Genomic Landscape and Mutational Spectrum of ADAMTS Family Genes in Mendelian Disorders Based on Gene Evidence Review for Variant Interpretation


Abstract
1. Introduction
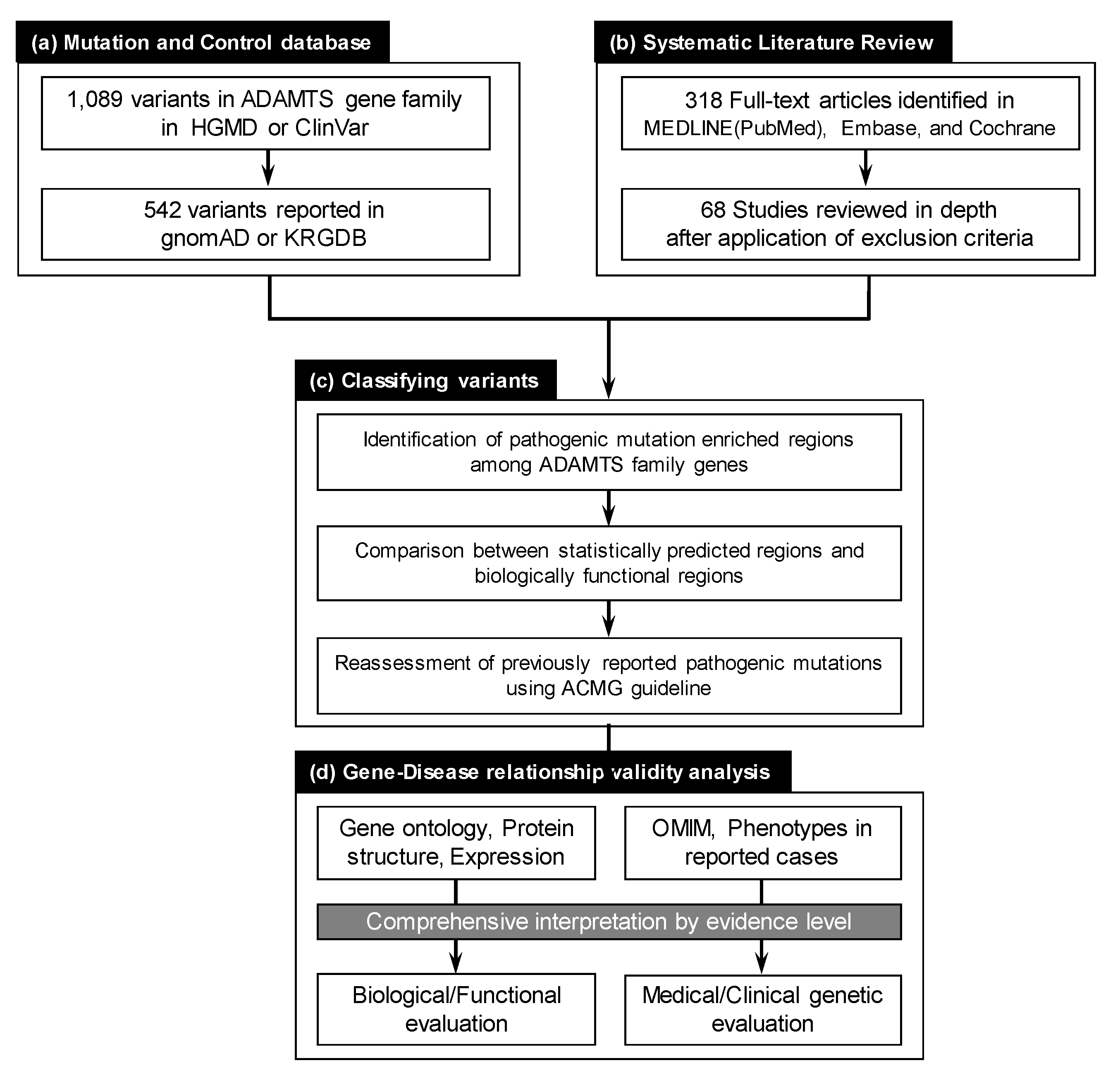
2. Materials and Methods
2.1. Collection of Reported ADAMTS Family Gene Variants
2.2. Systematic Literature Review for ADAMTS Family Genes on Mendelian Disorders
2.3. Evaluating ADAMTS Family Gene Disease Associations
2.4. Pathogenicity Interpretation for Variant and Gene Evaluation
3. Results
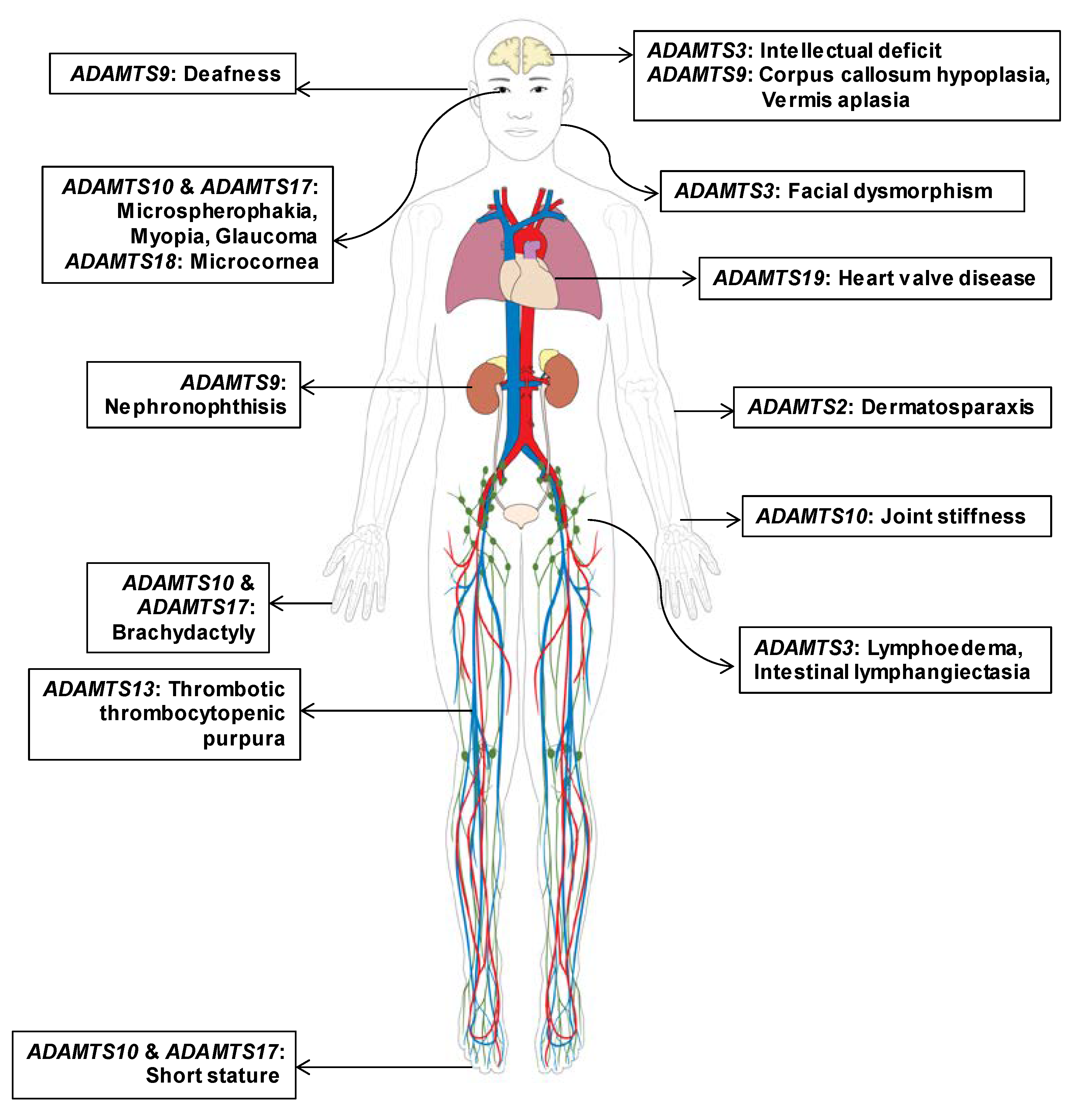
3.1. Gene-Disease Association of ADAMTS Family Genes Based on Pathogenic Mutations
3.2. Updated ADAMTS Family Genes Responsible for Mendelian Disorders; ADAMTS9 and ADAMTS19
3.3. Evidence-Based Evaluation for Gene-Disease Relationships in ADAMTS Genes
3.4. Mutational Spectrum of ADAMTS Family Genes in Current Databases
3.5. Reassessment of Previously Reported Pathogenic Mutations in ADAMTS Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Apte, S.S. Adamts proteins: Concepts, challenges, and prospects. Methods Mol. Biol. 2020, 2043, 1–12. [Google Scholar] [PubMed]
- Apte, S.S. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (adamts) superfamily: Functions and mechanisms. J. Biol. Chem. 2009, 284, 31493–31497. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The adamts (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Dubail, J.; Apte, S.S. Insights on adamts proteases and adamts-like proteins from mammalian genetics. Matrix Biol. 2015, 44, 24–37. [Google Scholar] [CrossRef]
- Mead, T.J.; Apte, S.S. Adamts proteins in human disorders. Matrix Biol. 2018, 71, 225–239. [Google Scholar] [CrossRef]
- Le Goff, C.; Cormier-Daire, V. The adamts(l) family and human genetic disorders. Hum. Mol. Genet. 2011, 20, R163–R167. [Google Scholar] [CrossRef]
- Adams, D.R.; Eng, C.M. Next-generation sequencing to diagnose suspected genetic disorders. New Engl. J. Med. 2018, 379, 1353–1362. [Google Scholar] [CrossRef]
- Grant, A.R.; Cushman, B.J.; Cave, H.; Dillon, M.W.; Gelb, B.D.; Gripp, K.W.; Lee, J.A.; Mason-Suares, H.; Rauen, K.A.; Tartaglia, M.; et al. Assessing the gene-disease association of 19 genes with the rasopathies using the clingen gene curation framework. Hum. Mutat. 2018, 39, 1485–1493. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. Clinvar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E. Improving sequence variant descriptions in mutation databases and literature using the mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. Omim.Org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the human phenotype ontology (hpo) knowledge base and resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef] [PubMed]
- The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef]
- UniProt Consortium. Uniprot: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Perez-Palma, E.; May, P.; Iqbal, S.; Niestroj, L.M.; Du, J.; Heyne, H.O.; Castrillon, J.A.; O’Donnell-Luria, A.; Nurnberg, P.; Palotie, A.; et al. Identification of pathogenic variant enriched regions across genes and gene families. Genome Res. 2020, 30, 62–71. [Google Scholar] [CrossRef]
- Silk, M.; Petrovski, S.; Ascher, D.B. Mtr-viewer: Identifying regions within genes under purifying selection. Nucleic Acids Res. 2019, 47, W121–W126. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Choi, Y.J.; Halbritter, J.; Braun, D.A.; Schueler, M.; Schapiro, D.; Rim, J.H.; Nandadasa, S.; Choi, W.I.; Widmeier, E.; Shril, S.; et al. Mutations of adamts9 cause nephronophthisis-related ciliopathy. Am. J. Hum. Genet. 2019, 104, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Wunnemann, F.; Ta-Shma, A.; Preuss, C.; Leclerc, S.; van Vliet, P.P.; Oneglia, A.; Thibeault, M.; Nordquist, E.; Lincoln, J.; Scharfenberg, F.; et al. Loss of adamts19 causes progressive non-syndromic heart valve disease. Nat. Genet. 2020, 52, 40–47. [Google Scholar] [CrossRef]
- Dagoneau, N.; Benoist-Lasselin, C.; Huber, C.; Faivre, L.; Megarbane, A.; Alswaid, A.; Dollfus, H.; Alembik, Y.; Munnich, A.; Legeai-Mallet, L.; et al. Adamts10 mutations in autosomal recessive weill-marchesani syndrome. Am. J. Hum. Genet. 2004, 75, 801–806. [Google Scholar] [CrossRef]
- Morales, J.; Al-Sharif, L.; Khalil, D.S.; Shinwari, J.M.; Bavi, P.; Al-Mahrouqi, R.A.; Al-Rajhi, A.; Alkuraya, F.S.; Meyer, B.F.; Al Tassan, N. Homozygous mutations in adamts10 and adamts17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet. 2009, 85, 558–568. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Alshammari, M.J.; Khan, A.O.; Mohamed, J.Y.; Alhabib, F.A.; Alkuraya, F.S. The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (mmcat) is caused by mutations in adamts18. Hum. Mutat. 2013, 34, 1195–1199. [Google Scholar] [CrossRef]
- Chandra, A.; Arno, G.; Williamson, K.; Sergouniotis, P.I.; Preising, M.N.; Charteris, D.G.; Thompson, D.A.; Holder, G.E.; Borman, A.D.; Davagnanam, I.; et al. Expansion of ocular phenotypic features associated with mutations in adamts18. JAMA Ophthalmol. 2014, 132, 996–1001. [Google Scholar] [CrossRef][Green Version]
- Van Damme, T.; Colige, A.; Syx, D.; Giunta, C.; Lindert, U.; Rohrbach, M.; Aryani, O.; Alanay, Y.; Simsek-Kiper, P.O.; Kroes, H.Y.; et al. Expanding the clinical and mutational spectrum of the ehlers-danlos syndrome, dermatosparaxis type. Genet. Med. 2016, 18, 882–891. [Google Scholar] [CrossRef]
- Brouillard, P.; Dupont, L.; Helaers, R.; Coulie, R.; Tiller, G.E.; Peeden, J.; Colige, A.; Vikkula, M. Loss of adamts3 activity causes hennekam lymphangiectasia-lymphedema syndrome 3. Hum. Mol. Genet. 2017, 26, 4095–4104. [Google Scholar] [CrossRef]
- Alwan, F.; Vendramin, C.; Liesner, R.; Clark, A.; Lester, W.; Dutt, T.; Thomas, W.; Gooding, R.; Biss, T.; Watson, H.G.; et al. Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood 2019, 133, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- van Dorland, H.A.; Taleghani, M.M.; Sakai, K.; Friedman, K.D.; George, J.N.; Hrachovinova, I.; Knobl, P.N.; von Krogh, A.S.; Schneppenheim, R.; Aebi-Huber, I.; et al. The international hereditary thrombotic thrombocytopenic purpura registry: Key findings at enrollment until 2017. Haematologica 2019, 104, 2107–2115. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | Disease | Inheritance Mode | Other Genes | Mutational Spectrum | Number of Reported Patients (Family) | Major References (PMID) |
|---|---|---|---|---|---|---|
| ADAMTS2 | Ehlers–Danlos syndrome, dermatosparaxis type | Autosomal recessive | none | nonsense, frameshift | 10 (10) | 26765342 |
| ADAMTS3 | Hennekam syndrome | Autosomal recessive | FAT4, CCBE1 | nonsense, missense | 3 (2) | 28985353 |
| ADAMTS9 | Nephronophthisis-related ciliopathy | Autosomal recessive | none | frameshift, missense | 2 (2) | 30609407 |
| ADAMTS10 | Weill–Marchesani syndrome | Autosomal recessive | LTBP2, FBN1 | nonsense, missense, splice site | 8 (7) | 18567016, 25469541 |
| ADAMTS13 | Thrombotic thrombocytopenic purpura (Upshaw–Schulman syndrome) | Autosomal recessive | none | missense, nonsense, frameshift, splice site | more than 200 | 30770395, 30792199 |
| ADAMTS17 | Weill–Marchesani-like syndrome (Weill–Marchesani syndrome 4) | Autosomal recessive | none | nonsense, frameshift, splice site | 4 (4) | 19836009 |
| ADAMTS18 | Microcornea, myopic chorioretinal atrophy and telecanthus | Autosomal recessive | none | missense, nonsense | 7 (5) | 23356391, 23818446 |
| ADAMTS19 | Nonsyndromic heart valve disease | Autosomal recessive | none | nonsense, exonic deletion | 4 (2) | 31844321 |
| Gene | Gene Ontology (Shared Biological Processes) | Gene Ontology (Shared Molecular Functions) * | Protein Domain (Major Functional Domain, ADAMTS Backbone Domain Shared) | Expression Database (Uniprot, Human Protein Atlas) | Functional Assay Availability |
|---|---|---|---|---|---|
| ADAMTS1 | Integrin-mediated signaling pathway Negative regulation of cell population Proliferation | Heparin binding Zinc ion binding | TSP type-1 repeats 2 | Ovary, Immune cells, Facial skeletal | Extracellular region or secreted |
| ADAMTS2 | Collagen catabolic process Collagen fibril organization Protein processing | Zinc ion binding | TSP type-1 repeats 3, Procollagen amino propeptidases | Connective tissue | NA |
| ADAMTS3 | Collagen catabolic process Collagen fibril organization Protein processing | Heparin binding Zinc ion binding | TSP type-1 repeats 3, Procollagen amino propeptidases | Extremities | Extracellular region or secreted, Immunoprecipitation |
| ADAMTS4 | Extracellular matrix Disassembly Proteolysis | Metal ion binding Metallopeptidase activity | None | Adipose tissue, CNS | NA |
| ADAMTS5 | Extracellular matrix Disassembly Proteolysis | Extracellular matrix binding Heparin binding Integrin binding Metallopeptidase activity Zinc ion binding | TSP type-1 repeats 1 | Breast, Placenta, Heart | NA |
| ADAMTS6 | NA | Metal ion binding Metallopeptidase activity | TSP type-1 repeats 4, PLAC | Placenta, Brain, Ovarian follicle cells | NA |
| ADAMTS7 | Cellular response to BMP stimulus Cellular response to tumor necrosis factor Negative regulation of chondrocyte Differentiation Proteolysis involved in cellular protein catabolic process | Metal ion binding Metallopeptidase activity | TSP type-1 repeats 7, Mucin-proteoglycans, PLAC | Heart muscle | NA |
| ADAMTS8 | Negative regulation of cell population proliferation | Heparin binding Integrin binding Metallopeptidase activity Zinc ion binding | TSP type-1 repeats 1 | Gallbladder, Lung | NA |
| ADAMTS9 | Endothelial cell–matrix adhesion Extracellular matrix organization Positive regulation of melanocyte Differentiation Proteolysis | Metallopeptidase activity Zinc ion binding | TSP type-1 repeats 14, GON-1 | Kidney, Adipose tissue | Extracellular region or secreted |
| ADAMTS10 | NA | Metal ion binding | TSP type-1 repeats 4, PLAC | Connective tissue, Skin | Extracellular region or secreted, N-linked deglycosylation assay |
| ADAMTS12 | Cell–matrix adhesion Cellular response to BMP stimulus Cellular response to tumor necrosis factor Negative regulation of chondrocyte differentiation Proteolysis involved in cellular protein catabolic process | Metal ion binding | TSP type-1 repeats 7, Mucin-proteoglycans, PLAC | NA | NA |
| ADAMTS13 | Cell-matrix adhesion Cellular response to tumor necrosis factor Integrin-mediated signaling pathway Protein processing Proteolysis | Integrin binding Metallopeptidase activity Zinc ion binding | TSP type-1 repeats 7, CUB domain | Liver, Blood | Extracellular region or secreted, Beta-galactosidase activity |
| ADAMTS14 | Collagen catabolic process Collagen fibril organization | Metal ion binding | TSP type-1 repeats 3, Procollagen amino propeptidases | Brain, Gallbladder, Placenta | NA |
| ADAMTS15 | NA | Extracellular matrix binding Heparin binding Zinc ion binding | TSP type-1 repeats 2 | Adipose tissue, Luminal membranes in the gastrointestinal tract | NA |
| ADAMTS16 | NA | Metal ion binding | TSP type-1 repeats 5, PLAC | Kidney, Brain, Ovary | NA |
| ADAMTS17 | NA | Metal ion binding | TSP type-1 repeats 4, PLAC | Lymphoid tissue, Connective tissue | Extracellular region or secreted |
| ADAMTS18 | NA | Metal ion binding | TSP type-1 repeats 5, PLAC | Eye, Adipose tissue, Brain, Placenta, Extravillous trophoblasts, CNS, Bone | Medaka fish model (ocular), In-situ hybridization |
| ADAMTS19 | NA | Metal ion binding | TSP type-1 repeats 4, PLAC | Cervix, Uterine, Endometrium, Smooth muscle, Ovary | NA |
| ADAMTS20 | Extracellular matrix organization Positive regulation of melanocyte differentiation | Zinc ion binding | TSP type-1 repeats 14, GON-1 | Brain, Placenta, Retina, Testis | NA |
| Gene | Disease Database | Prediction Algorithms | Population Database | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of SNV Mutations | Number of CNV Mutations | PER-Viewer | MTR-Viewer | gnomAD | |||||||
| HGMD * | ClinVar ** | Total *** | HGMD * | ClinVar ** | Total *** | Pathogenic Mutation Enriched Region | Missense Intolerance Regions | oe value_ Missense | oe value_ LoF | pLI Score | |
| ADAMTS1 | 4 | 0 | 4 | 0 | 0 | 0 | no (family1) | no | 0.92 | 0.2 | 0.72 |
| ADAMTS2 | 9 | 8 | 12 | 3 | 0 | 3 | no (family1) | no | 0.85 | 0.18 | 0.97 |
| ADAMTS3 | 4 | 3 | 4 | 0 | 0 | 0 | no (family1) | no | 0.94 | 0.44 | 0 |
| ADAMTS4 | 0 | 0 | 0 | 0 | 0 | 0 | no (family1) | no | 0.86 | 0.46 | 0 |
| ADAMTS5 | 1 | 0 | 1 | 0 | 0 | 0 | no (family1) | no | 0.91 | 0.46 | 0 |
| ADAMTS6 | 4 | 0 | 4 | 0 | 0 | 0 | no (family1) | Yes | 0.65 | 0.12 | 1 |
| ADAMTS7 | 1 | 0 | 1 | 0 | 0 | 0 | no (family2) | no | 0.94 | 0.49 | 0 |
| ADAMTS8 | 0 | 0 | 0 | 0 | 0 | 0 | no (family1) | no | 0.93 | 0.63 | 0 |
| ADAMTS9 | 3 | 0 | 3 | 0 | 0 | 0 | no (family3) | Yes | 0.92 | 0.29 | 0 |
| ADAMTS10 | 14 | 7 | 14 | 0 | 0 | 0 | no (family1) | Yes | 0.62 | 0.2 | 0.84 |
| ADAMTS12 | 0 | 0 | 0 | 0 | 0 | 0 | no (family2) | no | 0.91 | 0.53 | 0 |
| ADAMTS13 | 179 | 38 | 216 | 9 | 0 | 9 | no (family4) | no | 0.85 | 0.52 | 0 |
| ADAMTS14 | 0 | 0 | 0 | 0 | 0 | 0 | no (family1) | Yes | 0.97 | 0.53 | 0 |
| ADAMTS15 | 1 | 0 | 1 | 0 | 0 | 0 | no (family1) | no | 0.94 | 0.47 | 0 |
| ADAMTS16 | 2 | 0 | 2 | 0 | 0 | 0 | no (family1) | no | 0.96 | 0.29 | 0 |
| ADAMTS17 | 7 | 5 | 7 | 1 | 3 | 4 | no (family1) | no | 1.16 | 0.55 | 0 |
| ADAMTS18 | 14 | 4 | 14 | 1 | 4 | 5 | no (family1) | no | 1.38 | 0.74 | 0 |
| ADAMTS19 | 3 | 0 | 3 | 0 | 0 | 0 | no (family1) | no | 0.76 | 0.38 | 0 |
| ADAMTS20 | 0 | 0 | 0 | 0 | 0 | 0 | no (family3) | no | 1.06 | 0.69 | 0 |
| Gene | Tran- script | Nucleotide Change | Amino Acid Change | Conservation * | Population Database | Prediction Algorithms | ACMG Guideline | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mm | Gg | Xt | Dr | gnomAD_all | gnomAD_ maxP | db SNP | SIFT | PP2 | Final Class | Component | ||||
| ADAMTS2 | NM_014244.4 | c.2T>C | p.M1T | M | E | _ | _ | none | none | Pathogenic | PVS1, PM2, PM3 | |||
| ADAMTS2 | NM_014244.4 | c.673C>T | p.Q225* | na | na | na | na | 0.0150% | ASJ:0.30% | rs137853146 | Pathogenic | PVS1, PM1, PP4 | ||
| ADAMTS2 | NM_014244.4 | c.2384G>A | p.W795* | na | na | na | na | none | rs137853147 | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS2 | NM_014244.4 | c.3328C>G | p.P1110A | P | L | _ | _ | none | none | Tol (0.29) | Ben (0.001) | Likely pathogenic | PM2, PP1, PP2, PP4, PP5 | |
| ADAMTS2 | NM_014244.4 | c.2751-2A>T | NA | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS2 | NM_014244.4 | c.884_887del | p.M295Tfs*26 | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS2 | NM_014244.4 | c.2458-6_2458del | NA | na | na | na | na | none | rs1057517277 | Pathogenic | PVS1, PM2, PP1, PP4 | |||
| ADAMTS2 | NM_014244.4 | c.2927_2928del | p.P976Rfs*42 | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS2 | NM_014244.4 | c.669dup | p.P224Afs*42 | na | na | na | na | none | rs748037345 | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS2 | NM_014244.4 | c.1638dup | p.K547* | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS2 | NM_014244.4 | c.32del | p.L11Rfs*154 | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS3 | NM_014243.2 | c.280C>T | p.R94* | na | na | na | na | 0.0004% | AFR:0.0062% | rs747975445 | Pathogenic | PVS1, PP1, PP4 | ||
| ADAMTS3 | NM_014243.2 | c.503T>C | p.L168P | L | L | L | L | 0.0004% | NFE:0.00090% | rs1177851177 | Del (0.01) | Dam (1.000) | Pathogenic | PM2, PM3, PP3, PP4 |
| ADAMTS3 | NM_014243.2 | c.872T>C | p.I291T | I | I | I | I | none | rs61757480 | Del (0.01) | Dam (1.000) | Pathogenic | PM2, PM3, PP3, PP4 | |
| ADAMTS9 | NM_182920.1 | c.194C>G | p.T65R | T | T | S | T | 0.0240% | ASJ:0.096% | rs192420947 | Del (0.01) | Dam (0.559) | Likely pathogenic | PS3, PP1, PP3 |
| ADAMTS9 | NM_182920.1 | c.4575_4576del | p.Q1525Hfs*60 | na | na | na | na | none | none | Pathogenic | PVS1, PS3, PM2 | |||
| ADAMTS10 | NM_030957.3 | c.41T>A | p.L14Q | L | _ | _ | _ | none | none | Del (0.01) | Ben (0.090) | Pathogenic | PS3, PM2, PP1, PP2, PP4, PP5 | |
| ADAMTS10 | NM_030957.3 | c.73G>A | p.A25T | A | _ | _ | _ | 0.0032% | SAS:0.0098% | rs121434358 | Tol (0.05) | Ben (0.058) | Pathogenic | PS3, PM3, PP1, PP2, PP4, PP5 |
| ADAMTS10 | NM_030957.3 | c.709C>T | p.R237* | na | na | na | na | 0.0004% | EAS:0.0054% | rs121434357 | Pathogenic | PVS1, PM3, PP4 | ||
| ADAMTS10 | NM_030957.3 | c.952C>T | p.Q318* | na | na | na | na | none | rs121434359 | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS10 | NM_030957.3 | c.1553G>A | p.G518D | G | G | G | G | none | rs267606636 | Del (0) | Dam (1.000) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 | |
| ADAMTS10 | NM_030957.3 | c.1586G>A | p.G529E | G | G | G | G | 0.0004% | NFE:0.00090% | none | Del (0) | Ben (0.270) | Likely pathogenic | PM2, PP1, PP4, PP5 |
| ADAMTS10 | NM_030957.3 | c.2098G>T | p.G700C | G | G | G | G | none | rs267606637 | Del (0) | Dam (1.000) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 | |
| ADAMTS10 | NM_030957.3 | c.2485T>A | p.W829R | W | W | W | W | none | none | Del (0) | Dam (0.999) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 | |
| ADAMTS10 | NM_030957.3 | c.810+1G>A | NA | na | na | na | na | 0.0007% | ASJ:0.0097% | rs387906266 | Pathogenic | PVS1, PP1, PP4 | ||
| ADAMTS10 | NM_030957.3 | c.1190+1G>A | NA | na | na | na | na | none | rs431825170 | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS10 | NM_030957.3 | c.1797+2T>G | NA | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS10 | NM_030957.3 | c.2239+1G>A | NA | na | na | na | na | 0.0004% | AFR:0.0062% | rs782338897 | Pathogenic | PVS1, PP1, PP4 | ||
| ADAMTS17 | NM_139057.3 | c.760C>T | p.Q254* | na | na | na | na | none | rs267606638 | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS17 | NM_139057.3 | c.1051A>T | p.K351* | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS17 | NM_139057.3 | c.873+1G>T | NA | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS17 | NM_139057.3 | c.1721+1G>A | NA | na | na | na | na | 0.0032% | NFE:0.0071% | rs749116256 | Pathogenic | PVS1, PP1, PP4 | ||
| ADAMTS17 | NM_139057.3 | c.652delG | p.D218Tfs*41 | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS17 | NM_139057.3 | c.2458dupG | p.E820Gfs*23 | na | na | na | na | none | rs387906291 | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS18 | NM_199355.3 | c.97C>T | p.Q33* | na | na | na | na | none | rs397515469 | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS18 | NM_199355.3 | c.605T>C | p.L202P | L | L | L | I | none | rs397515468 | Del (0.01) | Dam (0.992) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 | |
| ADAMTS18 | NM_199355.3 | c.1067T>A | p.L356* | na | na | na | na | none | none | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS18 | NM_199355.3 | c.1298C>A | p.T433N | T | T | T | T | none | none | Del (0.02) | Dam (1.000) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 | |
| ADAMTS18 | NM_199355.3 | c.1731C>G | p.C577W | C | C | C | C | none | rs148319220 | Del (0) | Dam (1.000) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 | |
| ADAMTS18 | NM_199355.3 | c.1952G>A | p.R651Q | R | R | R | R | none | rs866074735 | Del (0) | Dam (0.921) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 | |
| ADAMTS18 | NM_199355.3 | c.2065G>T | p.E689* | na | na | na | na | none | rs397515467 | Pathogenic | PVS1, PM2, PP4 | |||
| ADAMTS18 | NM_199355.3 | c.2159G>C | p.C720S | C | C | C | C | 0.0004% | SAS:0.0033% | rs749517658 | Del (0) | Dam (1.000) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 |
| ADAMTS18 | NM_199355.3 | c.2546G>A | p.G849D | G | G | _ | G | 0.0004% | EAS:0.0054% | rs1417470741 | Tol (0.44) | Dam (0.838) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 |
| ADAMTS18 | NM_199355.3 | c.3235T>C | p.C1079R | C | C | C | C | 0.0004% | NFE:0.00090% | rs1268581022 | Del (0) | Dam (0.999) | Likely pathogenic | PM2, PP1, PP3, PP4, PP5 |
| ADAMTS19 | NM_133638.4 | c.1984C>T | p.R662* | na | na | na | na | 0.0008% | NFE:0.0018% | rs772148624 | Pathogenic | PVS1, PS3 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rim, J.H.; Choi, Y.J.; Gee, H.Y. Genomic Landscape and Mutational Spectrum of ADAMTS Family Genes in Mendelian Disorders Based on Gene Evidence Review for Variant Interpretation. Biomolecules 2020, 10, 449. https://doi.org/10.3390/biom10030449
Rim JH, Choi YJ, Gee HY. Genomic Landscape and Mutational Spectrum of ADAMTS Family Genes in Mendelian Disorders Based on Gene Evidence Review for Variant Interpretation. Biomolecules. 2020; 10(3):449. https://doi.org/10.3390/biom10030449
Chicago/Turabian StyleRim, John Hoon, Yo Jun Choi, and Heon Yung Gee. 2020. "Genomic Landscape and Mutational Spectrum of ADAMTS Family Genes in Mendelian Disorders Based on Gene Evidence Review for Variant Interpretation" Biomolecules 10, no. 3: 449. https://doi.org/10.3390/biom10030449
APA StyleRim, J. H., Choi, Y. J., & Gee, H. Y. (2020). Genomic Landscape and Mutational Spectrum of ADAMTS Family Genes in Mendelian Disorders Based on Gene Evidence Review for Variant Interpretation. Biomolecules, 10(3), 449. https://doi.org/10.3390/biom10030449