The Role of Torsin AAA+ Proteins in Preserving Nuclear Envelope Integrity and Safeguarding Against Disease


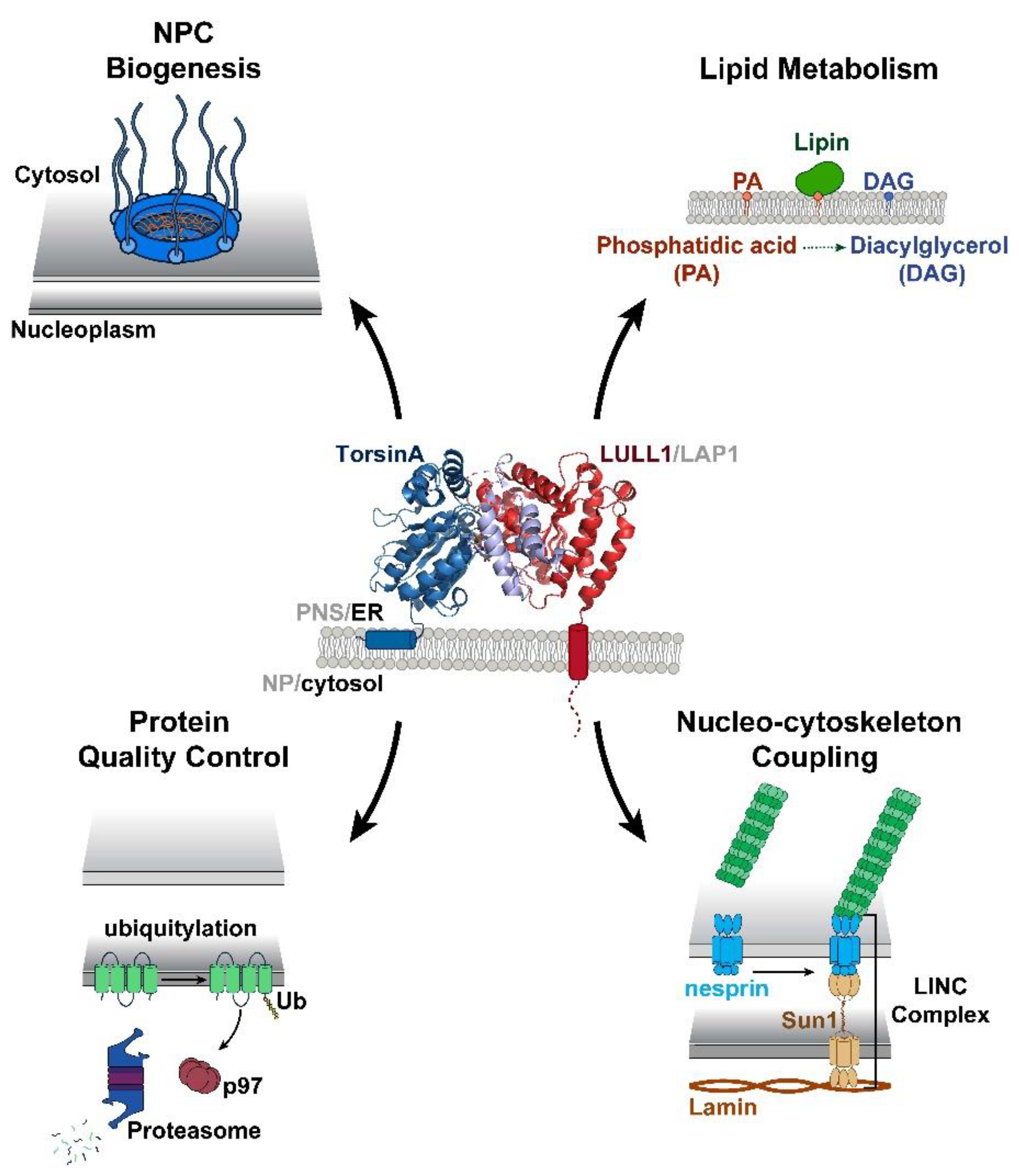{kind=link}
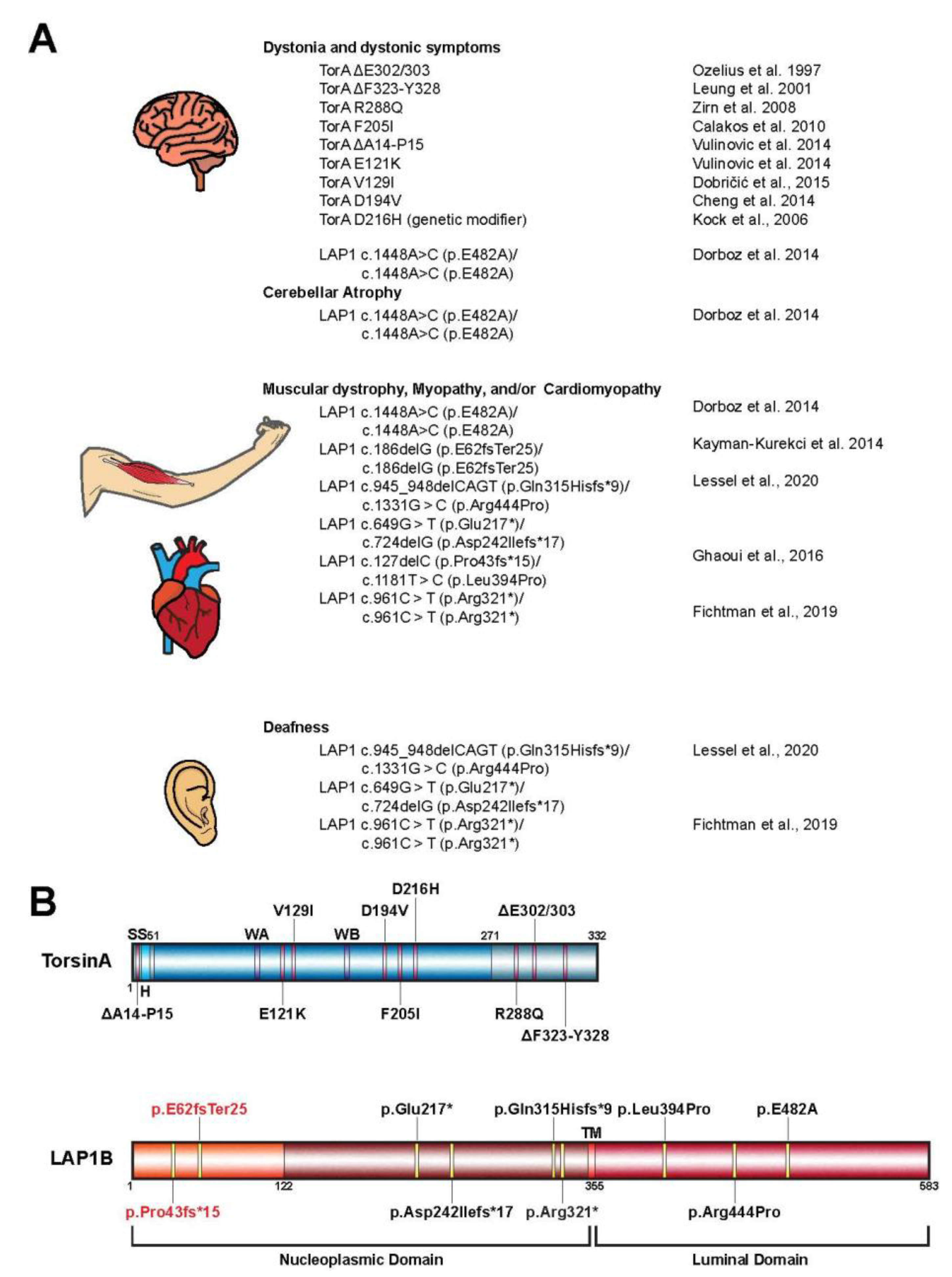{kind=link}
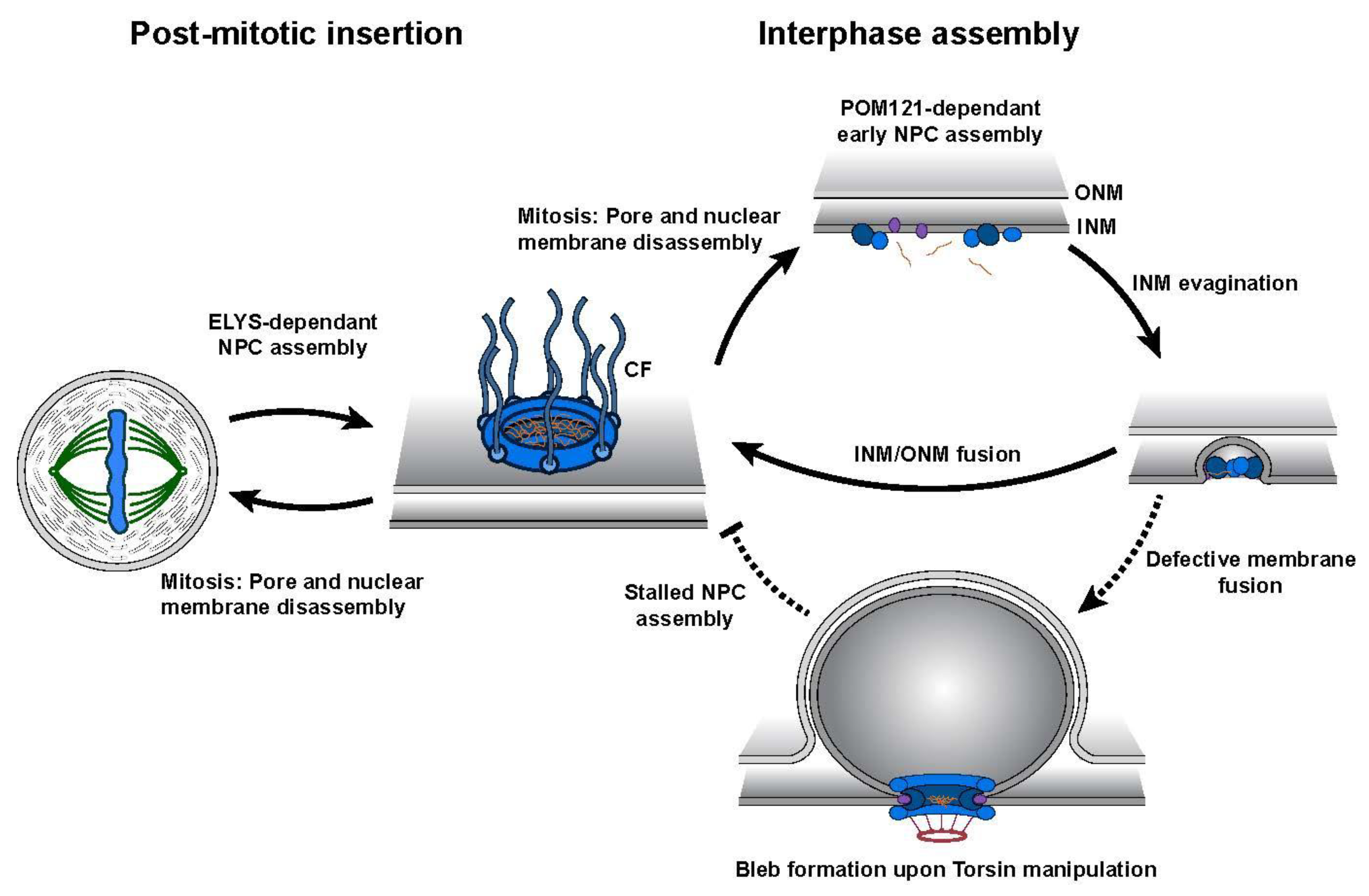{kind=link}
Abstract
1. Introduction
2. Structural and Biochemical Perspectives on Torsins
3. Torsin Assemblies and Dystonia Movement Disorders
4. Emerging Intersections between Torsins and Lipid Metabolism
5. The Role of Torsin ATPases in Nuclear Pore Biogenesis
6. Torsins ATPases Contribute to NPC Assembly during Interphase
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ozelius, L.J.; Hewett, J.W.; Page, C.E.; Bressman, S.B.; Kramer, P.L.; Shalish, C.; de Leon, D.; Brin, M.F.; Raymond, D.; Corey, D.P.; et al. The early-onset torsion dystonia gene (dyt1) encodes an atp-binding protein. Nat. Genet. 1997, 17, 40–48. [Google Scholar] [CrossRef]
- Ozelius, L.J.; Page, C.E.; Klein, C.; Hewett, J.W.; Mineta, M.; Leung, J.; Shalish, C.; Bressman, S.B.; de Leon, D.; Brin, M.F.; et al. The tor1a (dyt1) gene family and its role in early onset torsion dystonia. Genomics 1999, 62, 377–384. [Google Scholar] [CrossRef]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. Aaa+: A class of chaperone-like atpases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar]
- Rose, A.E.; Brown, R.S.; Schlieker, C. Torsins: Not your typical aaa+ atpases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 532–549. [Google Scholar] [CrossRef]
- Goodchild, R.E.; Dauer, W.T. The aaa+ protein torsina interacts with a conserved domain present in lap1 and a novel er protein. J. Cell Biol. 2005, 168, 855–862. [Google Scholar] [CrossRef]
- Goodchild, R.E.; Dauer, W.T. Mislocalization to the nuclear envelope: An effect of the dystonia-causing torsina mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wrabl, J.O.; Hayashi, A.P.; Rose, L.S.; Thomas, P.J. The torsin-family aaa+ protein ooc-5 contains a critical disulfide adjacent to sensor-ii that couples redox state to nucleotide binding. Mol. Biol. Cell 2008, 19, 3599–3612. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Brown, R.S.; Chase, A.R.; Eisele, M.R.; Schlieker, C. Regulation of torsin atpases by lap1 and lull1. Proc. Natl. Acad. Sci. USA 2013, 110, E1545–E1554. [Google Scholar] [CrossRef] [PubMed]
- Naismith, T.V.; Dalal, S.; Hanson, P.I. Interaction of torsina with its major binding partners is impaired by the dystonia-associated deltagag deletion. J. Biol. Chem. 2009, 284, 27866–27874. [Google Scholar] [CrossRef]
- Brown, R.S.; Zhao, C.; Chase, A.R.; Wang, J.; Schlieker, C. The mechanism of torsin atpase activation. Proc. Natl. Acad. Sci. USA 2014, 111, E4822–E4831. [Google Scholar] [CrossRef]
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.; Schwartz, T.U. How lamina-associated polypeptide 1 (lap1) activates torsin. Elife 2014, 3, e03239. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.E.; Sosa, B.A.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. Structures of torsina and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia. Elife 2016, 5, e17983. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Millen, L.; Mendoza, J.L.; Thomas, P.J. A unique redox-sensing sensor ii motif in torsina plays a critical role in nucleotide and partner binding. J. Biol. Chem. 2010, 285, 37271–37280. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.E.; Zheng, W.; McQuown, A.J.; Maier, N.K.; Watson, N.; Cheeseman, I.M.; Denic, V.; Egelman, E.H.; Schwartz, T.U. The aaa + atpase torsina polymerizes into hollow helical tubes with 8.5 subunits per turn. Nat. Commun. 2019, 10, 3262. [Google Scholar] [CrossRef] [PubMed]
- Fichtman, B.; Zagairy, F.; Biran, N.; Barsheshet, Y.; Chervinsky, E.; Ben Neriah, Z.; Shaag, A.; Assa, M.; Elpeleg, O.; Harel, A.; et al. Combined loss of lap1b and lap1c results in an early onset multisystemic nuclear envelopathy. Nat. Commun. 2019, 10, 605. [Google Scholar] [CrossRef] [PubMed]
- Dorboz, I.; Coutelier, M.; Bertrand, A.T.; Caberg, J.H.; Elmaleh-Berges, M.; Laine, J.; Stevanin, G.; Bonne, G.; Boespflug-Tanguy, O.; Servais, L. Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in tor1aip1. Orphanet J. Rare Dis. 2014, 9, 174. [Google Scholar] [CrossRef]
- Kayman-Kurekci, G.; Talim, B.; Korkusuz, P.; Sayar, N.; Sarioglu, T.; Oncel, I.; Sharafi, P.; Gundesli, H.; Balci-Hayta, B.; Purali, N.; et al. Mutation in tor1aip1 encoding lap1b in a form of muscular dystrophy: A novel gene related to nuclear envelopathies. Neuromuscul. Disord. NMD 2014, 24, 624–633. [Google Scholar] [CrossRef]
- Jungwirth, M.; Dear, M.L.; Brown, P.; Holbrook, K.; Goodchild, R. Relative tissue expression of homologous torsinb correlates with the neuronal specific importance of dyt1 dystonia-associated torsina. Hum. Mol. Genet. 2010, 19, 888–900. [Google Scholar] [CrossRef]
- Kim, C.E.; Perez, A.; Perkins, G.; Ellisman, M.H.; Dauer, W.T. A molecular mechanism underlying the neural-specific defect in torsina mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9861–9866. [Google Scholar] [CrossRef]
- Li, J.; Liang, C.-C.; Pappas, S.S.; Dauer, W.T. Torsinb overexpression prevents abnormal twisting in dyt1 dystonia mouse models. bioRxiv 2019, 836536. [Google Scholar] [CrossRef]
- Grillet, M.; Dominguez Gonzalez, B.; Sicart, A.; Pottler, M.; Cascalho, A.; Billion, K.; Hernandez Diaz, S.; Swerts, J.; Naismith, T.V.; Gounko, N.V.; et al. Torsins are essential regulators of cellular lipid metabolism. Dev. Cell 2016, 38, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Cascalho, A.; Foroozandeh, J.; Hennebel, L.; Klein, C.; Rous, S.; Gonzalez, B.D.; Pisani, A.; Meringolo, M.; Gallego, S.F.; Verstreken, P.; et al. Inhibition of lipin lipid phosphatase hyperactivity rescues torsina neurological disease. bioRxiv 2019, 606947. [Google Scholar] [CrossRef]
- Shin, J.Y.; Hernandez-Ono, A.; Fedotova, T.; Ostlund, C.; Lee, M.J.; Gibeley, S.B.; Liang, C.C.; Dauer, W.T.; Ginsberg, H.N.; Worman, H.J. Nuclear envelope-localized torsina-lap1 complex regulates hepatic vldl secretion and steatosis. J. Clin. Investig. 2019, 130, 4885–4900. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.A.; Harris, N.J.; Willey, P.T.; Woolums, B.M.; Wang, Y.; McQuown, A.J.; Schoenhofen, A.; Worman, H.J.; Dauer, W.T.; Gundersen, G.G.; et al. Torsina controls tan line assembly and the retrograde flow of dorsal perinuclear actin cables during rearward nuclear movement. J. Cell Biol. 2017, 216, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Dominguez Gonzalez, B.; Billion, K.; Rous, S.; Pavie, B.; Lange, C.; Goodchild, R. Excess linc complexes impair brain morphogenesis in a mouse model of recessive tor1a disease. Hum. Mol. Genet. 2018, 27, 2154–2170. [Google Scholar] [CrossRef] [PubMed]
- Gill, N.K.; Ly, C.; Kim, P.H.; Saunders, C.A.; Fong, L.G.; Young, S.G.; Luxton, G.W.G.; Rowat, A.C. Dyt1 dystonia patient-derived fibroblasts have increased deformability and susceptibility to damage by mechanical forces. Front. Cell Dev. Biol. 2019, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Jokhi, V.; Ashley, J.; Nunnari, J.; Noma, A.; Ito, N.; Wakabayashi-Ito, N.; Moore, M.J.; Budnik, V. Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep. 2013, 3, 988–995. [Google Scholar] [CrossRef]
- Rose, A.E.; Zhao, C.; Turner, E.M.; Steyer, A.M.; Schlieker, C. Arresting a torsin atpase reshapes the endoplasmic reticulum. J. Biol. Chem. 2014, 289, 552–564. [Google Scholar] [CrossRef]
- Zhao, C.; Brown, R.S.; Tang, C.H.; Hu, C.C.; Schlieker, C. Site-specific proteolysis mobilizes torsina from the membrane of the endoplasmic reticulum (er) in response to er stress and b cell stimulation. J. Biol. Chem. 2016, 291, 9469–9481. [Google Scholar] [CrossRef]
- Laudermilch, E.; Tsai, P.L.; Graham, M.; Turner, E.; Zhao, C.; Schlieker, C. Dissecting torsin/cofactor function at the nuclear envelope: A genetic study. Mol. Biol. Cell 2016, 27, 3964–3971. [Google Scholar] [CrossRef]
- Pappas, S.S.; Liang, C.C.; Kim, S.; Rivera, C.O.; Dauer, W.T. Torsina dysfunction causes persistent neuronal nuclear pore defects. Hum. Mol. Genet. 2018, 27, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Rampello, A.J.; Laudermilch, E.; Vishnoi, N.; Prohet, S.M.; Shao, L.; Zhao, C.; Lusk, C.P.; Schlieker, C. Torsin atpases are required to complete nuclear pore complex biogenesis in interphase. bioRxiv 2019, 821835. [Google Scholar] [CrossRef]
- Chen, P.; Burdette, A.J.; Porter, J.C.; Ricketts, J.C.; Fox, S.A.; Nery, F.C.; Hewett, J.W.; Berkowitz, L.A.; Breakefield, X.O.; Caldwell, K.A.; et al. The early-onset torsion dystonia-associated protein, torsina, is a homeostatic regulator of endoplasmic reticulum stress response. Hum. Mol. Genet. 2010, 19, 3502–3515. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.C.; Armata, I.A.; Farley, J.E.; Cho, J.A.; Yaqub, U.; Chen, P.; da Hora, C.C.; Wang, Q.; Tagaya, M.; Klein, C.; et al. Torsina participates in endoplasmic reticulum-associated degradation. Nat. Commun. 2011, 2, 393. [Google Scholar] [CrossRef] [PubMed]
- McLean, P.J.; Kawamata, H.; Shariff, S.; Hewett, J.; Sharma, N.; Ueda, K.; Breakefield, X.O.; Hyman, B.T. Torsina and heat shock proteins act as molecular chaperones: Suppression of alpha-synuclein aggregation. J. Neurochem. 2002, 83, 846–854. [Google Scholar] [CrossRef]
- Caldwell, G.A.; Cao, S.; Sexton, E.G.; Gelwix, C.C.; Bevel, J.P.; Caldwell, K.A. Suppression of polyglutamine-induced protein aggregation in caenorhabditis elegans by torsin proteins. Hum. Mol. Genet. 2003, 12, 307–319. [Google Scholar] [CrossRef]
- Burdette, A.J.; Churchill, P.F.; Caldwell, G.A.; Caldwell, K.A. The early-onset torsion dystonia-associated protein, torsina, displays molecular chaperone activity in vitro. Cell Stress Chaperones 2010, 15, 605–617. [Google Scholar] [CrossRef]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of aaa+ atpases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef]
- Hanson, P.I.; Whiteheart, S.W. Aaa+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef]
- Nagy, M.; Wu, H.C.; Liu, Z.; Kedzierska-Mieszkowska, S.; Zolkiewski, M. Walker-a threonine couples nucleotide occupancy with the chaperone activity of the aaa+ atpase clpb. Protein Sci. 2009, 18, 287–293. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Wittinghofer, A. Gtpase-activating proteins: Helping hands to complement an active site. Trends Biochem. Sci. 1998, 23, 257–262. [Google Scholar] [CrossRef]
- Schlieker, C.; Weibezahn, J.; Patzelt, H.; Tessarz, P.; Strub, C.; Zeth, K.; Erbse, A.; Schneider-Mergener, J.; Chin, J.W.; Schultz, P.G.; et al. Substrate recognition by the aaa+ chaperone clpb. Nat. Struct. Mol. Biol. 2004, 11, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.M.; Sauer, R.T.; Baker, T.A. Role of the processing pore of the clpx aaa+ atpase in the recognition and engagement of specific protein substrates. Genes Dev. 2004, 18, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Baker, T.A. Aaa+ proteases: Atp-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, R.E.; Kim, C.E.; Dauer, W.T. Loss of the dystonia-associated protein torsina selectively disrupts the neuronal nuclear envelope. Neuron 2005, 48, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Tanabe, L.M.; Jou, S.; Chi, F.; Dauer, W.T. Torsina hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J. Clin. Investig. 2014, 124, 3080–3092. [Google Scholar] [CrossRef] [PubMed]
- Pappas, S.S.; Darr, K.; Holley, S.M.; Cepeda, C.; Mabrouk, O.S.; Wong, J.M.; LeWitt, T.M.; Paudel, R.; Houlden, H.; Kennedy, R.T.; et al. Forebrain deletion of the dystonia protein torsina causes dystonic-like movements and loss of striatal cholinergic neurons. Elife 2015, 4, e08352. [Google Scholar] [CrossRef]
- DeSimone, J.C.; Pappas, S.S.; Febo, M.; Burciu, R.G.; Shukla, P.; Colon-Perez, L.M.; Dauer, W.T.; Vaillancourt, D.E. Forebrain knock-out of torsina reduces striatal free-water and impairs whole-brain functional connectivity in a symptomatic mouse model of dyt1 dystonia. Neurobiol. Dis. 2017, 106, 124–132. [Google Scholar] [CrossRef]
- Hewett, J.W.; Kamm, C.; Boston, H.; Beauchamp, R.; Naismith, T.; Ozelius, L.; Hanson, P.I.; Breakefield, X.O.; Ramesh, V. Torsinb--perinuclear location and association with torsina. J. Neurochem. 2004, 89, 1186–1194. [Google Scholar] [CrossRef]
- VanGompel, M.J.; Nguyen, K.C.; Hall, D.H.; Dauer, W.T.; Rose, L.S. A novel function for the caenorhabditis elegans torsin ooc-5 in nucleoporin localization and nuclear import. Mol. Biol. Cell 2015, 26, 1752–1763. [Google Scholar] [CrossRef]
- Breakefield, X.O.; Kamm, C.; Hanson, P.I. Torsina: Movement at many levels. Neuron 2001, 31, 9–12. [Google Scholar] [CrossRef]
- Fremont, R.; Tewari, A.; Angueyra, C.; Khodakhah, K. A role for cerebellum in the hereditary dystonia dyt1. Elife 2017, 6, e22775. [Google Scholar] [CrossRef] [PubMed]
- Vander Heyden, A.B.; Naismith, T.V.; Snapp, E.L.; Hodzic, D.; Hanson, P.I. Lull1 retargets torsina to the nuclear envelope revealing an activity that is impaired by the dyt1 dystonia mutation. Mol. Biol. Cell 2009, 20, 2661–2672. [Google Scholar] [CrossRef]
- Chase, A.R.; Laudermilch, E.; Wang, J.; Shigematsu, H.; Yokoyama, T.; Schlieker, C. Dynamic functional assembly of the torsin aaa+ atpase and its modulation by lap1. Mol. Biol. Cell 2017, 28, 2765–2772. [Google Scholar] [CrossRef] [PubMed]
- Kariminejad, A.; Dahl-Halvarsson, M.; Ravenscroft, G.; Afroozan, F.; Keshavarz, E.; Goullee, H.; Davis, M.R.; Faraji Zonooz, M.; Najmabadi, H.; Laing, N.G.; et al. Tor1a variants cause a severe arthrogryposis with developmental delay, strabismus and tremor. Brain J. Neurol. 2017, 140, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Reichert, S.C.; Gonzalez-Alegre, P.; Scharer, G.H. Biallelic tor1a variants in an infant with severe arthrogryposis. Neurol. Genet. 2017, 3, e154. [Google Scholar] [CrossRef] [PubMed]
- Isik, E.; Aykut, A.; Atik, T.; Cogulu, O.; Ozkinay, F. Biallelic tor1a mutations cause severe arthrogryposis: A case requiring reverse phenotyping. Eur. J. Med. Genet. 2019, 62, 103544. [Google Scholar] [CrossRef]
- Gonzalez-Alegre, P. Advances in molecular and cell biology of dystonia: Focus on torsina. Neurobiol. Dis. 2019, 127, 233–241. [Google Scholar] [CrossRef]
- Calakos, N.; Patel, V.D.; Gottron, M.; Wang, G.; Tran-Viet, K.N.; Brewington, D.; Beyer, J.L.; Steffens, D.C.; Krishnan, R.R.; Zuchner, S. Functional evidence implicating a novel tor1a mutation in idiopathic, late-onset focal dystonia. J. Med. Genet. 2010, 47, 646–650. [Google Scholar] [CrossRef]
- Zirn, B.; Grundmann, K.; Huppke, P.; Puthenparampil, J.; Wolburg, H.; Riess, O.; Muller, U. Novel tor1a mutation p.Arg288gln in early-onset dystonia (dyt1). J. Neurol. Neurosurg. Psychiatry 2008, 79, 1327–1330. [Google Scholar] [CrossRef]
- Leung, J.C.; Klein, C.; Friedman, J.; Vieregge, P.; Jacobs, H.; Doheny, D.; Kamm, C.; DeLeon, D.; Pramstaller, P.P.; Penney, J.B.; et al. Novel mutation in the tor1a (dyt1) gene in atypical early onset dystonia and polymorphisms in dystonia and early onset parkinsonism. Neurogenetics 2001, 3, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.B.; Feng, J.C.; Ma, L.Y.; Miao, J.; Ott, T.; Wan, X.H.; Grundmann, K. Combined occurrence of a novel tor1a and a thap1 mutation in primary dystonia. Mov. Disord. 2014, 29, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Vulinovic, F.; Lohmann, K.; Rakovic, A.; Capetian, P.; Alvarez-Fischer, D.; Schmidt, A.; Weissbach, A.; Erogullari, A.; Kaiser, F.J.; Wiegers, K.; et al. Unraveling cellular phenotypes of novel torsina/tor1a mutations. Hum. Mutat. 2014, 35, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Kock, N.; Naismith, T.V.; Boston, H.E.; Ozelius, L.J.; Corey, D.P.; Breakefield, X.O.; Hanson, P.I. Effects of genetic variations in the dystonia protein torsina: Identification of polymorphism at residue 216 as protein modifier. Hum. Mol. Genet. 2006, 15, 1355–1364. [Google Scholar] [CrossRef]
- Dobricic, V.; Kresojevic, N.; Zarkovic, M.; Tomic, A.; Marjanovic, A.; Westenberger, A.; Cvetkovic, D.; Svetel, M.; Novakovic, I.; Kostic, V.S. Phenotype of non-c.907_909delgag mutations in tor1a: Dyt1 dystonia revisited. Parkinsonism Relat. Disord. 2015, 21, 1256–1259. [Google Scholar] [CrossRef]
- Laudermilch, E.; Schlieker, C. Torsin atpases: Structural insights and functional perspectives. Curr. Opin. Cell Biol. 2016, 40, 1–7. [Google Scholar] [CrossRef]
- Lessel, I.; Chen, M.J.; Luttgen, S.; Arndt, F.; Fuchs, S.; Meien, S.; Thiele, H.; Jones, J.R.; Shaw, B.R.; Crossman, D.K.; et al. Two novel cases further expand the phenotype of tor1aip1-associated nuclear envelopathies. Hum. Genet. 2020, 139, 483–498. [Google Scholar] [CrossRef]
- Ghaoui, R.; Benavides, T.; Lek, M.; Waddell, L.B.; Kaur, S.; North, K.N.; MacArthur, D.G.; Clarke, N.F.; Cooper, S.T. Tor1aip1 as a cause of cardiac failure and recessive limb-girdle muscular dystrophy. Neuromuscul. Disord. NMD 2016, 26, 500–503. [Google Scholar] [CrossRef]
- Risch, N.J.; Bressman, S.B.; Senthil, G.; Ozelius, L.J. Intragenic cis and trans modification of genetic susceptibility in dyt1 torsion dystonia. Am. J. Hum. Genet. 2007, 80, 1188–1193. [Google Scholar] [CrossRef]
- Martino, D.; Gajos, A.; Gallo, V.; Cif, L.; Coubes, P.; Tinazzi, M.; Schneider, S.A.; Fiorio, M.; Zorzi, G.; Nardocci, N.; et al. Extragenetic factors and clinical penetrance of dyt1 dystonia: An exploratory study. J. Neurol. 2013, 260, 1081–1086. [Google Scholar] [CrossRef]
- Santos, M.; Domingues, S.C.; Costa, P.; Muller, T.; Galozzi, S.; Marcus, K.; da Cruz e Silva, E.F.; da Cruz e Silva, O.A.; Rebelo, S. Identification of a novel human lap1 isoform that is regulated by protein phosphorylation. PLoS ONE 2014, 9, e113732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; MacDonald, J.I.; Kent, C. Regulation of ctp:Phosphocholine cytidylyltransferase in hela cells. Effect of oleate on phosphorylation and intracellular localization. J. Biol. Chem. 1993, 268, 5512–5518. [Google Scholar] [PubMed]
- Watkins, J.D.; Kent, C. Immunolocalization of membrane-associated ctp:Phosphocholine cytidylyltransferase in phosphatidylcholine-deficient chinese hamster ovary cells. J. Biol. Chem. 1992, 267, 5686–5692. [Google Scholar] [PubMed]
- Hsieh, L.S.; Su, W.M.; Han, G.S.; Carman, G.M. Phosphorylation regulates the ubiquitin-independent degradation of yeast pah1 phosphatidate phosphatase by the 20s proteasome. J. Biol. Chem. 2015, 290, 11467–11478. [Google Scholar] [CrossRef]
- Peterfy, M.; Harris, T.E.; Fujita, N.; Reue, K. Insulin-stimulated interaction with 14-3-3 promotes cytoplasmic localization of lipin-1 in adipocytes. J. Biol. Chem. 2010, 285, 3857–3864. [Google Scholar] [CrossRef]
- Harris, T.E.; Huffman, T.A.; Chi, A.; Shabanowitz, J.; Hunt, D.F.; Kumar, A.; Lawrence, J.C., Jr. Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J. Biol. Chem. 2007, 282, 277–286. [Google Scholar] [CrossRef]
- Sundler, R.; Arvidson, G.; Akesson, B. Pathways for the incorporation of choline into rat liver phosphatidylcholines in vivo. Biochim. Biophys. Acta 1972, 280, 559–568. [Google Scholar] [CrossRef]
- Van Meer, G. Cellular lipidomics. EMBO J. 2005, 24, 3159–3165. [Google Scholar] [CrossRef]
- Han, G.S.; Wu, W.I.; Carman, G.M. The saccharomyces cerevisiae lipin homolog is a mg2+-dependent phosphatidate phosphatase enzyme. J. Biol. Chem. 2006, 281, 9210–9218. [Google Scholar] [CrossRef]
- Donkor, J.; Sariahmetoglu, M.; Dewald, J.; Brindley, D.N.; Reue, K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J. Biol. Chem. 2007, 282, 3450–3457. [Google Scholar] [CrossRef]
- Shelness, G.S.; Sellers, J.A. Very-low-density lipoprotein assembly and secretion. Curr. Opin. Lipidol. 2001, 12, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Prophet, S.M.; Schlieker, C. An unbiased approach de-livers unexpected insight into torsin biology. J. Clin. Investig. 2019, 129, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.R.; Laudermilch, E.; Schlieker, C. Torsin atpases: Harnessing dynamic instability for function. Front. Mol. Biosci. 2017, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Thaller, D.J.; Lusk, C.P. Fantastic nuclear envelope herniations and where to find them. Biochem. Soc. Trans. 2018, 46, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Hodgson, L.; Mantell, J.; Verkade, P.; Carlton, J.G. Escrt-iii controls nuclear envelope reformation. Nature 2015, 522, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; LaJoie, D.; Chen, O.S.; von Appen, A.; Ladinsky, M.S.; Redd, M.J.; Nikolova, L.; Bjorkman, P.J.; Sundquist, W.I.; Ullman, K.S.; et al. Lem2 recruits chmp7 for escrt-mediated nuclear envelope closure in fission yeast and human cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2166–E2175. [Google Scholar] [CrossRef]
- Webster, B.M.; Colombi, P.; Jager, J.; Lusk, C.P. Surveillance of nuclear pore complex assembly by escrt-iii/vps4. Cell 2014, 159, 388–401. [Google Scholar] [CrossRef]
- Webster, B.M.; Thaller, D.J.; Jager, J.; Ochmann, S.E.; Borah, S.; Lusk, C.P. Chm7 and heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. EMBO J. 2016, 35, 2447–2467. [Google Scholar] [CrossRef]
- Mackay, D.R.; Makise, M.; Ullman, K.S. Defects in nuclear pore assembly lead to activation of an aurora b-mediated abscission checkpoint. J. Cell Biol. 2010, 191, 923–931. [Google Scholar] [CrossRef]
- Carlton, J.G.; Caballe, A.; Agromayor, M.; Kloc, M.; Martin-Serrano, J. Escrt-iii governs the aurora b-mediated abscission checkpoint through chmp4c. Science 2012, 336, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Sandrin, V.; Chung, H.Y.; Morham, S.G.; Gygi, S.P.; Rodesch, C.K.; Sundquist, W.I. Human escrt and alix proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007, 26, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Rabut, G.; Lenart, P.; Ellenberg, J. Dynamics of nuclear pore complex organization through the cell cycle. Curr. Opin. Cell Biol. 2004, 16, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, S.; Bui, K.H.; Schorb, M.; Hossain, M.J.; Politi, A.Z.; Koch, B.; Eltsov, M.; Beck, M.; Ellenberg, J. Nuclear pore assembly proceeds by an inside-out extrusion of the nuclear envelope. Elife 2016, 5, e19071. [Google Scholar] [CrossRef]
- Doucet, C.M.; Talamas, J.A.; Hetzer, M.W. Cell cycle-dependent differences in nuclear pore complex assembly in metazoa. Cell 2010, 141, 1030–1041. [Google Scholar] [CrossRef]
- Dultz, E.; Ellenberg, J. Live imaging of single nuclear pores reveals unique assembly kinetics and mechanism in interphase. J. Cell Biol. 2010, 191, 15–22. [Google Scholar] [CrossRef]
- D’Angelo, M.A.; Anderson, D.J.; Richard, E.; Hetzer, M.W. Nuclear pores form de novo from both sides of the nuclear envelope. Science 2006, 312, 440–443. [Google Scholar] [CrossRef]
- Franz, C.; Walczak, R.; Yavuz, S.; Santarella, R.; Gentzel, M.; Askjaer, P.; Galy, V.; Hetzer, M.; Mattaj, I.W.; Antonin, W. Mel-28/elys is required for the recruitment of nucleoporins to chromatin and postmitotic nuclear pore complex assembly. EMBO Rep. 2007, 8, 165–172. [Google Scholar] [CrossRef]
- Maul, G.G.; Maul, H.M.; Scogna, J.E.; Lieberman, M.W.; Stein, G.S.; Hsu, B.Y.; Borun, T.W. Time sequence of nuclear pore formation in phytohemagglutinin-stimulated lymphocytes and in hela cells during the cell cycle. J. Cell Biol. 1972, 55, 433–447. [Google Scholar] [CrossRef]
- Otsuka, S.; Steyer, A.M.; Schorb, M.; Heriche, J.K.; Hossain, M.J.; Sethi, S.; Kueblbeck, M.; Schwab, Y.; Beck, M.; Ellenberg, J. Postmitotic nuclear pore assembly proceeds by radial dilation of small membrane openings. Nat. Struct. Mol. Biol. 2018, 25, 21–28. [Google Scholar] [CrossRef]
- Weberruss, M.; Antonin, W. Perforating the nuclear boundary - how nuclear pore complexes assemble. J. Cell Sci. 2016, 129, 4439–4447. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rampello, A.J.; Prophet, S.M.; Schlieker, C. The Role of Torsin AAA+ Proteins in Preserving Nuclear Envelope Integrity and Safeguarding Against Disease. Biomolecules 2020, 10, 468. https://doi.org/10.3390/biom10030468
Rampello AJ, Prophet SM, Schlieker C. The Role of Torsin AAA+ Proteins in Preserving Nuclear Envelope Integrity and Safeguarding Against Disease. Biomolecules. 2020; 10(3):468. https://doi.org/10.3390/biom10030468
Chicago/Turabian StyleRampello, Anthony J., Sarah M. Prophet, and Christian Schlieker. 2020. "The Role of Torsin AAA+ Proteins in Preserving Nuclear Envelope Integrity and Safeguarding Against Disease" Biomolecules 10, no. 3: 468. https://doi.org/10.3390/biom10030468
APA StyleRampello, A. J., Prophet, S. M., & Schlieker, C. (2020). The Role of Torsin AAA+ Proteins in Preserving Nuclear Envelope Integrity and Safeguarding Against Disease. Biomolecules, 10(3), 468. https://doi.org/10.3390/biom10030468