Role of p53 in the Regulation of Cellular Senescence




Abstract
1. Introduction
2. Senescence as Cellular Response to Stress
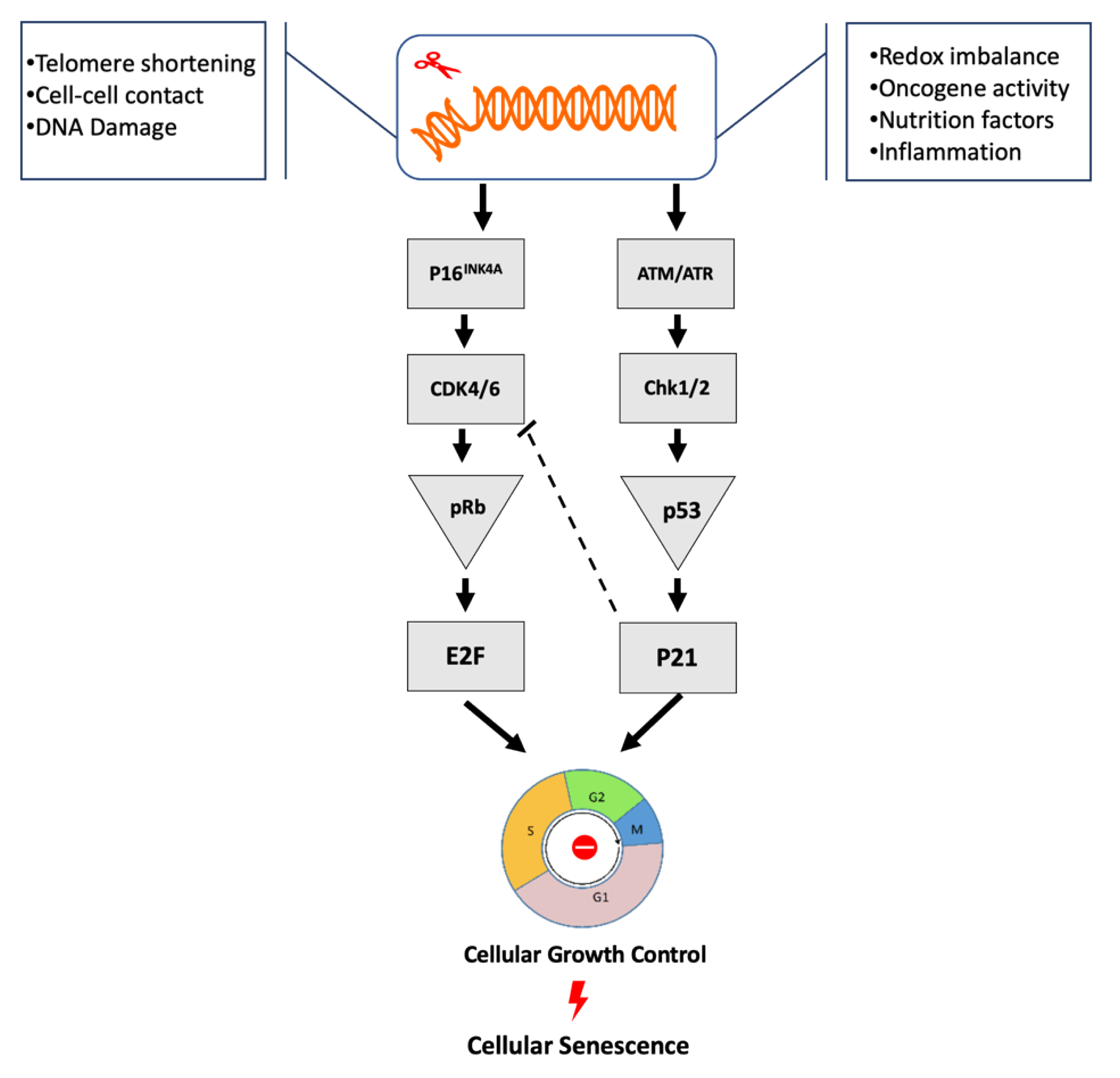
3. Critical Cellular Senescence Pathways
3.1. p53/p21cip1
3.2. p16INK4a/Rb
3.3. Senescence as a Multistep Process
4. Targeting the p53-Dependent Senescence in Different Pathophysiological Environment
4.1. Cancer
4.2. Metabolic Disorders
4.3. Inflammatory Responses and Inflammation-Associated Diseases
4.4. Neurodegenerative Diseases
5. Conclusion Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Pawlikowski, J.S.; Adams, P.D.; Nelson, D.M. Senescence at a glance. J. Cell Sci. 2013, 126, 4061–4067. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Idelchik, M.d.P.S.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.; Evangelou, K.; Gorgoulis, V.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhou, L. The Signaling of Cellular Senescence in Diabetic Nephropathy. Oxid. Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Au, B.; Kulkarni, M.; Shen, Y.; Lim, K.J.; Maimaiti, J.; Wong, C.K.; Luijten, M.N.H.; Chong, H.C.; Lim, E.H.; et al. Chromosomal instability-induced senescence potentiates cell non-autonomous tumourigenic effects. Oncogenesis 2018, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.P.; Cook, J.G. Cell cycle proliferation decisions: the impact of single cell analyses. FEBS J. 2017, 284, 362–375. [Google Scholar] [CrossRef]
- Lidzbarsky, G.; Gutman, D.; Shekhidem, H.A.; Sharvit, L.; Atzmon, G. Genomic Instabilities, Cellular Senescence, and Aging: In Vitro, In Vivo and Aging-Like Human Syndromes. Front. Med. 2018, 5, 104. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Vanzo, R.; Bartkova, J.; Merchut-Maya, J.M.; Hall, A.; Bouchal, J.; Dyrskjøt, L.; Frankel, L.B.; Gorgoulis, V.; Maya-Mendoza, A.; Jäättelä, M.; et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2019. [Google Scholar] [CrossRef]
- Giordano, A.; Macaluso, M. Fenofibrate triggers apoptosis of glioblastoma cells in vitro: New insights for therapy. Cell Cycle 2012, 11, 3154. [Google Scholar] [CrossRef]
- Surova, O.; Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 2013, 32, 3789–3797. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A. To clear, or not to clear (senescent cells)? That is the question: Clearance of senescent cells. BioEssays 2016, 38, S56–S64. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.M.; Kumar, S.; Moumen, A. A p53-independent role for the MDM2 antagonist Nutlin-3 in DNA damage response initiation. BMC Cancer 2011, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Talens, F.; Van Vugt, M.A.T.M. Inflammatory signaling in genomically instable cancers. Cell Cycle 2019, 18, 1830–1848. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Levine, A.J. The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes Cancer 2012, 3, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.Y.; Abedini, M.R.; Tsang, B.K. The oncogenic phosphatase PPM1D confers cisplatin resistance in ovarian carcinoma cells by attenuating checkpoint kinase 1 and p53 activation. Oncogene 2012, 31, 2175–2186. [Google Scholar] [CrossRef]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; Jeong, D.; Lee, K.; Park, H.J.; Ko, Y.-G.; Lee, J.-S. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2019, 38, 1639–1650. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Chen, J.; Huang, X.; Halicka, D.; Brodsky, S.; Avram, A.; Eskander, J.; Bloomgarden, N.A.; Darzynkiewicz, Z.; Goligorsky, M.S. Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: Permissive role of p53. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1575–H1586. [Google Scholar] [CrossRef]
- Malaquin, N.; Carrier-Leclerc, A.; Dessureault, M.; Rodier, F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.-G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [PubMed]
- Hobson, S.; Arefin, S.; Kublickiene, K.; Shiels, P.; Stenvinkel, P. Senescent Cells in Early Vascular Ageing and Bone Disease of Chronic Kidney Disease—A Novel Target for Treatment. Toxins 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Min, E.-Y.; Kim, I.-H.; Lee, J.; Kim, E.-Y.; Choi, Y.-H.; Nam, T.-J. The effects of fucodian on senescence are controlled by the p16INK4a-pRb and p14Arf-p53 pathways in hepatocellular carcinoma and hepatic cell lines. Int. J. Oncol. 2014, 45, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Spallarossa, P.; Altieri, P.; Aloi, C.; Garibaldi, S.; Barisione, C.; Ghigliotti, G.; Fugazza, G.; Barsotti, A.; Brunelli, C. Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H2169–H2181. [Google Scholar] [CrossRef]
- Chen, Q.M.; Liu, J.; Merrett, J.B. Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem. J. 2000, 347, 543. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Galluzzi, L.; Tajeddine, N.; Ortiz, C.; Criollo, A.; Tasdemir, E.; Morselli, E.; Ben Younes, A.; Maiuri, M.C.; Lavandero, S.; et al. Senescence, Apoptosis or Autophagy? Gerontology 2008, 54, 92–99. [Google Scholar] [CrossRef]
- Probin, V.; Wang, Y.; Bai, A.; Zhou, D. Busulfan selectively induces cellular senescence but not apoptosis in WI38 fibroblasts via a p53-independent but extracellular signal-regulated kinase-p38 mitogen-activated protein kinase-dependent mechanism. J. Pharmacol. Exp. Ther. 2006, 319, 551–560. [Google Scholar] [CrossRef]
- Fujita, K. p53 Isoforms in Cellular Senescence- and Ageing-Associated Biological and Physiological Functions. Int. J. Mol. Sci. 2019, 20, 6023. [Google Scholar] [CrossRef]
- Knights, C.D.; Catania, J.; Giovanni, S.D.; Muratoglu, S.; Perez, R.; Swartzbeck, A.; Quong, A.A.; Zhang, X.; Beerman, T.; Pestell, R.G.; et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 2006, 173, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: the critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.K.; Mungamuri, S.K.; Attie, O.; Kracikova, M.; Sachidanandam, R.; Manfredi, J.J.; Aaronson, S.A. p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes. Oncogene 2014, 33, 3959–3969. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, Y.; Zhang, G.; Huang, S.; Dong, Z.; Kong, C.; Su, D.; Du, J.; Zhu, S.; Liang, Q.; et al. DNMT3a plays a role in switches between doxorubicin-induced senescence and apoptosis of colorectal cancer cells. Int. J. Cancer 2011, 128, 551–561. [Google Scholar] [CrossRef]
- Martinez, L.A.; Yang, J.; Vazquez, E.S.; del Carmen Rodriguez-Vargas, M.; Olive, M.; Hsieh, J.-T.; Logothetis, C.J.; Navone, N.M. p21 modulates threshold of apoptosis induced by DNA-damage and growth factor withdrawal in prostate cancer cells. Carcinogenesis 2002, 23, 1289–1296. [Google Scholar] [CrossRef]
- Aliouat-Denis, C.-M. p53-Independent Regulation of p21Waf1/Cip1 Expression and Senescence by Chk2. Mol. Cancer Res. 2005, 3, 627–634. [Google Scholar] [CrossRef]
- Benson, E.K.; Zhao, B.; Sassoon, D.A.; Lee, S.W.; Aaronson, S.A. Effects of p21 deletion in mouse models of premature aging. Cell Cycle 2009, 8, 2002–2004. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Dolan, D.W.P.; Zupanic, A.; Nelson, G.; Hall, P.; Miwa, S.; Kirkwood, T.B.L.; Shanley, D.P. Integrated Stochastic Model of DNA Damage Repair by Non-homologous End Joining and p53/p21-Mediated Early Senescence Signalling. PLoS Comput. Biol. 2015, 11, e1004246. [Google Scholar] [CrossRef] [PubMed]
- Beausejour, C.M. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Helmbold, H.; Kömm, N.; Deppert, W.; Bohn, W. Rb2/p130 is the dominating pocket protein in the p53–p21 DNA damage response pathway leading to senescence. Oncogene 2009, 28, 3456–3467. [Google Scholar] [CrossRef]
- Indovina, P.; Marcelli, E.; Casini, N.; Rizzo, V.; Giordano, A. Emerging roles of RB family: New defense mechanisms against tumor progression. J. Cell. Physiol. 2013, 228, 525–535. [Google Scholar] [CrossRef]
- Alessio, N.; Capasso, S.; Ferone, A.; Di Bernardo, G.; Cipollaro, M.; Casale, F.; Peluso, G.; Giordano, A.; Galderisi, U. Misidentified Human Gene Functions with Mouse Models: The Case of the Retinoblastoma Gene Family in Senescence. Neoplasia 2017, 19, 781–790. [Google Scholar] [CrossRef]
- Galderisi, U.; Helmbold, H.; Squillaro, T.; Alessio, N.; Komm, N.; Khadang, B.; Cipollaro, M.; Bohn, W.; Giordano, A. In Vitro Senescence of Rat Mesenchymal Stem Cells is Accompanied by Downregulation of Stemness-Related and DNA Damage Repair Genes. Stem Cells Dev. 2009, 18, 1033–1042. [Google Scholar] [CrossRef]
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Kapić, A.; Helmbold, H.; Reimer, R.; Klotzsche, O.; Deppert, W.; Bohn, W. Cooperation between p53 and p130(Rb2) in induction of cellular senescence. Cell Death Differ. 2006, 13, 324–334. [Google Scholar] [CrossRef]
- Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence—Current Concepts and Open Lessons. Cells 2019, 8, 1446. [Google Scholar] [CrossRef]
- Dirac, A.M.G.; Bernards, R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J. Biol. Chem. 2003, 278, 11731–11734. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef]
- Mária, J.; Ingrid, Ž. Effects of bioactive compounds on senescence and components of senescence associated secretory phenotypes in vitro. Food Funct. 2017, 8, 2394–2418. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Bezzerri, V.; Piacenza, F.; Caporelli, N.; Malavolta, M.; Provinciali, M.; Cipolli, M. Is cellular senescence involved in cystic fibrosis? Respir. Res. 2019, 20, 32. [Google Scholar] [CrossRef]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette Smoke Induces Cellular Senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688. [Google Scholar] [CrossRef]
- He, L.; Chen, Y.; Feng, J.; Sun, W.; Li, S.; Ou, M.; Tang, L. Cellular senescence regulated by SWI/SNF complex subunits through p53/p21 and p16/pRB pathway. Int. J. Biochem. Cell Biol. 2017, 90, 29–37. [Google Scholar] [CrossRef]
- Schmitt, C.A. The persistent dynamic secrets of senescence. Nat. Cell Biol. 2016, 18, 913–915. [Google Scholar] [CrossRef]
- Galvis, D.; Walsh, D.; Harries, L.W.; Latorre, E.; Rankin, J. A dynamical systems model for the measurement of cellular senescence. J. R. Soc. Interface 2019, 16, 20190311. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.S.L.; Narita, M. Short-term gain, long-term pain: the senescence life cycle and cancer. Genes Dev. 2019, 33, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. Pathologies Associated with the p53 Response. Cold Spring Harb. Perspect. Biol. 2010, 2, a001180. [Google Scholar] [CrossRef] [PubMed]
- Northcott, J.M.; Czubryt, M.P.; Wigle, J.T. Vascular senescence and ageing: A role for the MEOX proteins in promoting endothelial dysfunction. Can. J. Physiol. Pharmacol. 2017, 95, 1067–1077. [Google Scholar]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. The TGF-β1/p53/PAI-1 Signaling Axis in Vascular Senescence: Role of Caveolin-1. Biomolecules 2019, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Nerstedt, A.; Smith, U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat. Commun. 2019, 10, 2757. [Google Scholar] [CrossRef]
- Krstic, J.; Reinisch, I.; Schupp, M.; Schulz, T.J.; Prokesch, A. p53 Functions in Adipose Tissue Metabolism and Homeostasis. Int. J. Mol. Sci. 2018, 19, 2622. [Google Scholar] [CrossRef]
- Burton, D.G.A.; Faragher, R.G.A. Obesity and type-2 diabetes as inducers of premature cellular senescence and ageing. Biogerontology 2018, 19, 447–459. [Google Scholar] [CrossRef]
- Palmer, A.K.; Gustafson, B.; Kirkland, J.L.; Smith, U. Cellular senescence: At the nexus between ageing and diabetes. Diabetologia 2019, 62, 1835–1841. [Google Scholar] [CrossRef]
- Qi, X.; Davis, B.; Chiang, Y.-H.; Filichia, E.; Barnett, A.; Greig, N.H.; Hoffer, B.; Luo, Y. Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson’s disease model. J. Neurochem. 2016, 138, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.H.; Seol, W.; Son, I. Upregulation of the p53-p21 pathway by G2019S LRRK2 contributes to the cellular senescence and accumulation of α-synuclein. Cell Cycle 2019, 18, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Belaidi, A.A.; Ayton, S.; Bush, A.I. Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease. Pharmaceuticals 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.; Lanni, C.; Govoni, S.; Uberti, D.; D’Orazi, G.; Racchi, M. Unfolded p53 in the pathogenesis of Alzheimer’s disease: is HIPK2 the link? Aging 2010, 2, 545–554. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.-Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Intihar, T.A.; Martinez, E.A.; Gomez-Pastor, R. Mitochondrial Dysfunction in Huntington’s Disease; Interplay Between HSF1, p53 and PGC-1α Transcription Factors. Front. Cell. Neurosci. 2019, 13, 103. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Ávila-Gálvez, M.Á.; Espín, J.C.; González-Sarrías, A. Conjugated Physiological Resveratrol Metabolites Induce Senescence in Breast Cancer Cells: Role of p53/p21 and p16/Rb Pathways, and ABC Transporters. Mol. Nutr. Food Res. 2019, 63, e1900629. [Google Scholar] [CrossRef]
- Sossey-Alaoui, K.; Pluskota, E.; Szpak, D.; Plow, E.F. The Kindlin2-p53-SerpinB2 signaling axis is required for cellular senescence in breast cancer. Cell Death Dis. 2019, 10, 539. [Google Scholar] [CrossRef]
- Li, H.; Petersen, S.; Garcia Mariscal, A.; Brakebusch, C. Negative Regulation of p53-Induced Senescence by N-WASP Is Crucial for DMBA/TPA-Induced Skin Tumor Formation. Cancer Res. 2019, 79, 2167–2181. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Brakebusch, C. Senescence regulation by nuclear N-WASP: a role in cancer? Oncoscience 2019, 6, 354–356. [Google Scholar] [PubMed]
- Kumar, R.; Gont, A.; Perkins, T.J.; Hanson, J.E.L.; Lorimer, I.A.J. Induction of senescence in primary glioblastoma cells by serum and TGFβ. Sci. Rep. 2017, 7, 2156. [Google Scholar] [CrossRef] [PubMed]
- Turnquist, C.; Beck, J.A.; Horikawa, I.; Obiorah, I.E.; Von Muhlinen, N.; Vojtesek, B.; Lane, D.P.; Grunseich, C.; Chahine, J.J.; Ames, H.M.; et al. Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro Oncol. 2019, 21, 474–485. [Google Scholar] [CrossRef] [PubMed]
- He, M.Y.; Xu, S.B.; Qu, Z.H.; Guo, Y.M.; Liu, X.C.; Cong, X.X.; Wang, J.F.; Low, B.C.; Li, L.; Wu, Q.; et al. Hsp90β interacts with MDM2 to suppress p53-dependent senescence during skeletal muscle regeneration. Aging Cell. 2019, 18, e13003. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, R.C.; Watts, A.C.; Murphy, R.J.; Snelling, S.J.; Carr, A.J.; Hulley, P.A. Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: in vivo and in vitro evidence. Ann. Rheum. Dis. 2014, 73, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Bar-Shai, A.; Levi, N.; Hatzav, M.; Zada, L.; Ovadya, Y.; Roitman, L.; Manella, G.; Regev, O.; Majewska, J.; et al. p53 in Bronchial Club Cells Facilitates Chronic Lung Inflammation by Promoting Senescence. Cell Rep. 2018, 22, 3468–3479. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Du, X.; Tang, S.; Wu, S.; Wang, L.; Xiang, Y.; Qu, X.; Liu, H.; Qin, X.; Liu, C. ITGB4 deficiency induces senescence of airway epithelial cells through p53 activation. FEBS J. 2019, 286, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Li, Y.; Liu, J.; Ying, X.; Liu, Y.; Yan, J.; Chen, C.; Zhou, H.; Cao, L.; Ma, Y. LncRNA-mediated SIRT1/FoxO3a and SIRT1/p53 signaling pathways regulate type II alveolar epithelial cell senescence in patients with chronic obstructive pulmonary disease. Mol. Med. Rep. 2017, 15, 3129–3134. [Google Scholar] [CrossRef]
- Kuwano, K.; Araya, J.; Hara, H.; Minagawa, S.; Takasaka, N.; Ito, S.; Kobayashi, K.; Nakayama, K. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir. Investig. 2016, 54, 397–406. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Collado, M.; Serrano, M. Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921. [Google Scholar] [CrossRef] [PubMed]
- Kreis, N.N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.-S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef]
- Vassilev, L.T. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Merkel, O.; Taylor, N.; Prutsch, N.; Staber, P.B.; Moriggl, R.; Turner, S.D.; Kenner, L. When the guardian sleeps: Reactivation of the p53 pathway in cancer. Mutat. Res. Mutat. Res. 2017, 773, 1–13. [Google Scholar] [CrossRef]
- Gonzalez-Meljem, J.M.; Apps, J.R.; Fraser, H.C.; Martinez-Barbera, J.P. Paracrine roles of cellular senescence in promoting tumourigenesis. Br. J. Cancer 2018, 118, 1283–1288. [Google Scholar] [CrossRef]
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef]
- Carracedo, J.; Alique, M.; Ramírez-Carracedo, R.; Bodega, G.; Ramírez, R. Endothelial Extracellular Vesicles Produced by Senescent Cells: Pathophysiological Role in the Cardiovascular Disease Associated with all Types of Diabetes Mellitus. Curr. Vasc. Pharmacol. 2019, 17, 447–454. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Boerma, M.; Zhou, D. Ionizing Radiation-Induced Endothelial Cell Senescence and Cardiovascular Diseases. Radiat. Res. 2016, 186, 153–161. [Google Scholar] [CrossRef]
- Rosso, A. p53 Mediates the Accelerated Onset of Senescence of Endothelial Progenitor Cells in Diabetes. J. Biol. Chem. 2006, 281, 4339–4347. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies. Oxid. Med. Cell. Longev. 2017, 2017, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Coussens, L.M.; Stewart, S.A. Senescence and cancer: An evolving inflammatory paradox. Biochim. Biophys. Acta BBA Rev. Cancer 2016, 1865, 14–22. [Google Scholar] [CrossRef]
- DiMauro, T.; David, G. Ras-Induced Senescence and its Physiological Relevance in Cancer. Curr. Cancer Drug Targets 2010, 10, 869–876. [Google Scholar] [CrossRef]
- Antony, V.B.; Thannickal, V.J. Cellular Senescence in Chronic Obstructive Pulmonary Disease: Multifaceted and Multifunctional. Am. J. Respir. Cell Mol. Biol. 2018, 59, 135–136. [Google Scholar] [CrossRef]
- Sundar, I.K.; Rashid, K.; Gerloff, J.; Li, D.; Rahman, I. Genetic Ablation of p16 INK4a Does Not Protect against Cellular Senescence in Mouse Models of Chronic Obstructive Pulmonary Disease/Emphysema. Am. J. Respir. Cell Mol. Biol. 2018, 59, 189–199. [Google Scholar] [CrossRef]
- Frey, N.; Venturelli, S.; Zender, L.; Bitzer, M. Cellular senescence in gastrointestinal diseases: From pathogenesis to therapeutics. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 81–95. [Google Scholar] [CrossRef]
- Uehara, I.; Tanaka, N. Role of p53 in the Regulation of the Inflammatory Tumor Microenvironment and Tumor Suppression. Cancers 2018, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. p53 and the Carcinogenicity of Chronic Inflammation. Cold Spring Harb. Perspect. Med. 2016, 6, a026161. [Google Scholar] [CrossRef] [PubMed]
- Salotti, J.; Johnson, P.F. Regulation of senescence and the SASP by the transcription factor C/EBPβ. Exp. Gerontol. 2019, 128, 110752. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef]
- Tan, F.C.C.; Hutchison, E.R.; Eitan, E.; Mattson, M.P. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology 2014, 15, 643–660. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Rane, A.; Demaria, M.; Campisi, J.; Andersen, J.K. Cellular senescence and the aging brain. Exp. Gerontol. 2015, 68, 3–7. [Google Scholar] [CrossRef]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]


{kind=link}
| Senescence Phase | Description of the Involved Pathways | Associated Markers | Reference |
|---|---|---|---|
| Primary senescence | Induction of p53/p21 pathway, induction of antiproliferative transcriptional program | BAF57, GADD45 NOTCH1/N1ICD, p21 | [62,69] |
| Developing senescence | p53/p21 and/or p16 pathway, SASP release, Morphological changes | p21, P19, p16, LIMA1, Ki-67 | [70,71] |
| Late senescence | Overproduction of SASP, chromatin remodeling, CCFs formation lysosomal activity, p16 pathway | Il-6, PGC-1β, SA-beta-galactosidase IFN-I, Ki-67 | [14,60] |
| Diseases Category | Diseases | p53 Regulation in Disease | Induction of Senescence | Pathological Characterization | Reference |
|---|---|---|---|---|---|
| Cardiovascular metabolic disorders | Cardiovascular injury | + | PAI-1; MEOX2 | caveolin-1 Inhibits VSMCs grow and promote senescence; Meox protein play a role in p53-mediated endothelial disfunction | [75,76] |
| Obesity | −/+ | HFD | Body weight phenotype and behavioral disorders; p53 represses the lipogenic Srebf1 pathway in Adipocytes | [77,78] | |
| Diabetes | + | hyperglycemia | p53 contributes to insulin resistance; decreased islet proliferation | [79,80] | |
| Neurodegenerative diseases | Parkinson’s disease | + | MPTP; αSyn fibrils | The KOp53 mice in Parkinson’s disease model | [81,82] |
| Alzheimer’s disease (AD) | + | Aβ (1-42) | SIRT3 rescues neurons from p53 mediated senescence; Aβ (1-42) induces senescence | [83,84] | |
| Huntington’s disease | + | CAG144 R6/2 | P53, miR-34a was disrupted in R6/2 mouse brain tissue; senescence involved in the striatum during HD development | [85,86] | |
| Cancer/ tumors | Breast cancer | + | RSV; Kindlin-2 | RSV metabolites induce senescence in breast cancer cells | [87,88] |
| Skin Tumor | + | N-WASP; Doxycycline | N-WASP is a negative regulator of senescence induction by p53 | [89,90] | |
| Brain cancer | + | TGFβ | Acute activation of senescence in Glioblastomas | [91,92] | |
| Inflammation | Musculoskeletal pain | + | Glucocorticoids; Hsp90β | Induction of irreversible senescence; Hsp90β/MDM2 induced p53-dependent senescence on muscle regeneration | [93,94] |
| Asthma/bronchitis | + | LPS; TNF-α | cell senescence promotes chronic lung inflammation; downregulation of ITGB4 induces senescence in inflammation | [95,96] | |
| COPD | + | lncRNA1 | accelerated cellular senescence in COPD | [97,98] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. https://doi.org/10.3390/biom10030420
Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules. 2020; 10(3):420. https://doi.org/10.3390/biom10030420
Chicago/Turabian StyleMijit, Mahmut, Valentina Caracciolo, Antonio Melillo, Fernanda Amicarelli, and Antonio Giordano. 2020. "Role of p53 in the Regulation of Cellular Senescence" Biomolecules 10, no. 3: 420. https://doi.org/10.3390/biom10030420
APA StyleMijit, M., Caracciolo, V., Melillo, A., Amicarelli, F., & Giordano, A. (2020). Role of p53 in the Regulation of Cellular Senescence. Biomolecules, 10(3), 420. https://doi.org/10.3390/biom10030420