1. Introduction
Flavonoids are the most effective and variable biologically active compounds in plants. Licochalcone D (LCD) is an active flavonoid isolated from the Chinese medicinal herb
Glycyrrhiza inflata [
1]. LCD is present in the roots and rhizomes of
G. inflata. The chemical name of LCD is (E)-3-(3,4-dihydroxy-2-methoxyphenyl)-1-[4-hydroxy-3-(3-methylbut-2-enyl)phenyl]prop-2-en-1-one. Various pharmacological actions, including anti-oxidant, anti-biotic, anti-ulcer, and anti-carcinogenic effects, have been described for LCD [
2]. Previous studies have demonstrated that LCD can induce cell apoptosis and suppress migration and invasion in skin cancer [
3]. In addition, LCD can suppress the proliferation in oral cancer cells [
4]. However, the mechanism by which LCD exerts its effects on lung cancer has not been fully determined yet.
Non-small cell lung cancer (NSCLC) is the major cause of death from cancer in the world. NSCLC accounts for approximately 85% of lung cancer cases [
5]. Chemotherapy remains marginally effective for NSCLC. Chemotherapy can slightly prolong the survival of patients with advanced lung cancer. However, it has clinically significant adverse effects [
6]. The current treatment approach includes surgical resection, radiation therapy, and chemotherapy alone or in combination [
6]. Despite these therapies, lung cancer is rarely curable, with an overall 5-year survival rate of only 15% [
7]. As a result of the low cure rate of NSCLC, it is important to find effective treatment, with a focus on new molecular and targeted therapies.
Epidermal growth factor receptor (EGFR) mutations lead to the outstanding activation of EGFR signaling and carcinogenicity transformation both in vitro and in vivo [
8]. Cancers with EGFR mutations (EGFR-mutated cancers) rely on EGFR signaling for growth. They are often sensitive to medical treatment with EGFR tyrosine kinase inhibitors (TKIs) [
8,
9]. Most patients with lung cancer have tumor-activating EGFR mutations. Treatment with EGFR-TKIs causes tumor reduction; however, the progression of cancer occurs at 6 to 12 months after treatment [
10]. Various mechanisms of resistance to EGFR-TKIs (such as erlotinib and gefitinib) have been identified. The comprehension of these mechanisms is critical to evolving treatment strategies in the setting of resistance development. One of the resistance mechanisms is EGFR T790M point mutation within exon 20 [
9]. hepatocyte growth factor receptor (MET) and human epidermal growth factor receptor (HER)2 overexpression with an upregulation of parallel signaling pathways have also been reported [
9]. A gefitinib-resistant HCC827GR (
MET-amplified) cell has been generated by exposing these cells to gefitinib for six months [
11]. Their results showed that lung cancer cell growth was inhibited by simultaneous treatment with gefitinib and MET inhibitor. Thus, the dual inhibition of EGFR and MET might be a means to overcome lung cancer resistance.
The objective of this study was to investigate whether LCD could inhibit cell proliferation through EGFR and MET dual targets in NSCLC using human gefitinib-sensitive or resistant NSCLC cells. We found that LCD could induce the apoptosis of HCC827 and HCC827GR cells by inhibiting both EGFR and the MET signaling pathway. To confirm whether LCD induced apoptosis, we carried out cell proliferation, cell cycle distribution, reactive oxygen species (ROS) production, mitochondrial membrane potential (MMP) depolarization, and multi-caspase activation assays. The results of this study might shed light on the mechanism involved in the effect of LCD on lung cancer. We expect that LCD for cancer treatment might give improved results.
2. Materials and Methods
2.1. Reagents and Antibodies
LCD was prepared by Professor Goo Yoon according to previous reports [
1]. Roswell Park Memorial Institute (RPMI)-1640 medium, phosphate-buffered saline (PBS), fetal bovine serum (FBS), penicillin/streptomycin, and trypsin were purchased from Hyclone (Logan, UT, USA). Dimethyl-sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and Basal Medium Eagle (BME) were purchased from Sigma Chemical Company (St. Louis, MO, USA). Gefitinib was purchased from Cayman Chemical (Ann Abor, MI, USA). Savolitinib was obtained from Selleckchem (Houston, TX, USA). Primary antibodies against cyclin B1, cdc2, p21, p27, β-actin, Bid, Bcl-xl, Mcl-1, Bad, cytochrome c (cyto c), α-tubulin, COX4, apoptotic protease activating factor-1 (Apaf-1), cleaved poly (ADP-Ribose) polymerase (C-PARP), and ERBB3 (HER3) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against phosphorylated (p)-EGFR (Tyr1068), EGFR, p-MET (Tyr1234/1235), MET, p-ERBB3 (Tyr1289), p-AKT (Ser473), and AKT were purchased from Cell Signaling Biotechnology (Beverly, MA, USA).
2.2. Cell Culture
The EGFR mutant (del E746_A750) NSCLC cell line HCC827 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). MET-amplified HCC827GR (gefitinib-resistant HCC827) cells were kindly contributed by professor Pasi A. Jänne (Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA). HCC827 and HCC827GR cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 100 U/ml penicillin/streptomycin at 37 °C in a humidified atmosphere of 5% CO2.
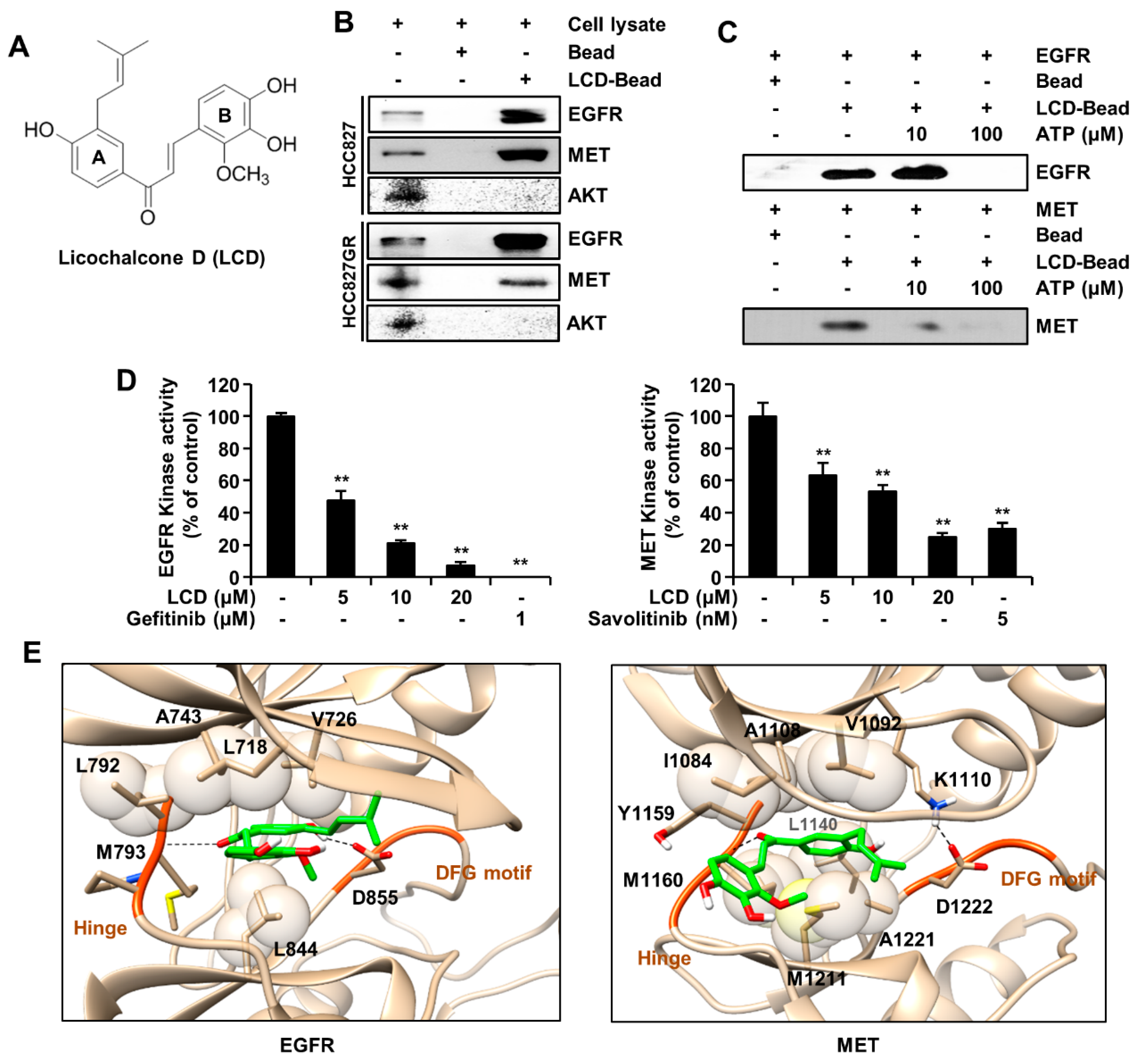
2.3. Pull-Down Assay
To confirm the interaction between LCD and EGFR or MET, HCC827 and HCC827GR cell lysates were mixed with Sepharose 4B or LCD-Sepharose 4B beads. The protein extract was incubated with LCD-Sepharose 4B beads or Sepharose 4B beads in reaction buffer [50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM/L dithiothreitol, 0.01% Nonidet P-40, 2 μg/mL bovine serum albumin, 0.02 mM phenylmethylsulfonyl fluoride, and 1X proteinase inhibitor] at 4 °C for 12 h. The mixture containing beads was washed six times with a washing buffer [50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM dithiothreitol, 0.01% Nonidet P-40, and 0.02 mM phenylmethylsulfonyl fluoride]. Then, bound proteins were eluted with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and subjected to Western blot analysis.
2.4. Western Blotting
Whole cells were lysed with Radio-Immunoprecipitation Assay (RIPA) buffer (iNtRON Biotechnology, Korea). Protein concentrations were determined using a Bio-Rad DC Protein Assay kit (Bio-Rad, Hercules, CA, USA). An equal amount of protein was loaded on 8%–15% SDS-PAGE gels. After separation, proteins were transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA), blocked with 3% or 5% skim milk in PBS containing 0.1% Tween-20 (PBST) at room temperature (RT) for 1 h or 2 h, and incubated with each primary antibody against its specific protein at 4 °C overnight. After washing six times, membranes were incubated with secondary antibodies for 2 h. Blots were scanned with an Image Quant LAS 500 (GE Healthcare, Uppsala, Sweden) using Western blotting luminol reagent (Santa Cruz, CA, USA).
2.5. ATP-Competitive Binding Assay
To determine whether LCD might compete with ATP, 100 ng of recombinant active EGFR or MET and ATP were pre-incubated at RT for 2 h. Subsequently, Sepharose 4B beads or LCD conjugated-Sepharose 4B beads were incubated at 4°C overnight and then washed with washing buffer. Bound proteins were subjected to Western blot analysis.
2.6. Kinase Assay
To determine the kinase activity of EGFR or MET in response to ATP, kinase reactions were incubated with EGFR (1.8 ng/μL; #3831), or MET (7 ng/μL; #3361), ATP (5 μΜ or 10 μΜ), substrates (0.2 μg/μL), LCD, gefitinib (1 μM), or savolitinib (5 nM) in a kinase reaction buffer containing 40 mM Tris (pH 7.5), 20 mM MgCl2, 0.1 mg/ml BSA, 50 μM dithiothreitol, 2 mM MnCl2, and 100 μM sodium vanadate using kinase enzyme systems (Promega, Madison, WI, USA). Before the plate was incubated at RT (22–25 °C) for 1 h, all kinase reactions were performed in 384-well plates with a volume of 5 μL. For the purpose of depleting the remaining ATP, 5 μL of ADP-Glo reagent (ADP-Glo kinase assay kit; Promega) was added to each well at RT for 40 min. Finally, 10 μL of kinase detection solution was added into each well of the 384-well plate. Luminescence reaction was proceeded and measured with a Centro LB 960 microplate luminometer (Berthold Technologies, Germany) for 0.5 s.
2.7. Molecular Modeling and Simulation
To predict the binding pose of the receptor tyrosine kinase, we performed a molecular docking simulation and a molecular dynamic (MD)s simulation. To perform the docking simulation, three-dimensional (3D) ligand and receptor structures were needed such as input files of the docking software, Vina. Receptor structures were downloaded from the protein database bank (PDB), including EGFR kinase in complex with gefitinib (PDB entry 2ITO) and MET kinase in complex with MK-2461 (PDB entry 3Q6W). LCD such as a ligand was made with Marvin sketch software. To run efficient searching, the assignment of the binding site was important for the docking simulation. Based on the binding interactions of the complex, the important ATP binding site was predicted to be Lys745 and Asp855 in EGFR and Lys1110 and Asp1222 in MET. Motifs of tyrosine kinase commonly included a glycine-rich nucleotide phosphate-binding loop, a G-loop (EGFR: Gly721-Gly724, MET: Gly1087-Gly1090), a hinge (EGFR: Leu792-Pro794, MET: Tyr1159-Lys1161), a Asp-Phe-Gly (DFG) motif (EGFR: Asp855-Gly857, MET: Asp1222-Gly1224), and an A-loop (EGFR: Asp855-Val876, MET: Asp1222-Leu1245). These binding poses were efficiently investigated for all important motifs. According to the result of the docking simulation, the top three binding poses were chosen based on the score of binding affinity. To confirm thermal stability in aqua solvent environment, MD simulation was performed using Gromacs software, which could overcome the lack of a rigid docking simulation. Complexes were solvated by a TIP3P water model and neutralized by adding ions. The Amber14SB force field (ff14SB) and general amber force field (GAFF) were applied for the protein and the ligand, respectively. Collected structures from the docking simulation were run in a physiological condition (310 K and 1 atm). After running for 500 ps in isothermal–isobaric (NPT) condition, canonical (NVT) ensemble was equilibrated for 30 ns. The stable structure was averaged in the converged state of MD simulation.
2.8. MTT Assay
HCC827 and HCC827GR cells were seeded into 96-well plates and incubated for 24 h. Then, these cells were treated with various concentrations of LCD for 24 h or 48 h. After incubation, MTT reagent was added to each well and incubated at 37 °C for 1 h. The culture medium and MTT were removed from each well, and the formazan in each well was dissolved with 100 μL of DMSO. The absorbance was measured at 570 nm using a Multiskan GO spectrophotometer (Thermo Scientific, Vantaa, Finland).
2.9. Anchorage-Independent Cell Growth Assay
Cells were seeded into 0.3% top agar over a layer of 0.6% bottom agar in a 6-well plate at a density of 8000 cells/well. Various concentrations of LCD and DMSO were added to the top and bottom layers containing culture medium (BME, 10% FBS, 2 mM L-glutamine and 5 μg/mL gentamicin). Plates were incubated at 37 °C for two weeks. The number of colonies was counted under a light microscope (Leica Microsystems, Wetzlar, Germany).
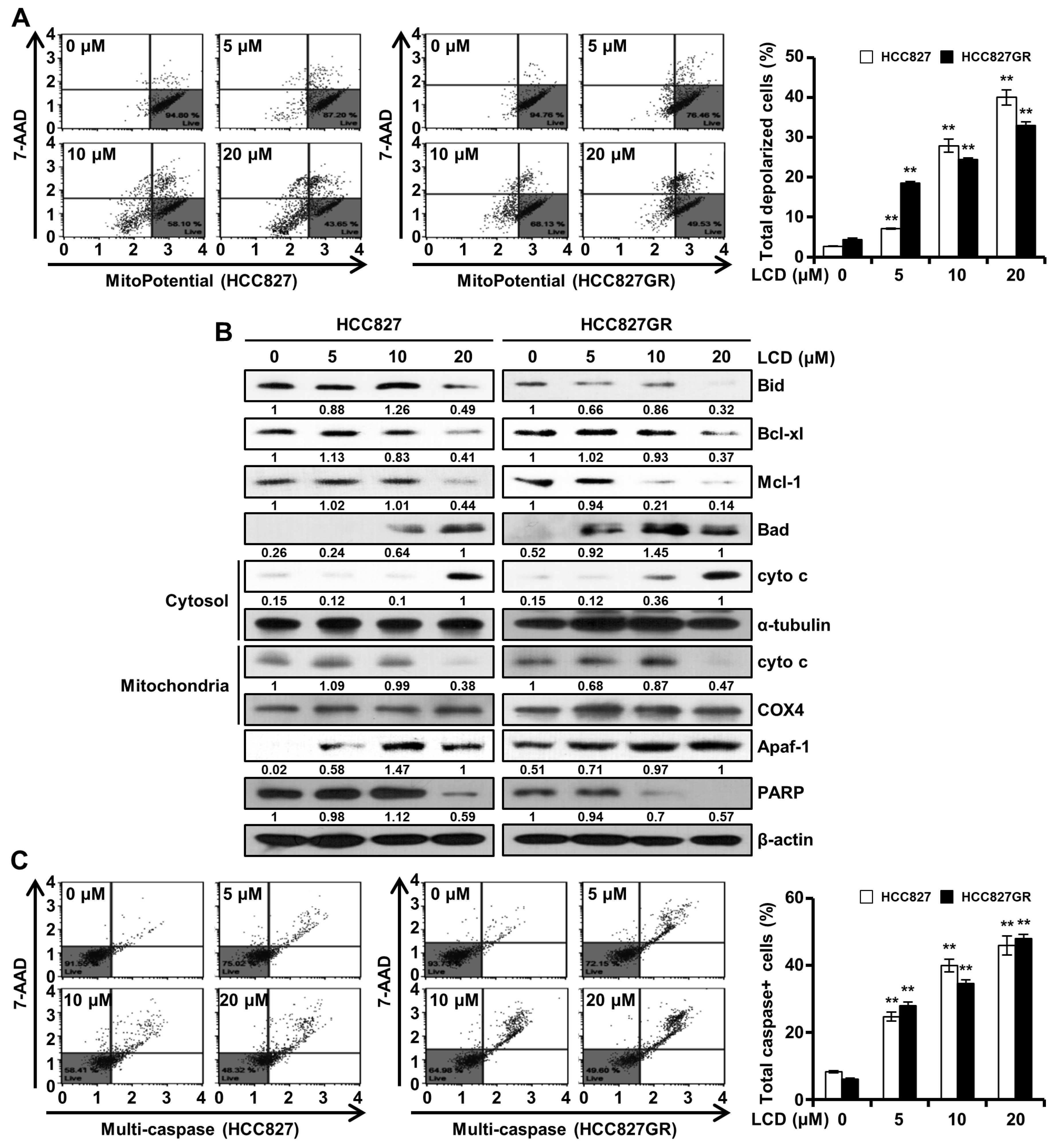
2.10. Annexin V/7-Minoactinomycin D (7-AAD) Staining
To evaluate NSCLC cell death with LCD treatment, Annexin V/7-AAD staining was performed using a Muse™ Annexin V and Dead Cell kit (MCH100105, Merck Millipore, Billerica, MA, USA). The HCC827 (1.95 × 105) and HCC827GR (1.8 × 105) cells were seeded onto a 6-well plate and treated with DMSO or LCD at different concentrations for 48 h. Cells were collected and subjected to Annexin V/7-AAD staining using 100 μL of Muse™ Annexin V and Dead Cell reagent according to the manufacturer’s protocol. Apoptotic cells were detected with a Muse™ Cell Analyzer (Merck Millipore).
2.11. Cell Cycle Analysis
A Muse™ Cell Cycle kit (MCH100106, Merck Millipore) was used to perform cell cycle analysis. Briefly, HCC827 and HCC827GR cells were collected by centrifugation at 4000 rpm for 5 min at 4 °C, washed three times with 1X PBS, and fixed with 70% cold ethanol at −20 °C for 24 h. These cells were collected by centrifugation at 4000 rpm for 10 min at 4 °C and washed once with PBS. Subsequently, Muse™ Cell Cycle Reagent was added to cell pellet followed by incubation at RT for 30 min in the dark. A Muse™ Cell Analyzer was used to obtain cell cycle data.
2.12. ROS Measurement
Intracellular ROS was measured with a Muse™ Oxidative Stress Kit (MCH100111, Merck Millipore). First, cells were grown in 6-well plates and treated with 5, 10, or 20 µM LCD for 48 h. Cells were washed with 1X assay buffer and incubated with a Muse™ Oxidative Stress Reagent working solution at 37 °C for 30 min. The level of fluorescence was determined with a Muse™ Cell Analyzer.
2.13. MMP Assay
MMP was measured using a Muse™ MitoPotential Kit (MCH100110, Merck Millipore). In brief, cells were exposed to 5, 10, or 20 µM of LCD for 48 h at 37 °C in a CO2 incubator. Cells were washed with 1× assay buffer, and fluorescence was then measured using Muse™ MitoPotential working solution. After incubation with 7-AAD for 5 min, the MMP was determined with a Muse™ Cell Analyzer.
2.14. Isolation of Cytosol and Mitochondrial Fractionation
Whole-cell extracts were obtained from LCD untreated or treated HCC827 and HCC827GR cells. Cells were resuspended in a plasma membrane extraction buffer containing 250 mM sucrose, 10 mM HEPES (pH 8.0), 10 mM KCl, 1.5 mM MgCl2∙6H2O, 1 mM EDTA, 1 mM EGTA, 0.1 mM phenylmethylsulfonyl fluoride, 0.01 mg/mL aprotinin, and 0.01 mg/mL leupeptin. Then, these cells were homogenized using 0.1% of digitonin and centrifuged at 13,000 rpm for 5 min. Supernatants were centrifuged at 13,000 rpm for 30 min to separate the cytosol fraction. The pellet was rinsed with plasma membrane extraction buffer and resuspended with 0.5 % of Triton X-100 in plasma membrane extraction buffer. Lysates were centrifuged at 13,000 rpm for 30 min to obtain supernatants as mitochondria fractions.
2.15. Multi-Caspase Assay
Multi-caspase (caspase-1, -3, -4, -5, -6, -7, -8, and -9) activity was analyzed with a Muse™ Multi-Caspase Kit (MCH100109, Merck Millipore). Briefly, HCC827 (1.95 × 105 cells/well) and HCC827GR (1.8 × 105 cells/well) cells were allowed to adhere for 24 h on 6-well plates. After treatment with LCD for 48 h, cells were harvested and washed with 1X caspase buffer. Then, these cells were incubated with Muse™ Multi-Caspase Reagent working solution at 37 °C for 30 min. After Muse™ Caspase 7-AAD working solution was added, flow cytometry analysis was carried out with a Muse™ Cell Analyzer.
2.16. Statistical Analysis
Statistical significance was evaluated using the software GraphPad Prism statistics (v5, GraphPad Software, USA, RRID: SCR_002798). Differences among multiple groups were tested using one-way or two-way ANOVA followed by Dunnett’s post hoc test. All data are expressed as mean ± standard deviation (SD). Differences were considered significant at p < 0.05.
4. Discussion
Chemotherapy drugs generally include cisplatin and taxanes in the treatment of lung cancer. Unfortunately, many patients acquire resistance to the drug either intrinsically or after medication [
6]. The occurrence of gefitinib resistance is a barrier to have effective clinical therapies for a number of solid tumors, including lung cancer. Various gefitinib resistance mechanisms, including EGFR mutations and
MET amplification, have been reported in several studies [
9,
10]. More than 90% of EGFR mutations occur in exons 19–21, with exon 19 mutations being the most frequent [
9]. The deletion of exon 19 and EGFR mutation of L858R of exon 21 have higher sensitivities than the TKI response in those with wild-type EGFR [
12]. Conversely, resistance to TKI can be induced through the acquisition of secondary mutations of EGFR (T790M, L747S, D761Y, and T854A) or the activation of other signaling bypass pathways [
9]. Resistance obtained after TKI treatment is almost unavoidable, and the success rate of treatment is low. Overcoming these resistances requires a strategy to target molecules related to resistance or in combination with other compounds.
Some clinical studies of EGFR- or MET-targeted inhibitors have been discontinued. However, others have shown encouraging results [
10]. The overexpression of EGFR and MET has been reported in NSCLC, which can activate various downstream signaling molecules involved in cell growth and survival [
8,
13]. Blocking EGFR or MET alone for tumor suppression can lead to cell survival by activating other alternative pathways. Previous reports have shown that treatment with a single inhibitor of EGFR and MET, respectively, has no effect on cell proliferation, although cell survival is suppressed when EGFR and MET inhibitors are combined [
11,
14]. A dual blockade of EGFR and MET can significantly inhibit the proliferation several carcinoma cells, including lung cancer cells [
14,
15], head and neck cancer cells [
16], and colon cancer cells [
17]. Similarly, our data showed that treatment with EGFR and MET alone did not affect cell viability. However, they inhibited cell survival upon combination treatment. LCD inhibited lung cancer cell proliferation through ATP competitive inhibition of EGFR and MET as a single drug. Other studies have reported that inhibitors of EGFR and MET alone could inhibit cell survival, whereas a combination treatment of EGFR and MET inhibitors can significantly inhibit cell survival, showing synergistic effects on the induction of apoptosis [
13].
In lung cancer cells, a signaling network exists between the same RTK family: EGFR, MET, and ERBB3. The results of the present study revealed that the levels of phosphorylated proteins of EGFR, MET, and ERBB3 decreased with an increasing concentration of LCD. Since the amplification of
MET can induce gefitinib resistance by inducing ERBB3-dependent activation [
11], it is thought that LCD targeting MET can also inhibit the expression of ERBB3. EGFR and MET can activate and share important downstream molecules involved in biological activities such as cell growth and survival [
18]. The activation of AKT in lung cancer cells is involved in imparting resistance to TKI as well as cell growth and proliferation [
10]. The reduction of p-AKT caused by the dual targeting of EGFR and MET might be a pathway to overcome resistance by preventing the conversion of compensation pathways. Thus, the dual targeting of EGFR and MET might be a promising therapeutic strategy to overcome lung cancer that is sensitive or resistant to TKI.
One of the meaningful findings in our study was that the mechanism underlying the apoptosis induced by LCD involved the inhibition of EGFR and MET. Apoptotic cell death is induced through two pathways: the extrinsic pathway and the intrinsic pathway. Apoptosis is caused by a variety of extrinsic and intrinsic factors such as ROS, DNA damage, and heat shock [
19]. ROS can regulate physiological functions such as cell proliferation and cell cycle progression. However, excessive ROS levels can cause cell damage and lead to apoptotic cell death [
19]. The exposure of NSCLC cells to LCD significantly increased ROS production, whereas ROS levels were restored to control levels by NAC. The expression of p-EGFR and p-MET, which were inhibited by LCD treatment, showed no significant difference by NAC. These results indicate that ROS is downstream of EGFR and MET, suggesting that ROS might be closely involved in the cell proliferation inhibition of LCD. ROS are known to affect EGFR signaling. ROS can induce TKI resistance by activating the EGFR signaling pathway [
20,
21]. High levels of ROS play an opposite role in tumor progression and drug resistance, leading to cell cycle arrest, apoptosis induction, and toxic effects on cancer cells [
21]. This indicates that an appropriate level of ROS can mediate drug resistance, whereas excessive ROS production can lead to cell death. LCD can induce the apoptosis of lung cancer cells through the accumulation of ROS. ROS is closely associated with the mitochondrial pathway. In fact, the location of most intracellular ROS production is mitochondria [
19]. LCD increased MMP loss and up- or down-regulated mitochondrial-related proteins, leading to apoptotic cell death through mitochondrial (intrinsic) pathways. Apoptosis molecular cascade by LCD led to the release of cyto c into the cytoplasm, the activation of Apaf-1 and caspase, and the cleavage of PARP. It is interesting to note that ROS plays an important role in the apoptosis-inducing mechanism of LCD because PARP, which was reduced by LCD, increased PARP expression by co-treatment with NAC.
ROS is also known to be involved in cell cycle progression, which is regulated by cyclin and cdk [
20]. The link between cell cycle and apoptosis has been demonstrated in many studies [
22]. It regulates cell proliferation. The cyclin B1/cdc2 complex is involved in the G2/M phase transition, and cdk activity is mediated by cdk inhibitors (CKI) such as p21 and p27 [
22,
23]. LCD decreased cyclin B1/cdc2 protein expression but increased CKI, causing G2/M cell cycle arrest. LCA and LCB of the same licorice family as LCD can also block G2/M cell cycle progression and induce the apoptosis of lung cancer cells [
14,
24]. Some agents that can induce apoptosis may also increase cytotoxicity in association with G2/M checkpoint arrest [
23].
Importantly, LCD shows anti-tumor activity without causing weight loss in vivo using a xenograft model of oral cancer cells [
4]. In addition, LCD (1 μg/ml) shows myocardial protective effects in the cardiac tissues of injured rats [
25]. These findings indicate that LCD is low in toxicity. In addition, while common flavonoids generally have low bioavailability in humans, LCs have been shown to be well absorbed through passive diffusion through the Caco-2 cell monolayer, which is a human intestinal cell line [
26]. Since intestinal permeability is an important factor influencing the bioavailability of drugs, LCs can be expected to have high bioavailability in the human body. Liquiritin, another component of licorice, enhanced the cell proliferation inhibitory effect of cisplatin in gastric cancer cells resistant to cisplatin, and showed synergistic effects on tumor growth inhibition in vivo [
27]. In the future, we will evaluate the anti-tumor efficacy of lung cancer cells with different resistance mechanisms for the combination of anti-cancer drugs and LCD.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}