A High-Throughput Single-Clone Phage Fluorescence Microwell Immunoassay and Laser-Driven Clonal Retrieval System

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Microwell Array Chip Fabrication
2.2. Library Preparation
2.3. Phage Display Preparation
2.4. On-Chip Phage Display Experiment
2.5. Image Acquisition
2.6. Image Analysis
2.7. Target Sample Retrieval and Sequencing
2.8. Clone Identification and Validation
3. Results and Discussion
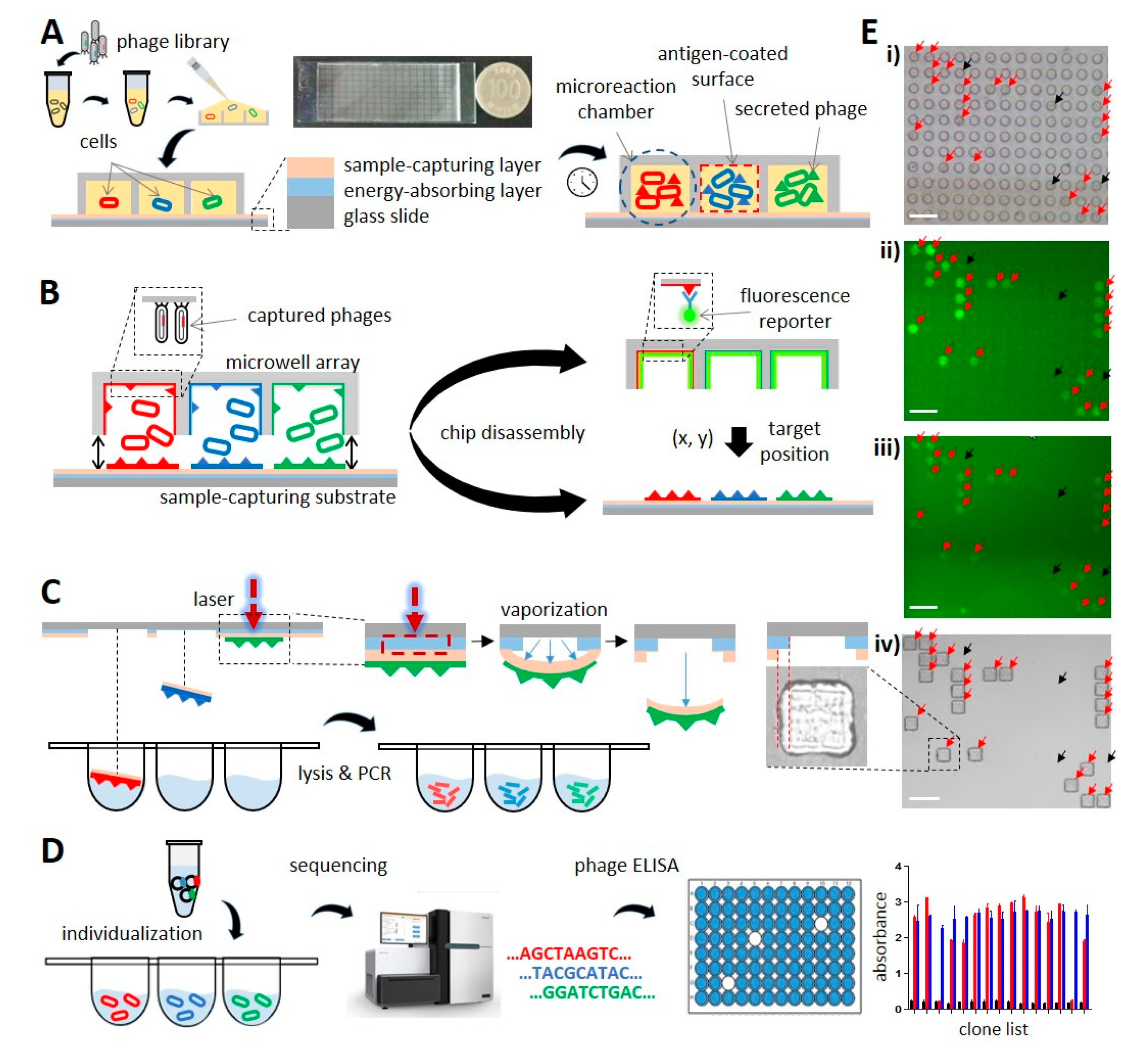
3.1. Cloning, Phage Fluorescence Microwell Immunoassay, and Laser-Driven Retrieval of Phage Clones
3.2. Designing the Microwell Array Chip and Establishment of the Cloning Procedure
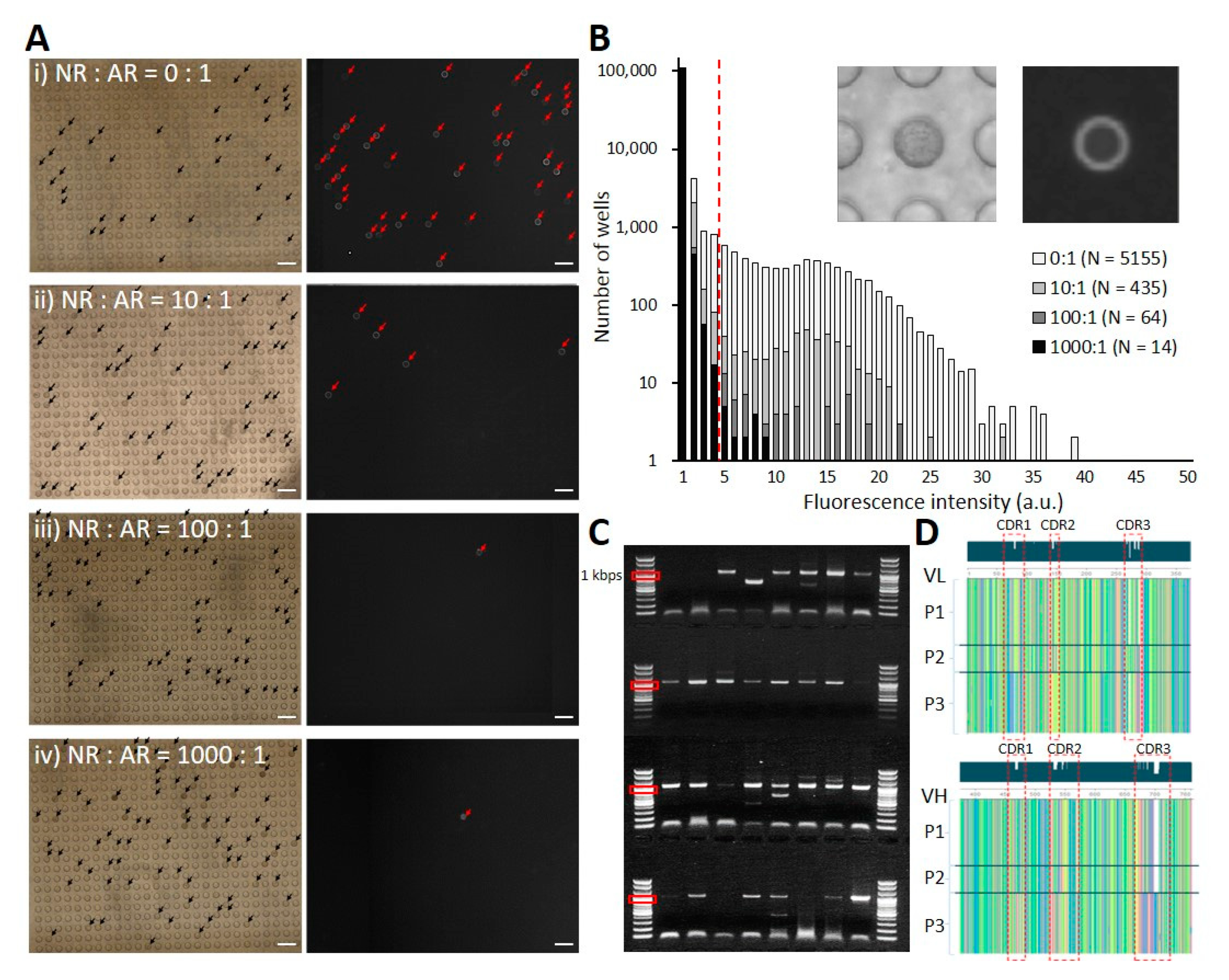
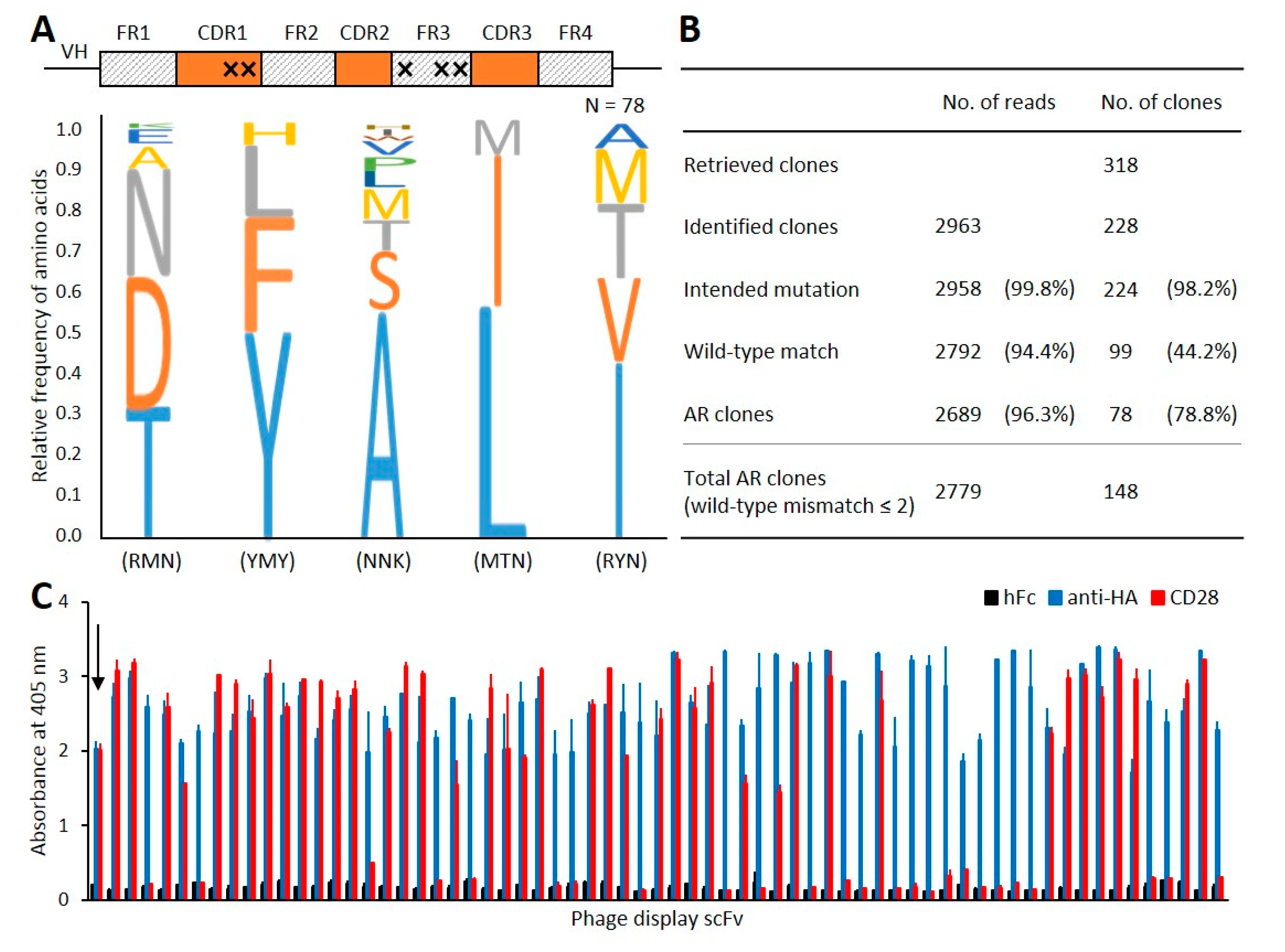
3.3. Validation Using Phage-Displayed scFv Clones
3.4. Application to the Phage-Displayed scFv Library
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoogenboom, H.R.; de Bruïne, A.P.; Hufton, S.E.; Hoet, R.M.; Arends, J.W.; Roovers, R.C. Antibody phage display technology and its applications. Immunotechnology 1998, 4, 1–20. [Google Scholar] [CrossRef]
- Lee, C.M.Y.; Iorno, N.; Sierro, F.; Christ, D. Selection of human antibody fragments by phage display. Nat. Protoc. 2007, 2, 3001–3008. [Google Scholar] [CrossRef]
- Bazan, J.; Całkosiñski, I.; Gamian, A. Phage display—A powerful technique for immunotherapy: 1. Introduction and potential of therapeutic applications. Hum. Vaccines Immunother. 2012, 8, 1817–1828. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage Display: An Overview in Context to Drug Discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef]
- Almagro, J.C.; Pedraza-Escalona, M.; Arrieta, H.I.; Pérez-Tapia, S.M. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Phage Display Libraries: From Binders to Targeted Drug Delivery and Human Therapeutics. Mol. Biotechnol. 2019, 61, 286–303. [Google Scholar] [CrossRef] [PubMed]
- Carmen, S.; Jermutus, L. Concepts in antibody phage display. Brief. Funct. Genom. Proteom. 2002, 1, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Frei, J.C.; Lai, J.R. Protein and Antibody Engineering by Phage Display. Methods Enzymol. 2016, 580, 45–87. [Google Scholar] [PubMed]
- Ceglarek, I.; Piotrowicz, A.; Lecion, D.; Miernikiewicz, P.; Owczarek, B.; Hodyra, K.; Harhala, M.; Górski, A.; Dabrowska, K. A novel approach for separating bacteriophages from other bacteriophages using affinity chromatography and phage display. Sci. Rep. 2013, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Kiss, M.M.; Batonick, M.; Weiner, M.P.; Kay, B.K. Generating Recombinant Antibodies to Membrane Proteins through Phage Display. Antibodies 2016, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Tian, T.; Liu, W.; Zhu, Z.J.; Yang, C.J. Advance in phage display technology for bioanalysis. Biotechnol. J. 2016, 11, 732–745. [Google Scholar] [CrossRef] [PubMed]
- ’t Hoen, P.A.C.; Jirka, S.M.G.; ten Broeke, B.R.; Schultes, E.A.; Aguilera, B.; Pang, K.H.; Heemskerk, H.; Aartsma-Rus, A.; van Ommen, G.J.; den Dunnen, J.T. Phage display screening without repetitious selection rounds. Anal. Biochem. 2012, 421, 622–631. [Google Scholar]
- Turunen, L.; Takkinen, K.; Söderlund, H.; Pulli, T. Automated panning and screening procedure on microplates for antibody generation from phage display libraries. J. Biomol. Screen. 2009, 14, 282–293. [Google Scholar] [CrossRef]
- Krebs, B.; Rauchenberger, R.; Reiffert, S.; Rothe, C.; Tesar, M.; Thomassen, E.; Cao, M.; Dreier, T.; Fischer, D.; Höß, A.; et al. High-throughput generation and engineering of recombinant human antibodies. J. Immunol. Methods 2001, 254, 67–84. [Google Scholar] [CrossRef]
- Schofield, D.J.; Pope, A.R.; Clementel, V.; Buckell, J.; Chapple, S.D.J.; Clarke, K.F.; Conquer, J.S.; Crofts, A.M.; Crowther, S.R.E.; Dyson, M.R.; et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007, 8, 1–18. [Google Scholar] [CrossRef]
- Derda, R.; Tang, S.K.Y.; Li, S.C.; Ng, S.; Matochko, W.; Jafari, M.R. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules 2011, 16, 1776–1803. [Google Scholar] [CrossRef]
- Matochko, W.L.; Li, S.C.; Tang, S.K.Y.; Derda, R. Prospective identification of parasitic sequences in phage display screens. Nucleic Acids Res. 2014, 42, 1784–1798. [Google Scholar] [CrossRef]
- Peltomaa, R.; Benito-Peña, E.; Barderas, R.; Moreno-Bondi, M.C. Phage display in the quest for new selective recognition elements for biosensors. ACS Omega 2019, 4, 11569–11580. [Google Scholar] [CrossRef]
- Pearce, T.M.; Williams, J.C. Microtechnology: Meet neurobiology. Lab Chip 2007, 7, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Neužil, P.; Giselbrecht, S.; Länge, K.; Huang, T.J.; Manz, A. Revisiting lab-on-a-chip technology for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 620–632. [Google Scholar]
- Wagner, V.; Dullaart, A.; Bock, A.K.; Zweck, A. The emerging nanomedicine landscape. Nat. Biotechnol. 2006, 24, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, V.; Manning, B.; O’Donnell, B.; O’Reilly, B.; O’Sullivan, D.; O’Kennedy, R.; Leonard, P. Exploiting highly ordered subnanoliter volume microcapillaries as microtools for the analysis of antibody producing cells. Anal. Chem. 2015, 87, 997–1003. [Google Scholar] [CrossRef]
- Kiss, M.M.; Babineau, E.G.; Bonatsakis, M.; Buhr, D.L.; Maksymiuk, G.M.; Wang, D.; Alderman, D.; Gelperin, D.M.; Weiner, M.P. Phage ESCape: An emulsion-based approach for the selection of recombinant phage display antibodies. J. Immunol. Methods 2011, 367, 17–26. [Google Scholar] [CrossRef]
- Buhr, D.L.; Acca, F.E.; Holland, E.G.; Johnson, K.; Maksymiuk, G.M.; Vaill, A.; Kay, B.K.; Weitz, D.A.; Weiner, M.P.; Kiss, M.M. Use of micro-emulsion technology for the directed evolution of antibodies. Methods 2012, 58, 28–33. [Google Scholar] [CrossRef]
- Cossarizza, A.; Chang, H.D.; Radbruch, A.; Akdis, M.; Andrä, I.; Annunziato, F.; Bacher, P.; Barnaba, V.; Battistini, L.; Bauer, W.M.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur. J. Immunol. 2017, 47, 1584–1797. [Google Scholar] [CrossRef]
- Plouffe, B.D.; Murthy, S.K.; Lewis, L.H. Fundamentals and Application of Magnetic Particles in Cell Isolation and Enrichment. Rep. Prog. Phys. 2015, 78, 1–76. [Google Scholar] [CrossRef]
- Kaminski, T.S.; Scheler, O.; Garstecki, P. Droplet microfluidics for microbiology: Techniques, applications and challenges. Lab Chip 2016, 16, 2168–2187. [Google Scholar] [CrossRef]
- Xia, Y.; Whitesides, G.M. Soft lithography. Annu. Rev. Mater. Sci. 1998, 28, 153–184. [Google Scholar] [CrossRef]
- Weibel, D.B.; Diluzio, W.R.; Whitesides, G.M. Microfabrication meets microbiology. Nat. Rev. Microbiol. 2007, 5, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.; Michel, B. Siloxane polymers for high-resolution, high-accuracy soft lithography. Macromolecules 2000, 33, 3042–3049. [Google Scholar] [CrossRef]
- Odom, T.W.; Love, J.C.; Wolfe, D.B.; Paul, K.E.; Whitesides, G.M. Improved pattern transfer in soft lithography using composite stamps. Langmuir 2002, 18, 5314–5320. [Google Scholar] [CrossRef]
- Choi, K.M.; Rogers, J.A. A photocurable poly(dimethylsiloxane) chemistry designed for soft lithographic molding and printing in the nanometer regime. J. Am. Chem. Soc. 2003, 125, 4060–4061. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.; Noh, J.; Lee, Y.; Han, H.; Yoo, D.K.; Kim, H.; Kwon, S.; Chung, J. Efficient selection of antibodies reactive to homologous epitopes on human and mouse hepatocyte growth factors by next-generation sequencing-based analysis of the B cell repertoire. Int. J. Mol. Sci. 2019, 20, 417. [Google Scholar] [CrossRef]
- Kim, S., II; Kim, S.; Kim, J.; Chang, S.Y.; Shim, J.M.; Jin, J.; Lim, C.; Baek, S.; Min, J.Y.; Park, W.B.; et al. Generation of a nebulizable CDR-modified MERS-CoV neutralizing human antibody. Int. J. Mol. Sci. 2019, 20, 5073. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Lee, J.C.; Choi, C.Y.; Chung, J. Production and characterization of monoclonal antibody to botulinum neurotoxin type B light chain by phage display. Hybridoma 2008, 27, 18–24. [Google Scholar] [CrossRef]
- Preibisch, S.; Saalfeld, S.; Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009, 25, 1463–1465. [Google Scholar] [CrossRef]
- Kim, S.; Kim, S.; Kim, J.; Kim, B.; Kim, S.I.; Kim, M.A.; Kwon, S.; Song, Y.S. Evaluating Tumor Evolution via Genomic Profiling of Individual Tumor Spheroids in a Malignant Ascites. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Kim, S.; Lee, A.C.; Lee, H.B.; Kim, J.; Jung, Y.; Ryu, H.S.; Lee, Y.; Bae, S.; Lee, M.; Lee, K.; et al. PHLI-seq: Constructing and visualizing cancer genomic maps in 3D by phenotype-based high-throughput laser-aided isolation and sequencing. Genome Biol. 2018, 19, 1–17. [Google Scholar] [CrossRef]
- Noh, J.; Kim, O.; Jung, Y.; Han, H.; Kim, J.E.; Kim, S.; Lee, S.; Park, J.; Jung, R.H.; Kim, S., II; et al. High-throughput retrieval of physical DNA for NGS-identifiable clones in phage display library. MAbs 2019, 11, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.; Jo, D.H.; Kim, J.H.; Kim, S.R.; Kang, D.; Hwang, D.; Chung, J. Bispecific anti-mPDGFRβ x cotinine scFv-Cκ-scFv fusion protein and cotinine-duocarmycin can form antibody-drug conjugate-like complexes that exert cytotoxicity against mPDGFRβ expressing cells. Methods 2019, 154, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Salvat, R.S.; Verma, D.; Parker, A.S.; Kirsch, J.R.; Brooks, S.A.; Bailey-Kellogg, C.; Griswold, K.E. Computationally optimized deimmunization libraries yield highly mutated enzymes with low immunogenicity and enhanced activity. Proc. Natl. Acad. Sci. USA 2017, 114, 5085–5093. [Google Scholar] [CrossRef] [PubMed]
- Therapeutic Antibodies Methods and Protocols, 1st ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 405–423.
- De Groot, A.S.; Goldberg, M.; Moise, L.; Martin, W. Evolutionary deimmunization: An ancillary mechanism for self-tolerance? Cell. Immunol. 2006, 244, 148–153. [Google Scholar] [CrossRef]
- Baker, M.P.; Reynolds, H.M.; Lumicisi, B.; Bryson, C.J. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self Nonself 2010, 1, 314–322. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Todhunter, D.; Oh, S.; Vallera, D.A. Mutagenic deimmunization of diphtheria toxin for use in biologic drug development. Toxins 2015, 7, 4067–4082. [Google Scholar] [CrossRef]
- Griswold, K.E.; Bailey-Kellogg, C. Design and engineering of deimmunized biotherapeutics. Curr. Opin. Struct. Biol. 2016, 39, 79–88. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, S.; Kim, S.; Han, J.; Ha, S.; Lee, H.; Song, S.W.; Lee, D.; Kwon, S.; Chung, J.; Kim, J. A High-Throughput Single-Clone Phage Fluorescence Microwell Immunoassay and Laser-Driven Clonal Retrieval System. Biomolecules 2020, 10, 517. https://doi.org/10.3390/biom10040517
Chang S, Kim S, Han J, Ha S, Lee H, Song SW, Lee D, Kwon S, Chung J, Kim J. A High-Throughput Single-Clone Phage Fluorescence Microwell Immunoassay and Laser-Driven Clonal Retrieval System. Biomolecules. 2020; 10(4):517. https://doi.org/10.3390/biom10040517
Chicago/Turabian StyleChang, Seohee, Soohyun Kim, Jerome Han, Suji Ha, Hyunho Lee, Seo Woo Song, Daewon Lee, Sunghoon Kwon, Junho Chung, and Junhoi Kim. 2020. "A High-Throughput Single-Clone Phage Fluorescence Microwell Immunoassay and Laser-Driven Clonal Retrieval System" Biomolecules 10, no. 4: 517. https://doi.org/10.3390/biom10040517
APA StyleChang, S., Kim, S., Han, J., Ha, S., Lee, H., Song, S. W., Lee, D., Kwon, S., Chung, J., & Kim, J. (2020). A High-Throughput Single-Clone Phage Fluorescence Microwell Immunoassay and Laser-Driven Clonal Retrieval System. Biomolecules, 10(4), 517. https://doi.org/10.3390/biom10040517