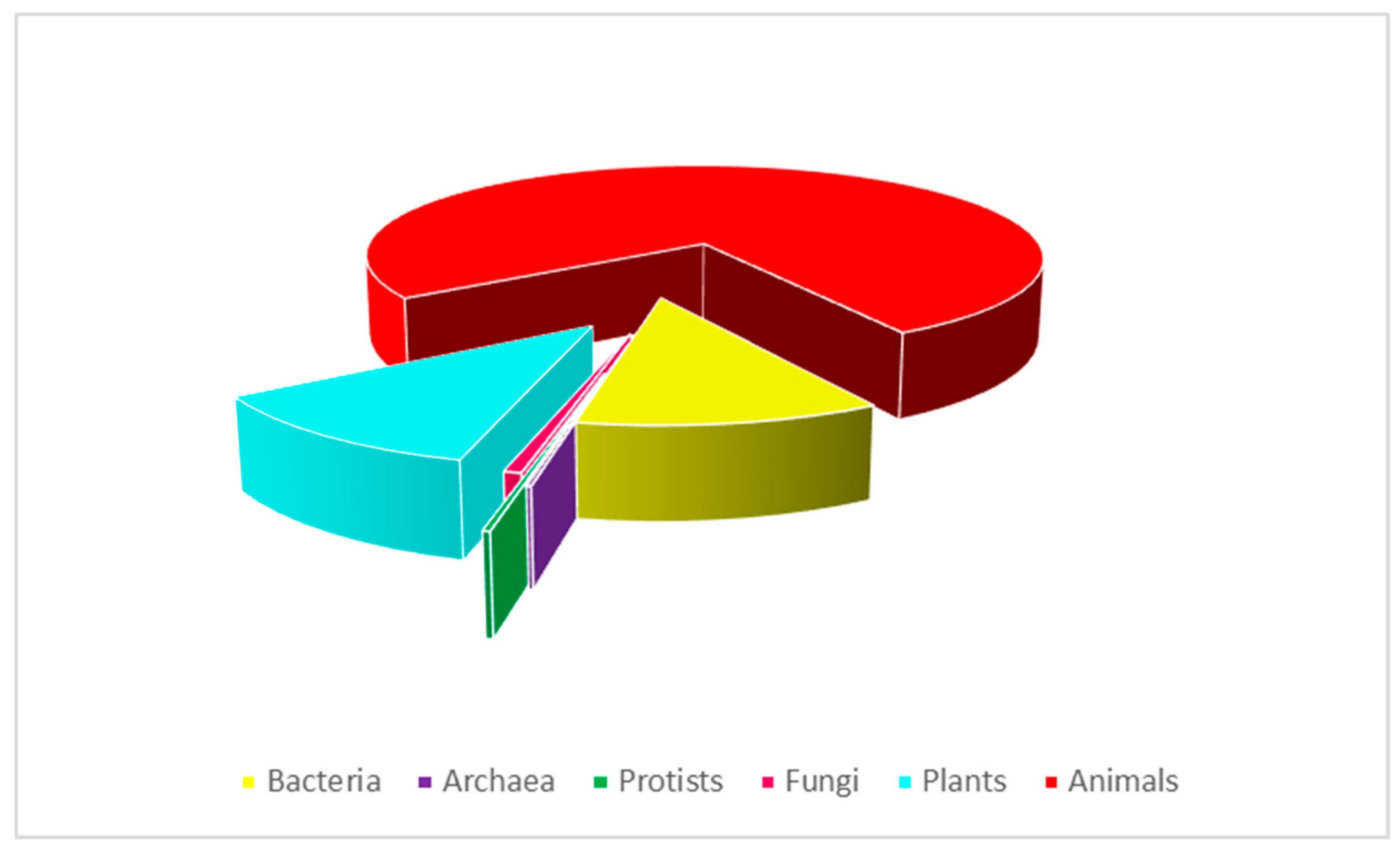
2. Antimicrobial Peptides
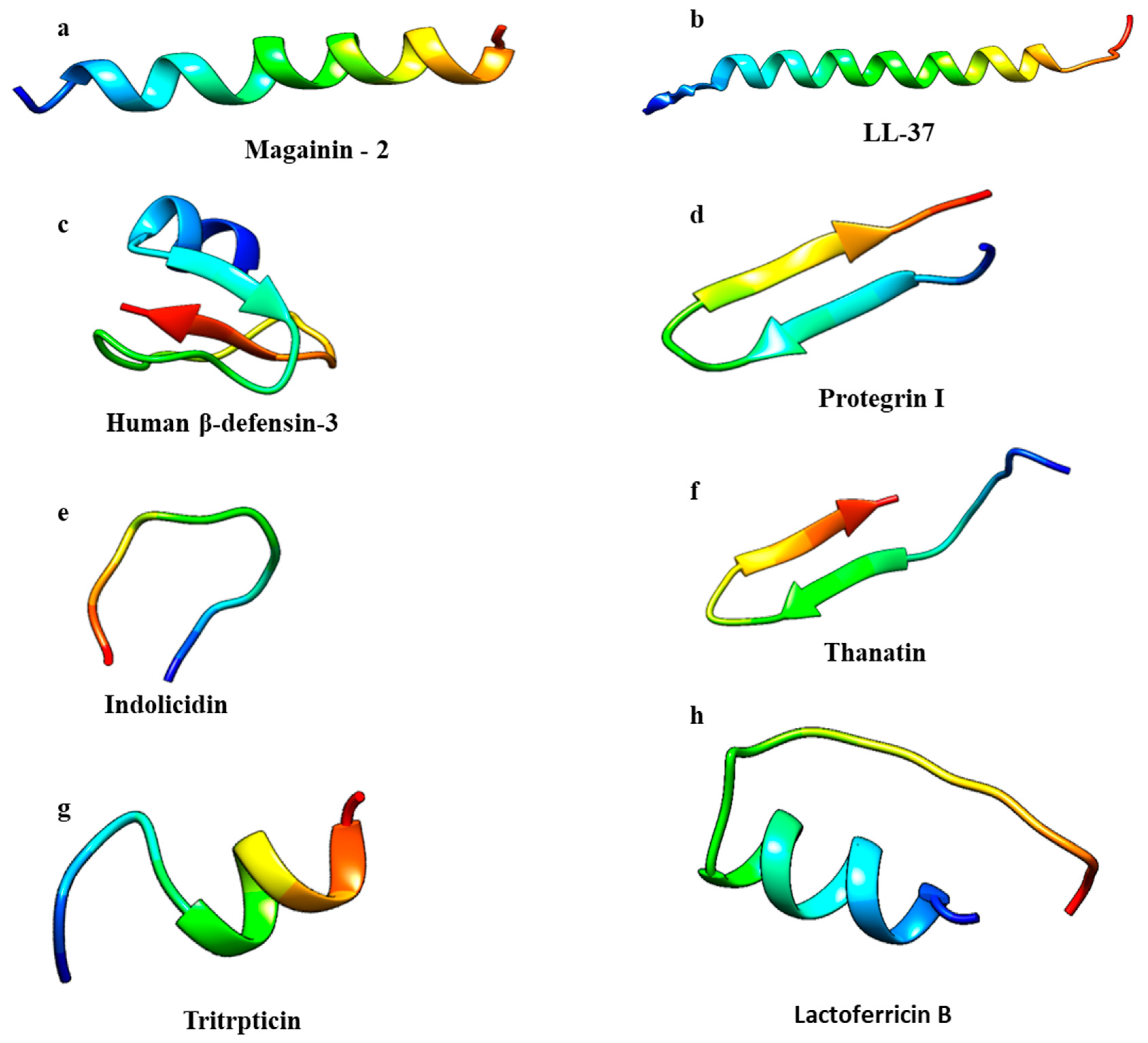
2.1. Structure
2.2. Antibacterial Peptides
2.3. Anticancer Peptides
2.4. Antiviral Peptides
2.5. Antifungal Peptides
2.6. Antiparasitic Peptides
3. Biofilm
3.1. Antimicrobial Peptides and Biofilm
3.2. Biofilm Resistance to Antimicrobial Peptides
4. Discussion and Future Considerations
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPs | Antimicrobial peptides |
| APD | Antimicrobial Peptide Database |
| ACPs | Anticancer peptides |
| MBIC | Minimum Biofilm Inhibitory Concentration |
| MBEC | Minimum Biofilm Eradication Concentration |
| MIC | Minimum Inhibitory Concentration |
| QS | Quorum Sensing |
| PIA | Polysaccharide Intracellular Adhesin |
| EPS | Extracellular Polymeric Substances |
| PS | Phosphatidylserine |
| PG | Phosphatidylglycerol |
| APG | Alanyl - Phosphatidylglycerol |
| LPG | Lysyl - Phosphatidylglycerol |
| TCS | Two Component Systems |
References
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 1929, 10, 226. [Google Scholar] [CrossRef]
- Zaffiri, L.; Gardner, J.; Toledo-Pereyra, L.H. History of antibiotics. From salvarsan to cephalosporins. J. Investig. Surg. 2012, 25, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R. Different roads to discovery; Prontosil (hence sulfa drugs) and penicillin (hence β-lactams). J. Ind. Microbiol. Biotechnol. 2009, 36, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Davies, J. Where have all the antibiotics gone? Can. J. Infect. Dis. Med Microbiol. 2006, 17, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Mueller, L.V.; Polyakov, M.; Weinstock, S.F. Where have all the antibiotic patents gone? Nat. Biotechnol. 2006, 24, 1529. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Guaní-Guerra, E.; Santos-Mendoza, T.; Lugo-Reyes, S.O.; Terán, L.M. Antimicrobial peptides: General overview and clinical implications in human health and disease. Clin. Immunol. 2010, 135, 1–11. [Google Scholar] [CrossRef]
- Harris, F.; Sarah, R.D.; David, A.P. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef]
- Malik, E.; Dennison, S.; Harris, F.; Phoenix, D. pH dependent antimicrobial peptides and proteins, their mechanisms of action and potential as therapeutic agents. Pharmaceuticals 2016, 9, 67. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389. [Google Scholar] [CrossRef] [PubMed]
- Soravia, E.; Martini, G.; Zasloff, M. Antimicrobial properties of peptides from Xenopus granular gland secretions. FEBS Lett. 1988, 228, 337–340. [Google Scholar] [CrossRef]
- Steiner, H.; Hultmark, D.; Engström, Å.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with dual antimicrobial and anticancer activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef]
- Jefferson, K.K. What drives bacteria to produce a biofilm? FEMS Microbiol. Lett. 2004, 236, 163–173. [Google Scholar] [CrossRef]
- Bahar, A.A.; Dacheng, R. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef]
- Powers, J.-P.S.; Hancock, R.E. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar] [CrossRef]
- Huang, Y.B.; Huang, J.F.; Chen, Y.X. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 143–152. [Google Scholar] [CrossRef]
- Lewies, A.; Wentzel, J.; Jacobs, G.; Du Plessis, L. The potential use of natural and structural analogues of antimicrobial peptides in the fight against neglected tropical diseases. Molecules 2015, 20, 15392–15433. [Google Scholar] [CrossRef] [PubMed]
- Yount, N.Y.; Bayer, A.S.; Xiong, Y.Q.; Yeaman, M.R. Advances in antimicrobial peptide immunobiology. Pept. Sci. Orig. Res. Biomol. 2006, 84, 435–458. [Google Scholar] [CrossRef]
- Bulet, P.; Stöcklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Hall, K.N.; Aguilar, M.I. Antimicrobial peptide structure and mechanism of action: A focus on the role of membrane structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Dhople, V.; Krukemeyer, A.; Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1499–1512. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Falla, T.J.; Karunaratne, D.N.; Hancock, R.E. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 1996, 271, 19298–19303. [Google Scholar] [CrossRef]
- Schibli, D.J.; Nguyen, L.T.; Kernaghan, S.D.; Rekdal, Ø.; Vogel, H.J. Structure-function analysis of tritrpticin analogs: Potential relationships between antimicrobial activities, model membrane interactions, and their micelle-bound NMR structures. Biophys. J. 2006, 91, 4413–4426. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Apostolov, O.; Seelig, J. Binding of the antibacterial peptide magainin 2 amide to small and large unilamellar vesicles. Biophys. Chem. 2000, 85, 187–198. [Google Scholar] [CrossRef]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Mankoci, S.; Ewing, J.; Dalai, P.; Sahai, N.; Barton, H.A.; Joy, A. Bacterial Membrane Selective Antimicrobial Peptide-Mimetic Polyurethanes: Structure—Property Correlations and Mechanisms of Action. Biomacromolecules 2019, 20, 4096–4106. [Google Scholar] [CrossRef] [PubMed]
- Alghalayini, A.; Garcia, A.; Berry, T.; Cranfield, C.G. The Use of Tethered Bilayer Lipid Membranes to Identify the Mechanisms of Antimicrobial Peptide Interactions with Lipid Bilayers. Antibiotics 2019, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238. [Google Scholar] [CrossRef] [PubMed]
- Cassone, M.; Frith, N.; Vogiatzi, P.; Wade, J.D.; Otvos, L. Induced resistance to the designer proline-rich antimicrobial peptide A3-APO does not involve changes in the intracellular target DnaK. Int. J. Pept. Res. Ther. 2009, 15, 121–128. [Google Scholar] [CrossRef]
- Shah, P.; Hsiao, F.S.H.; Ho, Y.H.; Chen, C.S. The proteome targets of intracellular targeting antimicrobial peptides. Proteomics 2016, 16, 1225–1237. [Google Scholar] [CrossRef]
- Graf, M.; Wilson, D.N. Intracellular Antimicrobial Peptides Targeting the Protein Synthesis Machinery. Adv. Exp. Med. Biol. 2019, 1117, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, M.; Sola, R.; Degasperi, M.; Scocchi, M. Search for Shorter Portions of the Proline-Rich Antimicrobial Peptide Fragment Bac5 (1–25) That Retain Antimicrobial Activity by Blocking Protein Synthesis. ChemMedChem 2019, 14, 343–348. [Google Scholar] [CrossRef]
- Barreto-Santamaría, A.; Curtidor, H.; Arévalo-Pinzón, G.; Herrera, C.; Suárez, D.; Pérez, W.H.; Patarroyo, M.E. A new synthetic peptide having two target of antibacterial action in E. coli ML35. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S.; Ifrah, D.; Lerche, S.; Gottlieb, C.T.; Cohn, M.T.; Hiasa, H.; Thomsen, L.E. The antimicrobial lysine-peptoid hybrid LP5 inhibits DNA replication and induces the SOS response in Staphylococcus aureus. BMC Microbiol. 2013, 13. [Google Scholar] [CrossRef]
- Rapaport, D.; Shai, Y. Interaction of fluorescently labeled pardaxin and its analogues with lipid bilayers. J. Biol. Chem. 1991, 266, 23769–23775. [Google Scholar] [PubMed]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Hong, T.; Waring, A.; Lehrer, R.I.; Hong, M. Solid-state NMR investigations of peptide-lipid interaction and orientation of a β-sheet antimicrobial peptide, protegrin. Biochemistry 2002, 41, 9852–9862. [Google Scholar] [CrossRef] [PubMed]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Pept. Sci. Orig. Res. Biomol. 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Sitaram, N.; Nagaraj, R. Interaction of antimicrobial peptides with biological and model membranes: Structural and charge requirements for activity. Biochim. Biophys. Acta (BBA)-Biomembr. 1999, 1462, 29–54. [Google Scholar] [CrossRef]
- Di Somma, A.; Avitabile, C.; Cirillo, A.; Moretta, A.; Merlino, A.; Paduano, L.; Duilio, A.; Romanelli, A. The antimicrobial peptide Temporin L impairs E. coli cell division by interacting with FtsZ and the divisome complex. Biochim. Biophys. Acta-Gen. Subj. 2020, 1864, 129606. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.A.; Maloy, W.L.; Zasloff, M.; Jacob, L.S. Anticancer efficacy of Magainin 2 and analogue peptides. Cancer Res. 1993, 53, 3052–3057. [Google Scholar] [PubMed]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Shahar, M.; Eisenbach, L.; Shai, Y. A novel lytic peptide composed of DL-amino acids selectively kills cancer cells in culture and in mice. J. Biol. Chem. 2003, 278, 21018–21023. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.C.; Mi, Z.; Kim, S.H.; Ng, B.; Robbins, P.D. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001, 61, 7709–7712. [Google Scholar] [PubMed]
- Bulet, P.; Hetru, C.; Dimarcq, J.L.; Hoffmann, D. Antimicrobial peptides in insects; structure and function. Dev. Comp. Immunol. 1999, 23, 329–344. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013, 33, 190–234. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.M.; Comfurius, P.; Zwaal, R.F.A. Regulatory mechanisms in maintenance and modulation of transmembrane lipid asymmetry: Pathophysiological implications. Lupus 1996, 5, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar] [PubMed]
- Van Zoggel, H.; Hamma-Kourbali, Y.; Galanth, C.; Ladram, A.; Nicolas, P.; Courty, J.; Amiche, M.; Delbé, J. Antitumor and angiostatic peptides from frog skin secretions. Amino Acids 2012, 42, 385–395. [Google Scholar] [CrossRef]
- Lin, W.J.; Chien, Y.L.; Pan, C.Y.; Lin, T.L.; Chen, J.Y.; Chiu, S.J.; Hui, C.F. Epinecidin-1, an antimicrobial peptide from fish (Epinephelus coioides) which has an antitumor effect like lytic peptides in human fibrosarcoma cells. Peptides 2009, 30, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Kołodziej, M.; Joniec, J.; Bartoszcze, M.; Mirski, T.; Gryko, R. Peptides—A new strategy for combating viral infections. Przeglad Epidemiologiczny 2011, 65, 477–482. [Google Scholar]
- Goodsell, D.S. Illustrations of the HIV life cycle. In The Future of HIV-1 Therapeutics; Springer: Cham, Switzerland, 2015; pp. 243–252. [Google Scholar] [CrossRef]
- Horne, W.S.; Wiethoff, C.M.; Cui, C.; Wilcoxen, K.M.; Amorin, M.; Ghadiri, M.R.; Nemerow, G.R. Antiviral cyclic d, l-α-peptides: Targeting a general biochemical pathway in virus infections. Bioorg. Med. Chem. 2005, 13, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.; Lima, L.A.; Miranda, V.; Dias, S.C.; Franco, O.L. Current scenario of peptide-based drugs: The key roles of cationic antitumor and antiviral peptides. Front. Microbiol. 2013, 4, 321. [Google Scholar] [CrossRef] [PubMed]
- Matanic, V.C.A.; Castilla, V. Antiviral activity of antimicrobial cationic peptides against Junin virus and herpes simplex virus. Int. J. Antimicrob. Agents 2004, 23, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.H.; Jenssen, H.; Sandvik, K.; Gutteberg, T.J. Anti-HSV activity of lactoferrin and lactoferricin is dependent on the presence of heparan sulphate at the cell surface. J. Med Virol. 2004, 74, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Andersen, J.H.; Uhlin-Hansen, L.; Gutteberg, T.J.; Rekdal, Ø. Anti-HSV activity of lactoferricin analogues is only partly related to their affinity for heparan sulfate. Antivir. Res. 2004, 61, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Rani, J. Sequential and structural aspects of antifungal peptides from animals, bacteria and fungi based on bioinformatics tools. Probiotics Antimicrob. Proteins 2016, 8, 85–101. [Google Scholar] [CrossRef]
- Aoki, W.; Ueda, M. Characterization of antimicrobial peptides toward the development of novel antibiotics. Pharmaceuticals 2013, 6, 1055–1081. [Google Scholar] [CrossRef] [PubMed]
- Van der Weerden, N.L.; Bleackley, M.R.; Anderson, M.A. Properties and mechanisms of action of naturally occurring antifungal peptides. Cell. Mol. Life Sci. 2013, 70, 3545–3570. [Google Scholar] [CrossRef] [PubMed]
- Gwadz, R.W.; Kaslow, D.; Lee, J.Y.; Maloy, W.L.; Zasloff, M.; Miller, L.H. Effects of magainins and cecropins on the sporogonic development of malaria parasites in mosquitoes. Infect. Immun. 1989, 579, 2628–2633. [Google Scholar] [CrossRef]
- Dagan, A.; Efron, L.; Gaidukov, L.; Mor, A.; Ginsburg, H. In vitro antiplasmodium effects of dermaseptin S4 derivatives. Antimicrob. Agents Chemother. 2002, 46, 1059–1066. [Google Scholar] [CrossRef]
- Conde, R.; Zamudio, F.Z.; Rodrı́guez, M.H.; Possani, L.D. Scorpine, an anti-malaria and anti-bacterial agent purified from scorpion venom. FEBS Lett. 2000, 471, 165–168. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Papo, N.; Saugar, J.M.; Barra, D.; Shai, Y.; Simmaco, M.; Rivas, L. Effect of natural L-to D-amino acid conversion on the organization, membrane binding, and biological function of the antimicrobial peptides bombinins H. Biochemistry 2006, 45, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Jang, S.H.; Lee, D.G.; Hahm, K.S. Antinematodal effect of antimicrobial peptide, PMAP-23, isolated from porcine myeloid against Caenorhabditis elegans. J. Pept. Sci. 2004, 10, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Maukonen, J.; Mättö, J.; Wirtanen, G.; Raaska, L.; Mattila-Sandholm, T.; Saarela, M. Methodologies for the characterization of microbes in industrial environments: A review. J. Ind. Microbiol. Biotechnol. 2003, 30, 327–356. [Google Scholar] [CrossRef] [PubMed]
- López, D.; Vlamakis, H.; Kolter, R. Biofilms. Cold Spring Harb. Perspect. Biol. 2010, 2, a000398. [Google Scholar] [CrossRef] [PubMed]
- Rabin, N.; Zheng, Y.; Opoku-Temeng, C.; Du, Y.; Bonsu, E.; Sintim, H.O. Biofilm formation mechanisms and targets for developing antibiofilm agents. Future Med. Chem. 2015, 7, 493–512. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ren, H.; Ding, L.; Geng, J.; Xu, K.; Zhang, Y. Aging biofilm from a full-scale moving bed biofilm reactor: Characterization and enzymatic treatment study. Bioresour. Technol. 2014, 154, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.E.; O’toole, G.A. Microbial biofilms: From ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 2000, 64, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.D.; Shah, S.; Tammela, P. Defining conditions for biofilm inhibition and eradication assays for Gram-positive clinical reference strains. BMC Microbiol. 2018, 18, 1–9. [Google Scholar] [CrossRef]
- Raheem, N.; Strauss, K. Mechanism of action for antimicrobial peptides with antibacterial and antibiofilm functions. Front. Microbiol. 2019, 10, 2866. [Google Scholar] [CrossRef]
- Morroni, G.; Simonetti, O.; Brenciani, A.; Brescini, L.; Kamysz, W.; Kamysz, E.; Neubauer, D.; Caffarini, M.; Orciani, M.; Giovanetti, E.; et al. In vitro activity of Protegrin-1, alone and in combination with clinically useful antibiotics, against Acinetobacter baumannii strains isolated from surgical wounds. Med Microbiol. Immunol. 2019, 208, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Tong, Z.; Lin, Y.; Xue, Y.; Wang, W.; Kuang, R.; Wang, P.; Tian, Y.; Ni, L. Antimicrobial and antibiofilm activity of pleurocidin against cariogenic microorganisms. Peptides 2011, 32, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 2160–2172. [Google Scholar] [CrossRef]
- De La Fuente-Núñez, C.; Cardoso, M.H.; De Souza Cândido, E.; Franco, O.L.; Hancock, R.E. Synthetic antibiofilm peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Blower, R.J.; Barksdale, S.M.; Van Hoek, M.L. Snake cathelicidin NA-CATH and smaller helical antimicrobial peptides are effective against Burkholderia thailandensis. PLoS Negl. Trop. Dis. 2015, 9. [Google Scholar] [CrossRef]
- Sutton, J.M.; Pritts, T.A. Human beta-defensin 3: A novel inhibitor of Staphylococcus-Produced biofilm production. Commentary on Human β-defensin 3 inhibits antibiotic-resistant Staphylococcus biofilm formation. J. Surg. Res. 2014, 186, 99. [Google Scholar] [CrossRef]
- Arslan, S.Y.; Leung, K.P.; Wu, C.D. The effect of lactoferrin on oral bacterial attachment. Oral Microbiol. Immunol. 2009, 24, 411–416. [Google Scholar] [CrossRef]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef]
- Okuda, K.I.; Zendo, T.; Sugimoto, S.; Iwase, T.; Tajima, A.; Yamada, S.; Sonomoto, K.; Mizunoe, Y. Effects of bacteriocins on methicillin-resistant Staphylococcus aureus biofilm. Antimicrob. Agents Chemother. 2013, 57, 5572–5579. [Google Scholar] [CrossRef]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef]
- Brancatisano, F.L.; Maisetta, G.; Di Luca, M.; Esin, S.; Bottai, D.; Bizzarri, R.; Campa, M.; Batoni, G. Inhibitory effect of the human liver-derived antimicrobial peptide hepcidin 20 on biofilms of polysaccharide intercellular adhesin (PIA)-positive and PIA-negative strains of Staphylococcus epidermidis. Biofouling 2014, 30, 435–446. [Google Scholar] [CrossRef]
- Pletzer, D.; Coleman, S.R.; Hancock, R.E. Anti-biofilm peptides as a new weapon in antimicrobial warfare. Curr. Opin. Microbiol. 2016, 33, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Tan, H.; Cheng, T.; Shen, H.; Shao, J.; Guo, Y.; Shi, S.; Zhang, X. Human β-defensin 3 inhibits antibiotic-resistant Staphylococcus biofilm formation. J. Surg. Res. 2013, 183, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.; Lee, J.H.; Kim, Y.G.; Kim, M.S.; Seo, C.H.; Park, Y. Anti-microbial, anti-biofilm activities and cell selectivity of the NRC-16 peptide derived from witch flounder, Glyptocephalus cynoglossus. Mar. Drugs 2013, 11, 1836–1852. [Google Scholar] [CrossRef]
- Anunthawan, T.; De La Fuente-Núñez, C.; Hancock, R.E.; Klaynongsruang, S. Cationic amphipathic peptides KT2 and RT2 are taken up into bacterial cells and kill planktonic and biofilm bacteria. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 1352–1358. [Google Scholar] [CrossRef]
- Butts, A.; Krysan, D.J. Antifungal drug discovery: Something old and something new. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- De Brucker, K.; Delattin, N.; Robijns, S.; Steenackers, H.; Verstraeten, N.; Landuyt, B.; Luyten, W.; Schoofs, L.; Dovgan, B.; Fröhlich, M.; et al. Derivatives of the mouse cathelicidin-related antimicrobial peptide (CRAMP) inhibit fungal and bacterial biofilm formation. Antimicrob. Agents Chemother. 2014, 58, 5395–5404. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; De la Fuente-Núñez, C.; Hancock, R.E.W. Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 2015, 21, 323–329. [Google Scholar] [CrossRef]
- De La Fuente-Núñez, C.; Reffuveille, F.; Mansour, S.C.; Reckseidler-Zenteno, S.L.; Hernández, D.; Brackman, G.; Coenye, T.; Hancock, R.E. D-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem. Biol. 2015, 22, 196–205. [Google Scholar] [CrossRef]
- Mataraci, E.; Dosler, S. In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant Staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 2012, 56, 6366–6371. [Google Scholar] [CrossRef]
- Otto, M. Bacterial evasion of antimicrobial peptides by biofilm formation. In Antimicrobial Peptides and Human Disease; Springer: Berlin/Heidelberg, Germany, 2006; pp. 251–258. [Google Scholar] [CrossRef]
- Vuong, C.; Voyich, J.M.; Fischer, E.R.; Braughton, K.R.; Whitney, A.R.; DeLeo, F.R.; Otto, M. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell. Microbiol. 2004, 6, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Burrows, L.L.; Deber, C.M. Helix induction in antimicrobial peptides by alginate in biofilms. J. Biol. Chem. 2004, 279, 38749–38754. [Google Scholar] [CrossRef]
- Kuo, H.H.; Chan, C.; Burrows, L.L.; Deber, C.M. Hydrophobic interactions in complexes of antimicrobial peptides with bacterial polysaccharides. Chem. Biol. Drug Des. 2007, 69, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.M.; Kuhn, S.; Slavetinsky, C.J.; Krismer, B.; Heilbronner, S.; Gekeler, C.; Kraus, D.; Peschel, A. The lipid-modifying multiple peptide resistance factor is an oligomer consisting of distinct interacting synthase and flippase subunits. MBio 2015, 6, e02340-14. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.M.; Staubitz, P.; Mishra, N.N.; Yang, S.J.; Hornig, G.; Kalbacher, H.; Bayer, A.S.; Kraus, D.; Peschel, A. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog. 2009, 5, e1000660. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Mishra, N.N.; Rubio, A.; Bayer, A.S. Causal role of single nucleotide polymorphisms within the mprF gene of Staphylococcus aureus in daptomycin resistance. Antimicrob. Agents Chemother. 2013, 57, 5658–5664. [Google Scholar] [CrossRef]
- Klein, S.; Lorenzo, C.; Hoffmann, S.; Walther, J.M.; Storbeck, S.; Piekarski, T.; Tindall, B.J.; Wray, V.; Nimtz, M.; Moser, J. Adaptation of Pseudomonas aeruginosa to various conditions includes tRNA-dependent formation of alanyl-phosphatidylglycerol. Mol. Microbiol. 2009, 71, 551–565. [Google Scholar] [CrossRef]
- Ramsey, M.M.; Whiteley, M. Pseudomonas aeruginosa attachment and biofilm development in dynamic environments. Mol. Microbiol. 2004, 53, 1075–1087. [Google Scholar] [CrossRef]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef]
- Dalebroux, Z.D.; Miller, S.I. Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr. Opin. Microbiol. 2014, 17, 106–113. [Google Scholar] [CrossRef]
- Guo, L.; Lim, K.B.; Poduje, C.M.; Daniel, M.; Gunn, J.S.; Hackett, M.; Miller, S.I. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998, 95, 189–198. [Google Scholar] [CrossRef]
- Trent, M.S.; Pabich, W.; Raetz, C.R.; Miller, S.I. A PhoP/PhoQ-induced Lipase (PagL) That Catalyzes 3-O-Deacylation of Lipid A Precursors in Membranes of Salmonella typhimurium. J. Biol. Chem. 2001, 276, 9083–9092. [Google Scholar] [CrossRef] [PubMed]
- Groisman, E.A.; Kayser, J.; Soncini, F.C. Regulation of polymyxin resistance and adaptation to low-Mg2+ environments. J. Bacteriol. 1997, 179, 7040–7045. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, E.L.; Kwasnicka, A.; Hancock, R.E. Role of Pseudomonas aeruginosa PhoP-PhoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 2000, 146, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, S.K.; Chowdhury, I.; Singh, R. Understanding the mechanism of bacterial biofilms resistance to antimicrobial agents. Open Microbiol. J. 2017, 11, 53–62. [Google Scholar] [CrossRef]
- Naves, P.; Del Prado, G.; Huelves, L.; Rodriguez-Cerrato, V.; Ruiz, V.; Ponte, M.C.; Soriano, F. Effects of human serum albumin, ibuprofen and N-acetyl-l-cysteine against biofilm formation by pathogenic Escherichia coli strains. J. Hosp. Infect. 2010, 76, 165–170. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Source | Reference |
|---|---|---|---|
| Protegrin 1 | RGGRLCYCRRRFCVCVGR | leukocytes; Pig, Sus scrofa | [82] |
| Pleurocidin | GWGSFFKKAAHVGKHVGKAALTHYL | skin mucous secretions, Winter flounder, Pleuronectes americanus | [83] |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | neutrophils, monocytes; mast cells; lymphocytes, Mesenchymal Stem Cells; islets; skin, sweat; airway surface liquid, saliva; Homo sapiens; Also Pan troglodytes | [84] |
| Indolicidin | ILPWKWPWWPWRR | bovine neutrophils, Bos taurus | [85] |
| SMAP-29 | RGLRRLGRKIAHGVKKYGPTVLRIIRIAG | sheep leukocytes; Ovis aries | [86] |
| Human β defensin 3 | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK | skin, tonsils, oral/saliva, Homo sapiens | [87] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Somma, A.; Moretta, A.; Canè, C.; Cirillo, A.; Duilio, A. Antimicrobial and Antibiofilm Peptides. Biomolecules 2020, 10, 652. https://doi.org/10.3390/biom10040652
Di Somma A, Moretta A, Canè C, Cirillo A, Duilio A. Antimicrobial and Antibiofilm Peptides. Biomolecules. 2020; 10(4):652. https://doi.org/10.3390/biom10040652
Chicago/Turabian StyleDi Somma, Angela, Antonio Moretta, Carolina Canè, Arianna Cirillo, and Angela Duilio. 2020. "Antimicrobial and Antibiofilm Peptides" Biomolecules 10, no. 4: 652. https://doi.org/10.3390/biom10040652
APA StyleDi Somma, A., Moretta, A., Canè, C., Cirillo, A., & Duilio, A. (2020). Antimicrobial and Antibiofilm Peptides. Biomolecules, 10(4), 652. https://doi.org/10.3390/biom10040652