1. Introduction
Plant-biomass is a major source of renewable energy in the form of biofuels and bio-produced chemicals, but the recalcitrance of lignocellulose is a major obstacle to cost-effective saccharification. Fungal copper-dependent lytic polysaccharide monooxygenases (LPMOs, EC 1.14.99.53, - 56) have been found to boost the overall effectiveness of lignocellulolytic secretomes in the decomposition of insoluble recalcitrant polysaccharide structures [
1]. Initially described in bacteria in 2010 [
2], LPMOs have since been identified in fungi and insects and have been classified by the curators of the carbohydrate-active enzyme (CAZy) database (
www.cazy.org) into several auxiliary activity (AA) families [
3]. Fungal LPMOs have so far been classified as AA9 (activity on soluble and insoluble beta-glucans), AA11 (chitin-active), AA13 (starch-active) [
4], AA14 (xylan-active) [
5], AA15, and AA16 [
6]. LPMOs differ from common endo- and exo-glycosyl hydrolases by employing an oxidative mechanism to cleave glycosidic bonds in polysaccharides, such as cellulose, hemicellulose, starch or chitin, to produce access points for exo-acting hydrolases. After prolonged incubation, they also release soluble oligosaccharide products [
7,
8,
9,
10]. The ability to cleave polymers is enabled by a characteristic, flat binding-site consisting of aromatic and hydrophilic amino acids for the interaction with sugar moieties. An exposed active-site copper is held in place by a conserved, “histidine brace” motif [
11,
12,
13]. These structural features enable LPMOs to attack polysaccharide surfaces inaccessible to hydrolases [
13,
14].
The catalytic mechanism of LPMOs is still under investigation, but several cornerstones of LPMO catalysis have been elucidated. The first step in the catalytic cycle is the reduction of the Cu(II) in the catalytic site to Cu(I), either by small molecular reductants [
15,
16,
17], such as ascorbic acid (Asc) or gallic acid or by the fungal redox enzyme cellobiose dehydrogenase (CDH) [
18]. CDH most likely requires the dissociation of LPMO from the substrate in order to contact the otherwise buried copper center of LPMO [
19]. The reduction of the copper ion is also accompanied by a conformational change in LPMO, as was observed by using NMR [
20] or electronic circular dichroism [
21], and it also increases the affinity to its carbohydrate substrate [
21,
22]. As co-substrate for the LPMO reaction, either O
2 [
23] or H
2O
2 [
24] have been suggested. Whether the binding and reduction in an oxygen species happen preferentially in the free or substrate-bound state is not settled, but a recent publication suggests that hydrogen peroxide can access the active side of substrate-bound LPMO [
25]. The catalytically competent LPMO abstracts hydrogens from its polysaccharide substrate and thereby breaks the glycosidic bond [
23,
26,
27]. Oxidation on both C1 and C4 positions of the polysaccharide substrate was reported, with different resulting products. C1 oxidation leads to the formation of a lactone which spontaneously hydrolyzes into gluconic acid, while C4 oxidation forms a C4 ketone that hydrolyzes into a gemdiol in aqueous solution [
7,
27,
28]. Several studies have shown that H
2O
2 increased the reaction rate relative to O
2 by an order of magnitude [
24,
29,
30] while the measured affinity of LPMO for H
2O
2 was in the micromolar range [
24,
29]. The proposed reaction mechanism based on H
2O
2 as cosubstrate would also require only one electron to initiate the catalytic reaction, which aligns well with CDH’s capability of transferring single electrons via its cytochrome domain. Several studies reported that the presence of H
2O
2-scavengers, such as peroxidase or catalase, inhibited the LPMO reaction, which implies that H
2O
2 is required for the LPMO reaction [
21,
24,
30]. By providing a mix of different H
2O
2 and O
2 isotopes, Bissaro et al. showed that the oxygen atom inserted into the LPMO reaction products originated from the H
2O
2 [
24]. A recent paper showed that potentially both O
2 and H
2O
2 can serve as co-substrates for LPMO, resulting in polysaccharide oxidative cleavage, but concluded that different molecular mechanisms are probably employed [
30].
LPMOs were reported to be notoriously unstable under non-optimal reaction conditions. Especially, the observed higher catalytic rates in the presence of H
2O
2 were accompanied by a rapid inactivation of LPMO [
30]. Employing LPMO in a commercial cellulase cocktail using industrially relevant substrates, therefore, requires fine-tuning of the reaction rate by controlling the H
2O
2 supply [
31]. The observed instability was mainly attributed to autoxidative damage caused by oxygen radicals released from the active site, which lead to the oxidation of amino acids surrounding and forming the active site of LPMO [
21,
24,
32]. The apparent T
m of the fungal LPMO9C from
Neurospora crassa decreased from 61.5 ± 0.4 °C to 48.8 ± 1.1 °C when the active site copper was reduced by Asc. Under these conditions, H
2O
2 accumulates via the oxidation of O
2 by Asc [
33,
34]. In addition, the reduced LPMO may also generate low amounts of H
2O
2 through an uncoupling reaction [
35]. In the presence of suitable substrates, however, the apparent T
m of
NcLPMO9C remained relatively unchanged at 60.4 ± 0.5 °C [
21]. The stabilization of reduced LPMO by the substrate can be explained by the prevention of freely diffusing oxygen radicals due to the catalytic reaction [
36], which may also consume the generated H
2O
2.
In addition to autooxidative damage, another form of destabilization was observed upon removal of the active site copper, e.g., by incubation of LPMO with an excess of the metal chelator - ethylenediaminetetraacetic acid (EDTA) [
21,
37]. Several studies showed reduced temperature stability of copper-depleted LPMOs [
21,
38]. For example, removal of copper reduced the apparent T
m of
NcLPMO9C from 61.5 to 52.7 °C [
21], which indicates that the histidine brace motif and the presence of the copper ion stabilize the overall protein fold. The thermal stability of the apoenzyme was unaffected by the presence of reducing agents and substrates, showing that an intact active site is required for substrate recognition and catalysis. While it is possible that the loss of the active-site copper ion contributes to the initial LPMO destabilization following active-site reduction and subsequent oxidation, it was demonstrated that the vast majority of LPMO molecules retained the copper ion upon unfolding.
In this work, we study the destabilizing effect of reducing agent and copper ion removal on NcLPMO9C in temporal and structural detail by hydrogen/deuterium exchange mass spectrometry (HDX-MS). We also investigate the stabilizing effect of carbohydrate substrates during catalysis. The employed HDX-MS methods are well suited for the detection of structural changes involving the rearrangement of hydrogen bonds and changes in solvent accessibility in proteins, and they allow studying heterogeneous reactions with insoluble components, as long as they can be quickly removed before MS analysis. This is crucial for studying LPMOs, as their typical natural substrates, e.g., cellulose, are inherently insoluble. We observed a significant increase in peptide solvent accessibility throughout the LPMO molecule when incubated with a reducing agent, starting at peptides close to the copper ion active site and then propagating further. This effect was found to be slowed down in the presence of cellulose. Removal of the active site copper ion caused a temperature-induced unfolding beginning at lower temperatures, which affected the histidine brace peptides first and then propagated to the rest of the molecule upon longer incubation times.
Thus far, amino acid modifications resulting from autooxidative damage of LPMOs have only been reported in bacterial LPMOs [
24]. We, therefore, aimed to verify that the same modifications occur in fungal LPMOs. Using LC-MS/MS analysis, we show that various oxidative alterations of peptides located around the copper active site occurred in a fungal LPMO. Oxidative peptide bond cleavages and, thus, direct degradation of the protein was detected for the first time in a fungal LPMO. The peptide signal intensity observed in our HDX-MS experiments provides a measure of the given peptide abundance. We observed a decrease in the intensity for unmodified peptides located in the vicinity of the active-site, indicating their cleavage or modification.
3. Results
A recent report from Kracher et al. described the interaction of the fungal
NcLPMO9C with various substrates, the enhancement of substrate binding upon active-site copper reduction, and the protective/stabilizing role of substrates on LPMO under reducing conditions. Spectroscopic techniques, such as circular dichroism, indicated conformational changes in the LPMO upon active-site copper reduction and copper depletion but did not provide insight into the structural details of the various
NcLPMO9C states [
38,
45]. In order to gain accurate structurally resolved answers underlying these phenomena, we performed a systematic hydrogen/deuterium (H/D) exchange study, which allowed us to extend the previous conclusions.
In the first step, we optimized the HDX-MS workflow, which included tuning of digestion conditions and rapid separation of proteins from the insoluble substrate during the post-labeling step. In our previous studies, which focused on cellobiose dehydrogenase, an enzyme closely cooperating with LPMO during the cellulolytic activity, we showed that strong reducing and denaturing conditions are required for efficient digestion. As these enzymes are adapted to a harsh extracellular environment, it is not surprising that LPMO digestion required denaturation. However, in contrast to the previously used guanidine hydrochloride, we switched to urea, which provided similar sequence coverage but avoided adverse effects on LC analysis. In addition, from the studies published so far, it is evident that in contrast to guanidine hydrochloride, even a high concentration of urea has much less deleterious or even enhancing effects on the protease activity [
46,
47,
48]. We also tested various proteolytic setups (protease columns, flow rates) with serial coupling of nepenthesin-1 and rhizopuspepsin columns operated at 200 µL min
−1, providing the best sequence coverage and spatial resolution. Finally, we had to cope with the heterogeneous, insoluble substrate (microcrystalline cellulose) present in the sample. We found the use of 0.45 µm spin filters as an optimal solution, adding approximately 50 s to the sample processing time while ensuring complete removal of insoluble matter. The harsh denaturing conditions also prevented possible adherence of LPMO to the substrate via its interaction surface and/or carbohydrate-binding module (CBM). The additional post-quench time of 90 s was also required to achieve the reduction in the disulfide bonds in
NcLPMO9C. We observed that raising the TCEP concentration or prolonging the incubation time did not lead to more efficient digestion and, therefore, we used the lowest possible TCEP concentration and reduction time. As a result, we fully covered the catalytic domain (except for residues 41–44) and the CBM. The linker peptide connecting the catalytic domain to the CBM (residues 225–309) could not be resolved due to its high and heterogeneous O-glycosylation (
Figure S1).
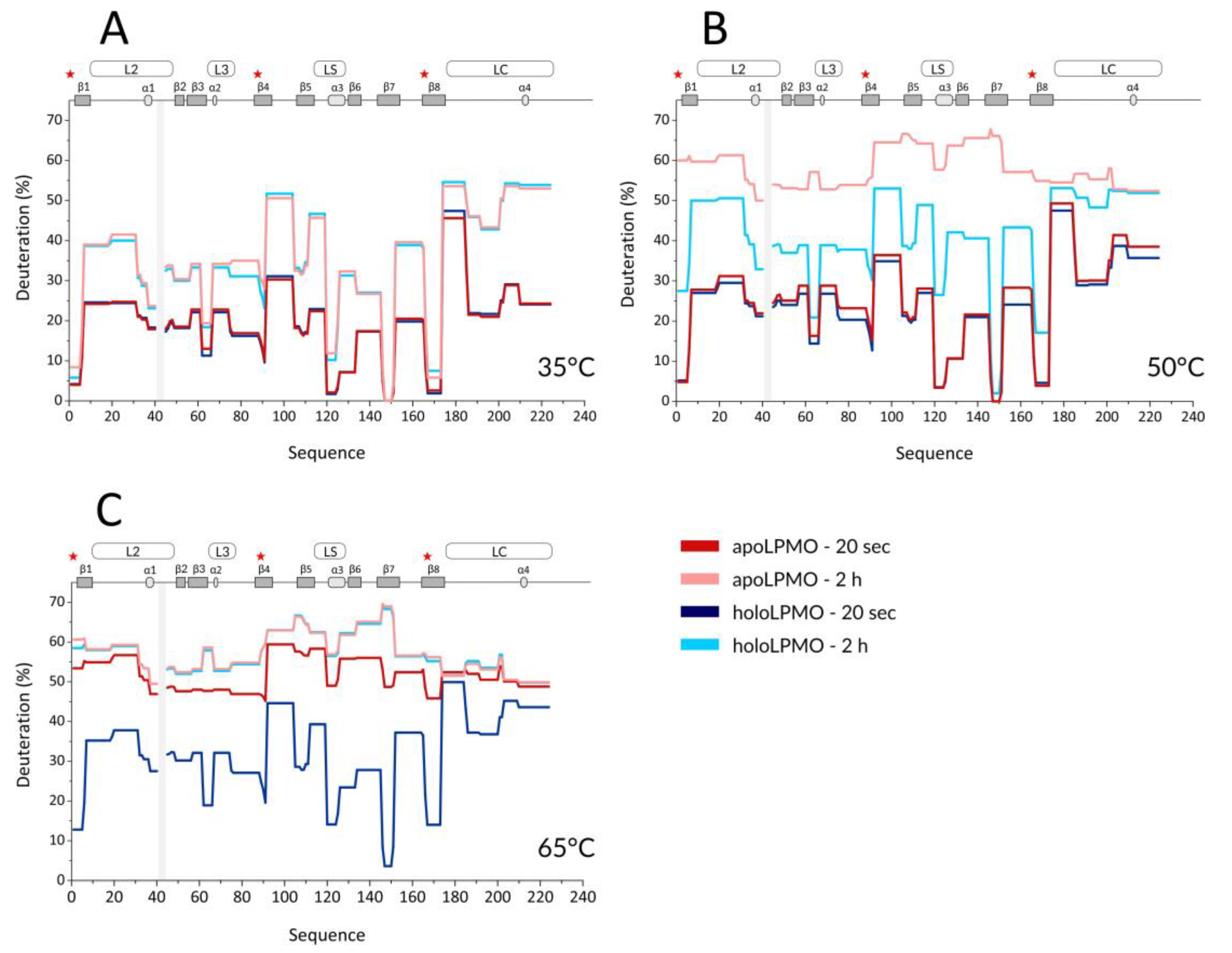
With the optimized workflow, we performed an initial set of HDX-MS measurements in which we focused on several factors. First, building upon the data from Kracher et al., we looked at the effect of high temperature on the free, oxidized LPMO and its apo form prepared by EDTA treatment. Here, the exchange was followed at 35, 50, and 65 °C, and the HDX kinetics covered time points at 0.33, 2, 20, 120, and 360 min. A summary of selected HDX data is provided in the protection plots in
Figure 1. While this presentation focuses on the catalytic domain and representative exchange times only, we provide full data covering the whole LPMO sequence, including the CBM and all exchange times in the form of mirror plots (
Figure S2) [
43]. Peptides in mirror plots are represented on the
X-axis by their “midpoint” value, which is calculated as an average value between the N- and C- terminal residue sequence position of a given peptide and thus allows for easy and reproducible sorting of the overlapping peptides generated and analyzed in the HDX-MS experiment.
Since we observed no difference induced by cofactor absence or active site copper reduction at the CBM (
Figure S2) and we missed a large part of the linker, we avoided these structural features in
Figure 1 and
Figure 2. It should be stressed that the intrinsic exchange rate was influenced by the temperature and, thus, the data acquired at different temperatures are not directly comparable. We, therefore, plotted these data separately (
Figure 1A–C). However, normalization using correction factors calculated based on the Arrhenius equation can be applied to compensate for the different exchange rates. The exchange rate at 50 °C was multiplied by a factor of 3.628 and those at 65 °C by 11.739, which led to normalization to the lowest temperature of the dataset (35 °C). How this affects the data interpretation is shown in several selected deuterium uptake curves (
Figure S3). From these graphs, it can be inferred that the deuterium uptake curves of the LPMO holoenzyme obtained at 35 °C and at 50 °C were either fully or partially overlapping for the whole protein and differences between these two temperatures could be seen only after longer exchange times ( > 20 min). The main differences were observed in peptides covering the histidine brace residues (His1, His88, and Tyr166) or the closely neighboring regions. This shows that even at 50 °C, the structural integrity of
NcLPMO9C was well preserved, and only weak destabilization occurred around the active site. In contrast, at 65 °C, the majority of the uptake curves had a much steeper slope, reaching equilibrium deuteration already after 2 min of exchange, except for unstructured and freely accessible regions (regions/peptides 92–104, 174–185, 201–224. and the CBM: 310–343). These observations clearly show that structural destabilization at 65 °C was strongly accelerated when compared with the lower temperatures. These trends were further amplified (
Figure S2) when the copper was removed from the active site using EDTA treatment. When incubating the apoprotein at 35 °C, the LPMO was slightly perturbed around the active site as the deuteration increase could be observed for the peptides covering the copper-binding residues (
Figure 1A and
Figure S3). Interestingly, virtually no difference was detected for the region around Tyr166 (see peptides 152–165 and 166–173 in
Figure S3—black and grey curves). The importance of the copper ion on the overall structure stabilization is demonstrated by a significant structure opening at elevated temperatures. At 50 °C, the apo-protein was much more accessible for deuteration (
Figure 1B and
Figure S3), and this was further enhanced at the highest temperature monitored (65 °C) where the protein was readily deuterated even after 20 s of deuteration (
Figure 1C,
Figures S2C and S3). To position these effects onto the structure, we colored the LPMO structure using the difference in HDX between the apo- and holoenzyme (
Figure S4), which illustrates the destabilization of the protein core upon copper ion removal.
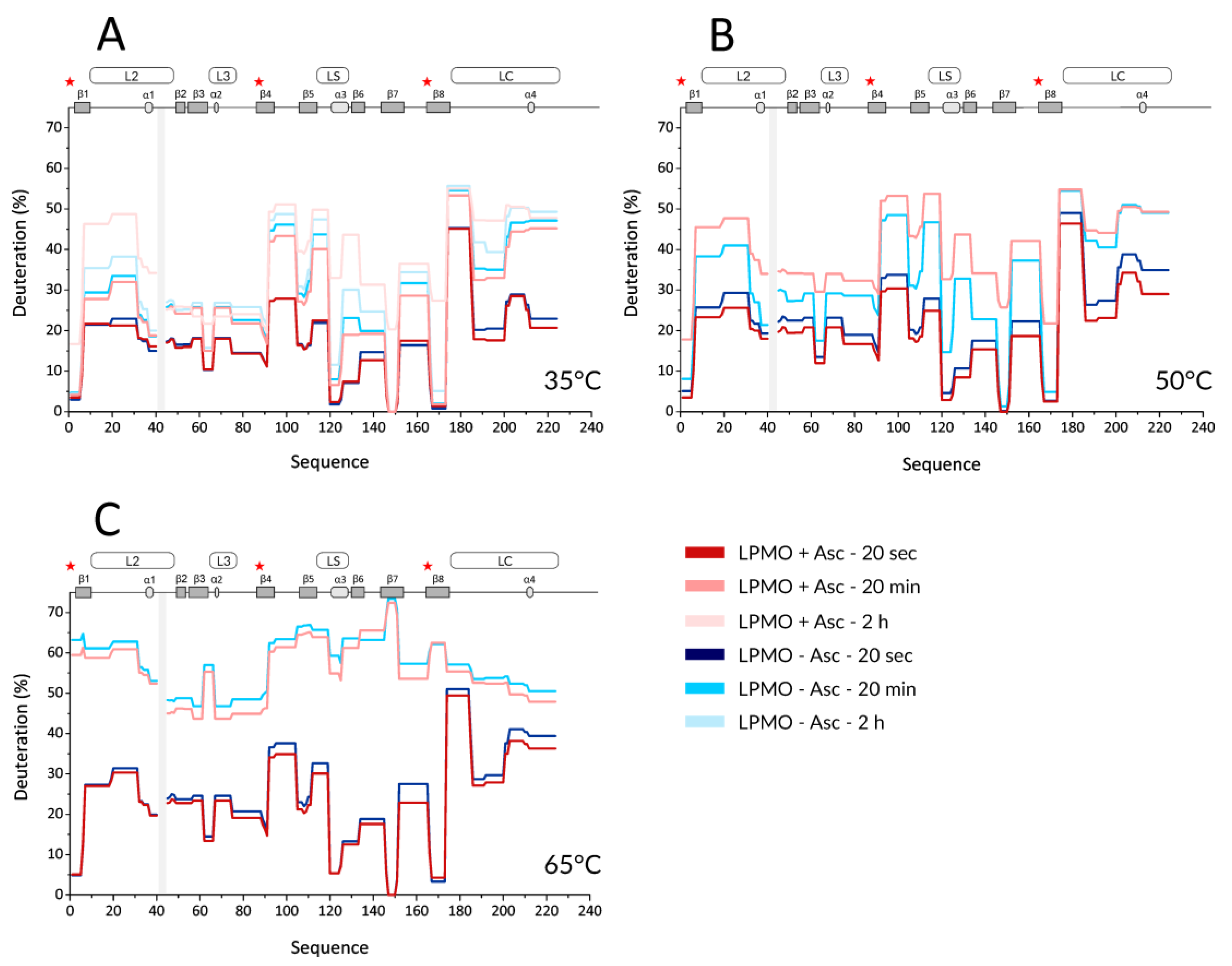
Next, we performed HDX experiments targeting effects induced by ascorbic acid (Asc). To allow for time-resolved analysis of the effect of LPMO reduction, we added Asc at the same time when the deuteration was started. To be able to compare the results to holo- and apo-form experiments, the reduction by Asc was also followed at three different temperatures (35, 50, and 65 °C). Key data are shown in the form of protection plots in
Figure 2, while the whole dataset was visualized using mirror plots (
Figure S5). The most obvious change observed at 35 °C was a generally higher deuteration of the reduced LPMO at longer incubation times (> 2 h,
Figure 2A—the lightest colors). This was observed for peptides covering β1 (including the N-terminus with His1), loop L2, β2, β3-α2 linker (in L3), β5, α3 in LS, β6, β7, β8 (including Tyr166), and the middle part of loop LC. Interestingly, no such effects were detected for peptides covering the third His brace residue, His83. The protection plots showed that the peptides covering His83 were slightly less deuterated in the presence of Asc and at longer incubation times. Besides the above-described effects detected at longer time scales (> 2 h), there were also indications of lower deuteration of the reduced enzyme at earlier time points (
Figure 2A-light and dark colors). However, these differences were small and cannot be considered as significant without further supporting observations. Such additional validation was obtained through inspection of data acquired at 50 °C (
Figure 2B and
Figure S5B). Here, the lower deuteration at 20 s and 2 min in the presence of Asc was more obvious and manifested through the peptides covering the His brace residues, including His83. Lower deuteration was also observed in neighboring peptides (e.g., L2, LS with α3, N-term part of β6, β7) or parts distant from the active site (e.g., β2, β4- β5, N-term part of LC) of
NcLPMO9C. On the other hand, the destabilizing effect of Asc was observed as a much higher deuteration occurring already after 20 min of exchange/reduction. Intriguingly, we observed a significant decrease in deuteration at longer incubation times beyond 2 h, which was specific for certain peptides, including the His brace residues (
Figures S5 and S6A).
We observed such unusual effects if protein degradation or precipitation occurred during the H/D experiment. Indeed, an inspection of the summed up peak intensity plots of the individual peptides (alternative data visualization in DeutEx,
Figure S6B) showed a signal decrease that correlated with the trend of deuteration decrease. Hence, we conclude that at 50 °C and in the presence of Asc, LPMO is likely oxidized and/or degraded, which leads to the signal loss. At 65 °C, the effects induced by copper ion reduction were fading, and the only prominent event we observed was degradation after longer reduction/deuteration times (
Figure 2C and
Figure S5C). This is likely due to the destabilization of LPMO at this temperature, which we also detected for oxidized LPMO. However, considering that we still detected signal loss on peptides close to the active site even at 65 °C (see
Figure S5C), we speculate that even at this temperature, the copper ion could remain bound to the His brace and the enzyme at least partially exerts its activity. To put the Asc-induced effect into a structural perspective, the difference in deuteration between reduced and oxidized
NcLPMO9C was plotted onto the structure (
Figure S7).
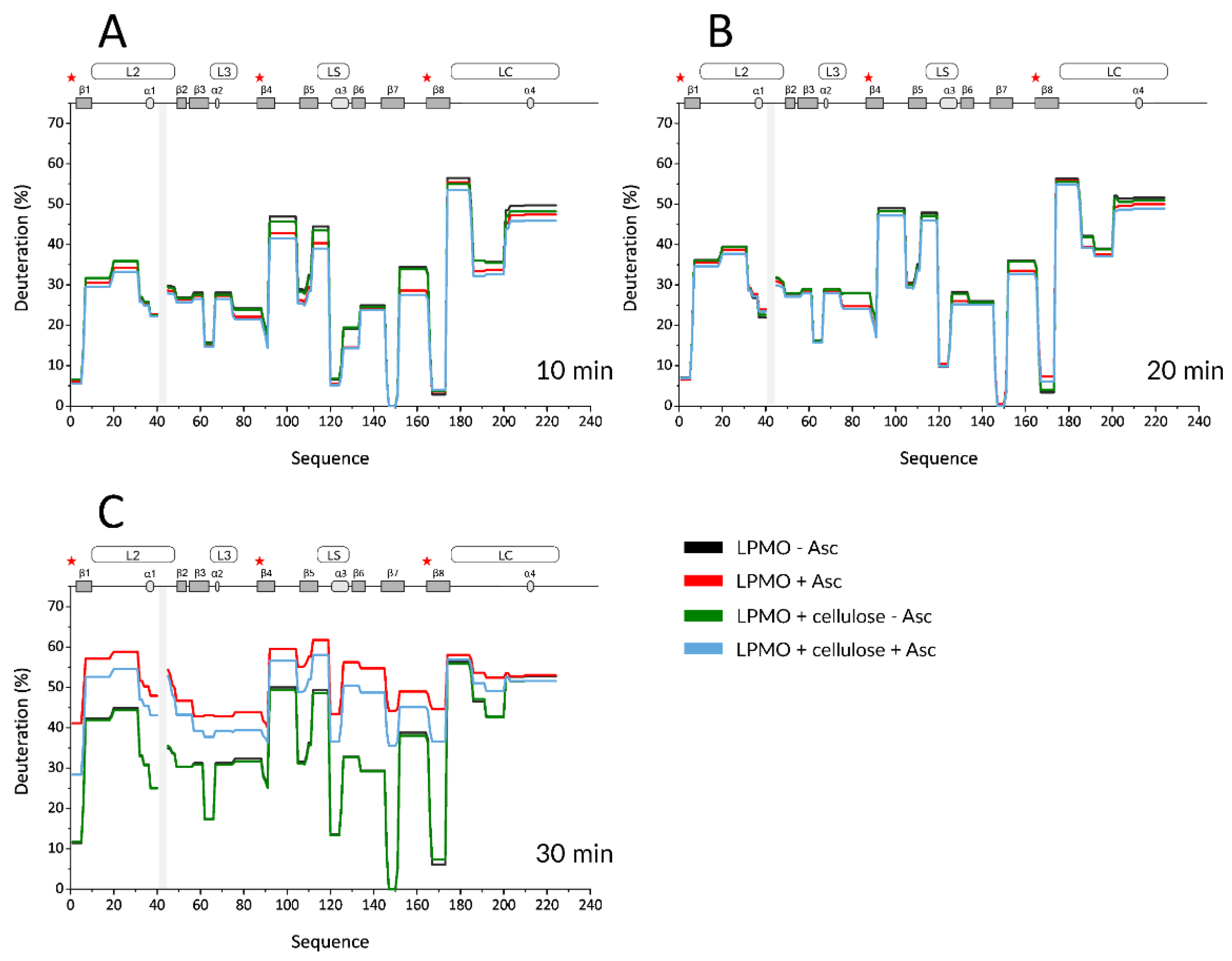
Based on these data, we then set up the final experiment. A temperature of 50 °C was chosen, as the kinetics of H/D changes were slow at 35 °C and, on the other hand, the protein was significantly perturbed at 65 °C. In addition, given the autooxidative damage detected in the experiments with Asc, the maximum exchange time was reduced to 45 min. We increased the temporal resolution of these experiments by collecting a higher number of samples and focused on the substrate binding and its protective role on the LPMO. The experimental conditions included oxidized and Asc-reduced LPMO, either alone in solution or mixed with microcrystalline cellulose. Representative HDX data are shown in the form of protection plots in
Figure 3, and the whole dataset is shown as mirror plots in
Figure S8. The profile of the oxidized LPMO was virtually indistinguishable from that obtained in the presence of cellulose, which implies that there is no tight binding that would alter the H/D exchange of the protein (
Figure 3: black vs. green and
Figure S8A). Similar to previous experiments, reduction by Asc led to two distinct effects (
Figure 3—black vs. red and
Figure S8B).
First, lower deuteration was detected for reduced LPMO on the peptides covering the His brace and the neighboring regions, mainly L2, L3, and its extension to β4, LS, and LC. This effect lasted until 10–15 min of deuteration. Second, the difference in deuteration between oxidized and reduced LPMO vanished after 15–20 min, and the reduced LPMO became more deuterated at later time points. This was likely linked to the oxidation-induced LPMO damage, which, after 35 min, also led to an apparent decrease in the deuteration of the His brace-covering peptides. Finally, the trends observed for free, reduced LPMO were also observed when cellulose was added.
Although it was shown that LPMO binds to microcrystalline cellulose under reducing conditions, albeit weakly compared to more amorphous phosphoric acid swollen cellulose (PASC) [
21], we observed no decrease in deuteration in the first data points due to the presence of cellulose. However, the transition point at which the reduced protein switched from a more protected to a more deuterated state was shifted, indicating that oxidation and degradation are slowed down due to the protective role of the cellulose substrate. The last data points of the kinetics showed a deuteration decrease (described above for free reduced LPMO), which can be considered as an indicator of protein degradation. This effect was observed for both the free and cellulose-bound and Asc-reduced LPMO, which shows that the autooxidative damage at a certain point overrode the protective effect of the cellulose. We also observed protection from exchange on the CBM upon cellulose addition. This effect is not affected by the presence of Asc and justifies the binding of LPMO to the substrate. However, the extent of this protection was very small and at the border of significance. All these effects were visualized on the LPMO structure (
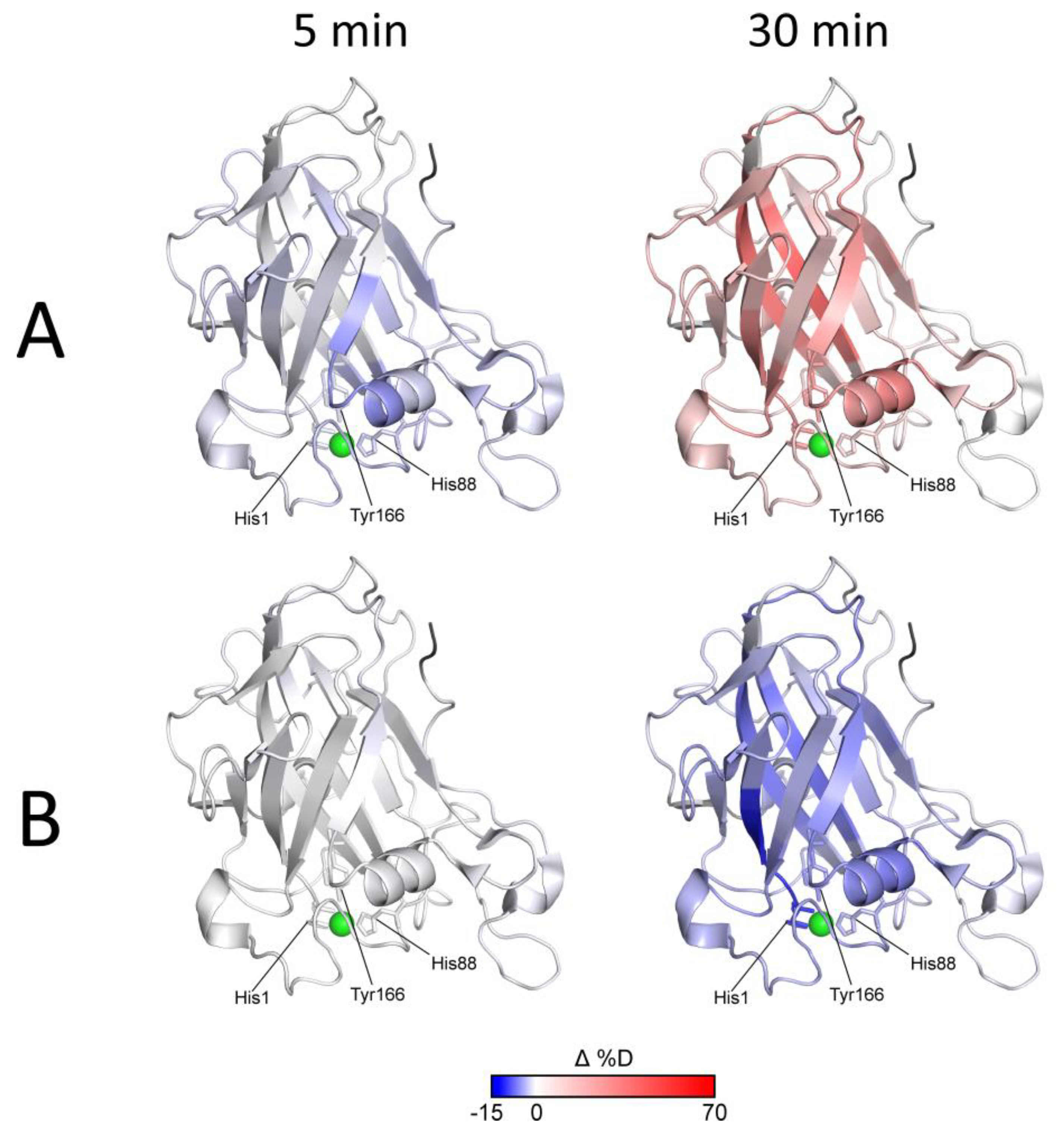
Figure 4—selected time points and
Figure S9—whole dataset, excluding oxidized LPMO conditions where no difference was detected).
The effects of Asc reduction (protection/lower deuteration) and subsequent autooxidative damage to LPMO (higher deuteration) are evident from the blue-to-red transition shown in
Figure 4A and
Figure S9A.
Figure 4B and
Figure S9B show that the expected tighter LPMO binding to cellulose did not cause detectable alterations in the HDX kinetics and that only protection from autooxidative damage could be captured using HDX-MS. It should be noted that this protective effect aligned well with the timing of increased oxidative damage (detectable after 15 min of exchange/reduction) and was diminished after 30 min when oxidative damage prevailed. Oxidation and/or degradation and the protective effect of the cellulose were also supported by the plots following signal intensity throughout the experiment. These are shown in
Figure S10 together with the respective deuterium uptake plots for the peptides covering the His brace and several representative parts of the protein structure. It is evident that in cases where the deuteration drop occurred during the last time points, the signal intensity also decreased (
Figure S10A,B). The protective effect of the cellulose substrate under reducing conditions then manifested as slower deuteration, and it also slowed-down the degradation. The structural and spatial dependences of these effects could be deduced from the localization of the peptides on the
NcLPMO9C structure. Here, it is evident that the proximity of peptides to the catalytic center was the major factor behind the signal loss (amino acid oxidation extent,
Figure S10C).
In order to verify that the mechanism underlying signal loss and deuteration decrease is indeed protein oxidation caused by reactive oxygen species generated by the reduced LPMO, we searched for oxidized versions of the peptides detected in HDX-MS. However, this approach was largely unsuccessful, with only a few significant hits. This can be demonstrated by peptide 1–6 (
Figure S11) for which the singly oxidized version (oxidation of His1, part of the His brace) was found. The deuterium uptake (
Figure S11A) had a different shape when compared with the unmodified peptide and increased rapidly starting from the early time-points on. In addition, the time-dependent signal intensity (
Figure S11B) showed the opposite trend. While the unmodified peptide vanished, the intensity of the oxidized peptide increased, showing that the oxidation was ongoing throughout the experiment.
A detailed description of oxidative modifications in LPMO was done using a classical proteomic bottom-up approach based on the digestion of the protein with specific proteases.
NcLPMO9C was subjected to reduction using Asc, and protein samples were taken at different incubation times and digested using Trypsin or Asp-N proteases. The breakdown products were analyzed by LC-MS/MS, followed by a database search and manual validation. Due to the sheer amount of variability in oxidative modifications present, we only focused on the most abundant peptide forms, which yielded intense fragment spectra of sufficient quality to verify the modification type and residue position. An overview of the identified oxidative modifications is provided in
Table S1. The analysis confirmed that similar to a previous report on
ScLPMOC [
24],
NcLPMO9C underwent extensive autocatalyzed oxidation in the presence of Asc. When searching our dataset, we found not only known oxidative modifications of amino acid side chains but also generally overlooked peptide bond cleavages [
49,
50]. These peptide bond cleavages result in either +25,980 Da or -1,030 Da mass changes in the N-terminus of resulting shorter cleaved peptides and -30,010 Da or -0,985 Da mass change in the C-terminus, depending on the cleavage mechanism. An example shown in
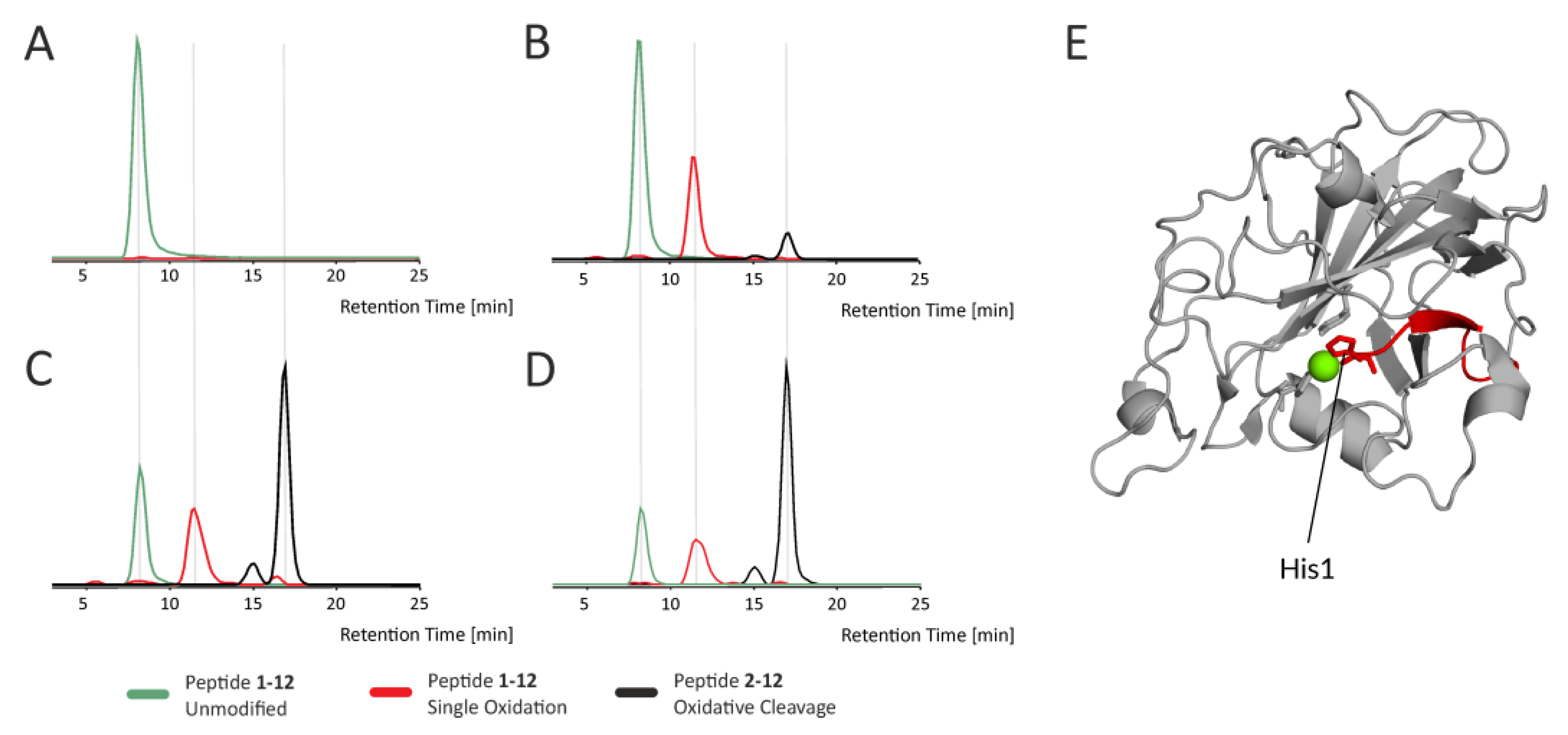
Figure 5 shows the extracted ion chromatograms of various forms of the N-terminal peptide bearing His1 (part of His brace), which belongs to a group of rapidly oxidized regions undergoing further oxidation-induced events, such as oxidative cleavage.
Generally, the extent and kinetics of oxidation depended on the proximity of the peptide to the catalytic center and on the amino-acid composition of the peptide, as shown in
Figure S12. It is apparent from both
Figures S10 and S12 that residues close to the active site were oxidized faster than those located on the opposite side of the protein.
4. Discussion
Previous reports described thermal aspects of LPMO stability in relation to its cofactor and substrates [
21,
45]. Other publications focused on the structural differences between the apo- and the holoenzyme and substrate binding effects. These employed protein X-ray crystallography or NMR providing excellent spatial resolution and detailed answers about key amino acids and their side chains involved in these interactions. Here, we employed hydrogen/deuterium exchange, which is a lower-resolution structural biology method but offers a more detailed view on the dynamics of thermal- and ROS-induced destabilization of proteins. It also complements the crystallography and NMR data by allowing the monitoring of different conditions not easily amenable by either of the two classical techniques. We also employed hydrogen/deuterium exchange to follow the deactivation of reduced LPMO in a time-dependent manner. Thereby, we shed light on the nature of the destabilization, which is important in the context of effective industrial utilization of LPMOs.
Our first experiment aimed to elucidate the structural differences between the apo- and holo-forms of
NcLPMO9C and their stability at different temperatures. A previous NMR study followed the titration of the apoLPMO by Cu
2+ and provided evidence about changes mainly at the copper coordinating residues and their surroundings [
20]. Other studies based on thermal unfolding assays then showed that the absence of the copper ion results in structural unfolding occurring at lower temperatures when compared to the holo form [
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38]. Using HDX-MS, we further extended these findings by providing spatially resolved data on
NcLPMO9C. We selected three temperatures representing important points on the
NcLPMO9C unfolding pathway. At 35 °C, a very mild structure perturbation was detected (
Figure 1A and
Figure S4A). Slightly higher deuteration of the apo form was detected for peptides covering His1, His83, and Tyr166 after 2 h of incubation. Kinetics of deuteration varied between the individual amino acids forming the His brace. Changes in His83 manifested mostly between 2 and 20 min of deuteration, while changes in His1 and Tyr166 were observed only at longer deuteration times between 2 and 6 h (
Figure S3). This is likely related to the location of these residues in the protein structure. His1 and Tyr166 are both linked to the core beta-sandwich, which could result in slower deuteration despite the fact that both amino acids are solvent-exposed. In contrast, His83 is positioned in the loop L3 connecting helices 3 and 4 and is thus less protected from an exchange. At 50 °C, the crucial role of the copper ion in the stabilization of the overall protein fold manifested (
Figure 1B and
Figure S4B). Detected deuteration differences were similar to those at 35 °C, albeit much more pronounced, mainly in regions covering the histidine brace residues (especially His1 and Tyr166) but also for regions 61–66, 120–125, and 146–151. These segments are either forming the beta-sandwich core (146–151) or are positioned close to the catalytic center (61–66, 120–125). Interestingly, the region 105–111, which is distant from the catalytic center, showed a high degree of perturbation as well, which suggests the overall “disassembly” of the LPMO core upon removal of the copper ion. Finally, we observed that the highest employed temperature of 65 °C leads to very rapid deuteration of both LPMO states with slower kinetics present in the holo-form (
Figure 1C and
Figure S3). This indicates that the copper ion was probably still bound to the enzyme, as was previously observed for fungal and bacterial LPMOs [
21,
38].
The function of LPMOs depends on the reduction state of their active-site metal ion. The reduction and subsequent catalytic activity, however, leads to rapid inactivation of the enzyme in the absence of suitable substrates. This inactivation was attributed to the formation of oxygen radicals and subsequent oxidative modifications of amino acid residues lining the active site [
19,
22,
28]. Here, the accompanying structural changes were investigated through another set of HDX-MS experiments, and the oxidation pathways were further explored by LC-MS/MS analyses. Among the three temperatures followed in this experiment, 50 °C was the most informative temperature since the differences at 35 °C were only observable after long incubation times and both states (oxidized and reduced) were highly perturbed at a higher temperature of 65 °C (
Figure 2 and
Figure S5). Two types of changes in the Asc-reduced LPMO structure could be observed at 50 °C (
Figure 2 and
Figure S6). At short incubation times, we observed a low deuteration of peptides covering the histidine brace motif and of some of the loops surrounding the active site (
Figure S7). This change in structure could be responsible for the increased affinity of the reduced LPMO to the polysaccharide substrate [
21]. Similar loops were also found to be responsible for substrate binding of a bacterial LPMO [
20] and could correspond to structural changes observed in
NcLPMO9C using circular dichroism [
21]. A second important observation was an extensive increase in deuteration of reduced
NcLPMO9C at longer incubation times. Deuteration started on peptides bearing the histidine brace residues and propagated to the rest of the molecule during the incubation time. At a temperature of 50 °C, even the innermost parts of the protein were highly deuterated after 30 min, clearly showing enzyme unfolding and probably partial protein degradation. Therefore, we concluded that the optimal conditions to follow the Asc-induced reduction and LPMO substrate binding are best represented by 50 °C and shorter incubation/deuteration times, likely not exceeding 1 hr.
Based on these findings, we set up the final experiment in which the protein was followed in its oxidized and reduced state either in solution or bound to a substrate. Finer sampling clearly supported the conclusions of the previous experiment, which showed that Asc reduction (
Figure 4A,
Figures S9A and S10) caused a lower deuteration at shorter incubation times, transitioning towards increased deuteration at longer incubation times. Finally, we observed signal loss events in amino acids localized mostly around the histidine brace. Inclusion of microcrystalline cellulose as a substrate showed no deuteration effects under oxidizing conditions, which agrees well with the previous findings [
21]. However, when the cellulose was added to the reduced LPMO, a protective effect was detected via slower deuteration. As can be seen in
Figure 3C,
Figures S9A and S10, the substrate failed to stop the destructive pathways at some point (time > 35min), and the oxidation-driven LPMO damage overrode the protective abilities of the cellulose. This can likely be attributed to the weaker binding of
NcLPMO9C to crystalline cellulose when compared to amorphous substrates, such as PASC [
21]. However, it should be noted that our initial trials with PASC provided poorly reproducible results (data not shown) and, thus, we switched to crystalline cellulose. Interestingly, we failed to observe any further decrease in deuteration induced by substrate binding. This can be again attributed to the weaker binding of the LPMO to microcrystalline cellulose. However, another and more likely reason might be the inability of the H/D exchange approach to capture these interactions since they are side chain mediated, and the HDX targets back-bone amides only [
51,
52]. The same reasoning could explain the negligible effect of crystalline cellulose on CBM. It is known from the literature that CBM binding to cellulose surface is mediated via side chains of aromatic residues [
53,
54]. Since the overall fold of CBM is highly compact, it is not likely that side-chain mediated cellulose-binding may induce significant changes in CBM hydrogen bonding or solvent protection detectable by HDX-MS. Hence, while we repeatedly detected Asc-independent protection of the CBM in the presence of cellulose, the effect was at the level of insignificance. Nevertheless, our experiments clearly demonstrate that the substrate protects the enzyme from degradation and that this protective effect correlates well with the mechanism through which the enzyme is structurally perturbed in its free, Asc-reduced form (compare
Figure S9A,B). This protection can be due to either scavenging of the reactive species, LPMO reoxidation, or active-site protection due to substrate binding.
An additional effect we observed throughout the experiments with Asc-reduced LPMO was a time-dependent decrease in signal intensity for peptides around the active site or nearby loops. Loss of peptide signal intensity can result from the modification of some of its amino acids, leading to mass shifts and, thus, to the vanishing of the peptide from HDX-MS analysis. Such effects were used as an indirect measure of oxidative peptide modification by Loose at al. [
34]. In addition to the signal intensity loss, we here attempted to identify increases in signal intensity of oxidatively modified peptides. This approach was largely unsuccessful with the reported example (
Figure S11) being the only unambiguous one. It should be stressed that looking for oxidatively modified peptides in HDX-MS data is not ideal for several reasons. First, the nonspecific proteases (here a combination of nepenthesin I and rhizopuspepsin) create many overlapping peptides that cause signal intensity splitting. Second, oxidative modifications are often heterogeneous and create a variety of subsequent reaction products adding variations to the multiple peptides covering each region. Finally, the signal intensity is further decreased due to deuteration, which usually widens the isotopic structure of the peptide ions. These combined effects make all but the most common peptide forms undetectable. We, therefore, used a separate LC-MS/MS analysis step to obtain detailed information about protein modifications occurring in Asc-treated LPMO digested by either trypsin or Asp-N. This approach also helped to verify that the introduced oxidative modifications caused the observed decrease in signal intensity. However, in contrast to results obtained with
ScLPMO10C by Bissaro et al., we were not able to get a comprehensive list of all modifications, since
NcLPMO9C was not amenable by any typical proteomic-based proteolysis approach [
24]. Even the use of two different proteases (trypsin and Asp-N) failed to provide fragments of reasonable size covering the entire protein sequence or at least the catalytic unit. Instead of relying on a pure search engine scoring, we performed manual validation of the MS/MS assignments and listed only those modifications where the exact oxidation position could be deciphered. This may also lead to a shortening of the list of modified residues. However, even with a less comprehensive listing, we provide clear proof that the
NcLPMO9C is extensively oxidized on many sites (
Table S1). Besides the oxidations and the known subsequent products of the oxidation pathways, we also found prominent oxidation leading to peptide bond cleavages. These were mainly located on amino acids close to the active site (
Table S1). These peptide cleavages have been identified before in other proteins [
32,
33], but are not very well-known and have not been connected to LPMO oxidative damage so far. Using extracted ion chromatograms from our LC-MS/MS data, we also followed the abundance of oxidatively modified peptides during incubation of LPMO with ascorbic acid (
Figure 5 and
Figure S12). These data show a clear increase in the level of protein oxidation over time, resulting also in peptide bond cleavages, as illustrated by peptide H1-A12 in
Figure 5. In addition, there is a clear dependency between the level of oxidation at particular sites and their proximity to the active site. We highlight this in
Figure S12, where side chains containing amino acids prone to oxidation are shown as sticks. Considering their surface accessibility and the rate of oxidation, which can be inferred from the extracted ion chromatograms, it is clear that the driving factor affecting the extent of oxidation in Asc-reduced LPMO is the proximity to the active center, specifically the involvement in copper ion binding (compare peptides. 94–109 and 22–45 versus 58–84).

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




