Beta Cell Therapies for Preventing Type 1 Diabetes: From Bench to Bedside


{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Autoimmune Type 1 Diabetes
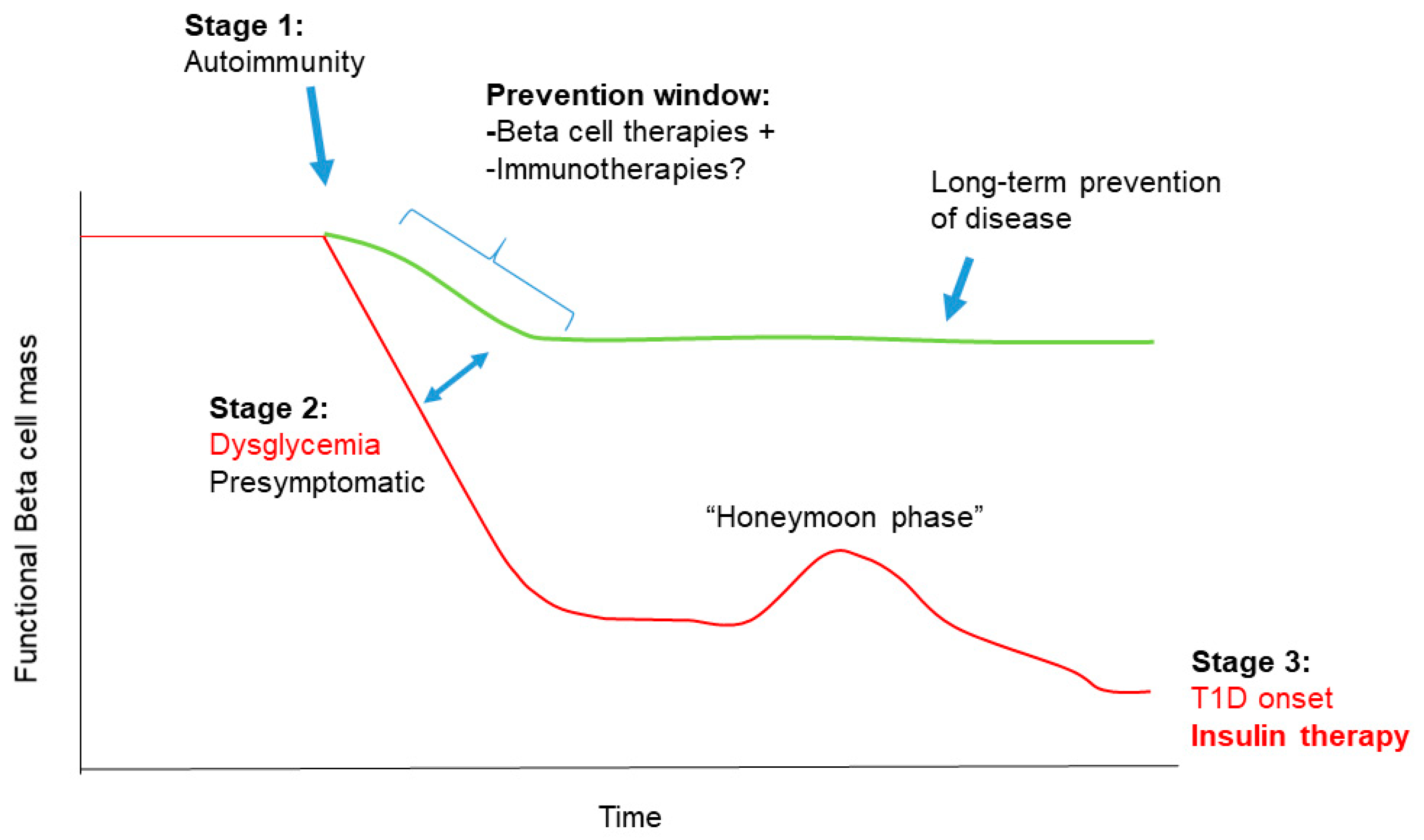
1.2. Stages in T1D Pathogenesis
1.3. T1D as a Disease of the Immune System and Beta Cells
2. Beta Cell Dysfunction in T1D
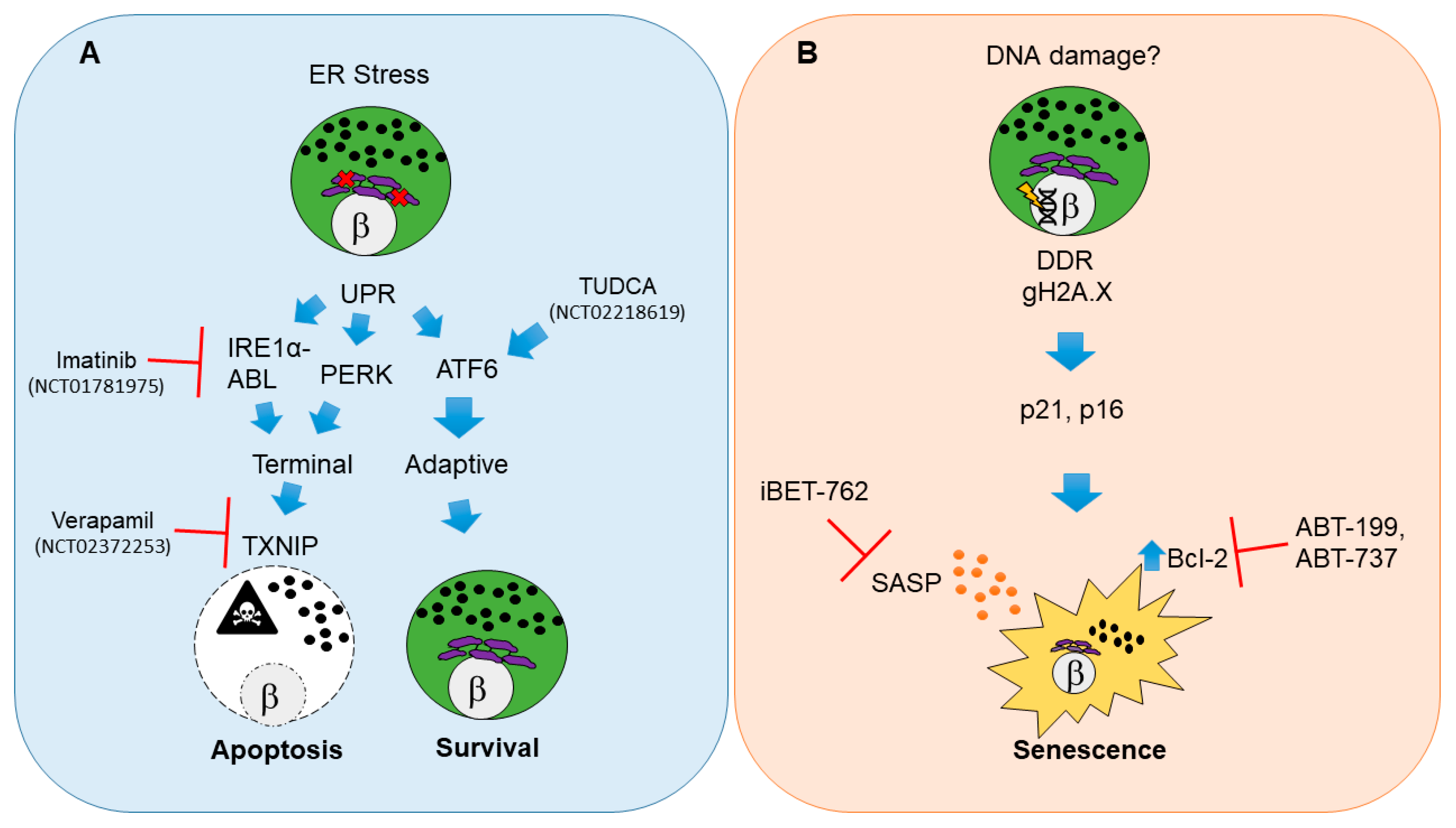
2.1. Beta Cell Endoplasmic Reticulum Stress, Unfolded Protein Response and Apoptosis
2.1.1. Pathways for UPR-Mediated Beta Cell Apoptosis
2.1.2. Clinical Trials for UPR Therapies in T1D
2.2. Damage-Induced Beta Cell Senescence
2.2.1. Molecular Pathways of Damage-Induced Beta Cell Senescence
2.2.2. Potential for Clinical Translation of Senescence-Targeting Therapies in T1D
2.3. Other States of Beta Cell Dysfunction: Defective Proinsulin Processing and Bihormonal Beta/Islet Cells
2.4. Additional Mechanisms of Beta Cell Dysfunction
3. Combining Beta Cell Therapy with Immunotherapy for T1D Prevention
3.1. Strengths and Weaknesses of a Combination Therapy Approach to T1D Prevention
3.2. Combination Therapy and the Re-Evaluation of T1D Etiology
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Prim. 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Catarino, D.; Silva, D.; Guiomar, J.; Ribeiro, C.; Ruas, L.; Cardoso, L.; Paiva, I. Non-immune-mediated versus immune-mediated type 1 diabetes: Diagnosis and long-term differences—Retrospective analysis. Diabetol. Metab. Syndr. 2020, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ilonen, J.; Lempainen, J.; Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Mayer-Davis, E.J.; Saydah, S.; Imperatore, G.; Linder, B.; Divers, J.; Bell, R.; Badaru, A.; Talton, J.W.; Crume, T.; et al. Prevalence of Type 1 and Type 2 Diabetes Among Children and Adolescents from 2001 to 2009. JAMA 2014, 311, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Beran, D.; Mirza, Z.; Dong, J. Access to insulin: Applying the concept of security of supply to medicines. Bull. World Health Organ. 2019, 97, 358–364. [Google Scholar] [CrossRef]
- The Lancet Diabetes & Endocrinology. The bare essentials: Ensuring affordable access to insulin. Lancet Diabetes Endocrinol. 2017, 5, 151. [Google Scholar] [CrossRef]
- Sharma, H.; Lencioni, M.; Narendran, P. Cardiovascular disease in type 1 diabetes. Cardiovasc. Endocrinol. Metab. 2019, 8, 28–34. [Google Scholar] [CrossRef]
- Norris, J.M.; Johnson, R.K.; Stene, L.C. Type 1 diabetes—Early life origins and changing epidemiology. Lancet Diabetes Endocrinol. 2020, 8, 226–238. [Google Scholar] [CrossRef]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, A.; et al. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef]
- Eisenbarth, G.S. Type I Diabetes Mellitus. N. Engl. J. Med. 1986, 314, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Lampasona, V.; Liberati, D. Islet Autoantibodies. Curr. Diabetes Rep. 2016, 16, 53. [Google Scholar] [CrossRef]
- Regnell, S.E.; Lernmark, Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to Multiple Islet Autoantibodies and Risk of Progression to Diabetes in Children. JAMA 2013, 309, 2473–2479. [Google Scholar] [CrossRef]
- Ferrat, L.A.; Vehik, K.; Sharp, S.A.; Lernmark, Å.; Rewers, M.J.; She, J.X.; Ziegler, A.G.; Toppari, J.; Akolkar, B.; Krischer, J.P.; et al. A combined risk score enhances prediction of type 1 diabetes among susceptible children. Nat. Med. 2020, 26, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Sims, E.K.; Mirmira, R.G.; Evans-Molina, C. The role of beta-cell dysfunction in early type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 215–224. [Google Scholar] [CrossRef]
- Nwosu, B.U. Partial Clinical Remission of Type 1 Diabetes Mellitus in Children: Clinical Applications and Challenges with its Definitions. Eur. Med. J. Diabetes 2019, 4, 89–98. [Google Scholar]
- Zhong, T.; Tang, R.; Gong, S.; Li, J.; Li, X.; Zhou, Z. The remission phase in type 1 diabetes: Changing epidemiology, definitions, and emerging immuno-metabolic mechanisms. Diabetes/Metab. Res. Rev. 2020, 36, 1–7. [Google Scholar] [CrossRef]
- Sims, E.K.; Bahnson, H.T.; Nyalwidhe, J.; Haataja, L.; Davis, A.K.; Speake, C.; DiMeglio, L.A.; Blum, J.; Morris, M.A.; Mirmira, R.G.; et al. Proinsulin Secretion Is a Persistent Feature of Type 1 Diabetes. Diabetes Care 2019, 42, 258–264. [Google Scholar] [CrossRef]
- Wang, Y.J.; Traum, D.; Schug, J.; Gao, L.; Liu, C.; Atkinson, M.A.; Powers, A.C.; Feldman, M.D.; Naji, A.; Chang, K.M.; et al. Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab. 2019, 29, 769–783.e4. [Google Scholar] [CrossRef]
- Damond, N.; Engler, S.; Zanotelli, V.R.T.; Schapiro, D.; Wasserfall, C.H.; Kusmartseva, I.; Nick, H.S.; Thorel, F.; Herrera, P.L.; Atkinson, M.A.; et al. A Map of Human Type 1 Diabetes Progression by Imaging Mass Cytometry. Cell Metab. 2019, 29, 755–768.e5. [Google Scholar] [CrossRef]
- Lam, C.J.; Jacobson, D.R.; Rankin, M.M.; Cox, A.R.; Kushner, J.A. β Cells Persist in T1D Pancreata Without Evidence of Ongoing β-Cell Turnover or Neogenesis. J. Clin. Endocrinol. Metab. 2017, 102, 2647–2659. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Gale, E.A.M. The discovery of type 1 diabetes. Diabetes 2001, 50, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.D.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D.; et al. Teplizumab (Anti-CD3 mAb) Treatment Preserves C-Peptide Responses in Patients with New-Onset Type 1 Diabetes in a Randomized Controlled Trial: Metabolic and Immunologic Features at Baseline Identify a Subgroup of Responders. Diabetes 2013, 62, 3766–3774. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Quattrin, T.; Haller, M.J.; Steck, A.K.; Felner, E.I.; Li, Y.; Xia, Y.; Leu, J.H.; Zoka, R.; Hedrick, J.A.; Rigby, M.R.; et al. Golimumab and Beta-Cell Function in Youth with New-Onset Type 1 Diabetes. N. Engl. J. Med. 2020, 383, 2007–2017. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Roep, B.O.; Posgai, A.; Wheeler, D.C.S.; Peakman, M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 52–64. [Google Scholar] [CrossRef]
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2020. [Google Scholar] [CrossRef]
- Pociot, F. Type 1 diabetes genome-wide association studies: Not to be lost in translation. Clin. Transl. Immunol. 2017, 6, e162. [Google Scholar] [CrossRef]
- Evans-Molina, C.; Sims, E.K.; DiMeglio, L.A.; Ismail, H.M.; Steck, A.K.; Palmer, J.P.; Krischer, J.P.; Geyer, S.; Xu, P.; Sosenko, J.M. β Cell dysfunction exists for more than 5 years prior to type 1 diabetes diagnosis. JCI Insight 2018, 3, e120877. [Google Scholar] [CrossRef] [PubMed]
- Oram, R.A.; McDonald, T.J.; Shields, B.M.; Hudson, M.M.; Shepherd, M.H.; Hammersley, S.; Pearson, E.R.; Hattersley, A.T.; Sanders, T.; Tiley, S.; et al. Most People with Long-Duration Type 1 Diabetes in a Large Population-Based Study Are Insulin Microsecretors. Diabetes Care 2015, 38, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M.; Fu, A.; Kaddis, J.S.; Wasserfall, C.; Schatz, D.A.; Pugliese, A.; Atkinson, M.A. Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016, 65, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, N.S.; Rui, J.; Hebrok, M.; Herold, K.C. Life and death of Beta cells in Type 1 diabetes: A comprehensive review. J. Autoimmun. 2016, 71, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27, S60–S68. [Google Scholar] [CrossRef]
- Szabat, M.; Page, M.M.; Panzhinskiy, E.; Skovsø, S.; Mojibian, M.; Fernandez-Tajes, J.; Bruin, J.E.; Bround, M.J.; Lee, J.T.C.; Xu, E.E.; et al. Reduced Insulin Production Relieves Endoplasmic Reticulum Stress and Induces β Cell Proliferation. Cell Metab. 2016, 23, 179–193. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 2016, 66, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Amrani, A.; Verdaguer, J.; Anderson, B.; Utsugi, T.; Bou, S.; Santamaria, P. Perforin-independent β-cell destruction by diabetogenic CD8+ T lymphocytes in transgenic nonobese diabetic mice. J. Clin. Investig. 1999, 103, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Kägi, B.D.; Odermatt, B.; Seiler, P.; Zinkernagel, R.M.; Mak, T.W.; Hengartner, H. Perforin-deficient Nonobese Diabetic Mice. J. Exp. Med. 1997, 186, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Mohamood, A.S.; Guler, M.L.; Xiao, Z.; Zheng, D.; Hess, A.; Wang, Y.; Yagita, H.; Schneck, J.P.; Hamad, A.R.A. Protection from Autoimmune Diabetes and T-Cell Lymphoproliferation Induced by FasL Mutation Are Differentially Regulated and Can Be Uncoupled Pharmacologically. Am. J. Pathol. 2007, 171, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.E.; Kay, T.W. Intracellular pathways of pancreatic β-cell apoptosis in type 1 diabetes. Diabetes/Metab. Res. Rev. 2011, 27, 790–796. [Google Scholar] [CrossRef]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.H.; Atkinson, M.A.; Roep, B.O.; Von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Panzer, J.K.; Hiller, H.; Cohrs, C.M.; Almaça, J.; Enos, S.J.; Beery, M.; Cechin, S.; Drotar, D.M.; Weitz, J.R.; Santini, J.; et al. Pancreas tissue slices from organ donors enable in situ analysis of type 1 diabetes pathogenesis. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Graham, K.L.; Sutherland, R.M.; Mannering, S.I.; Zhao, Y.; Chee, J.; Krishnamurthy, B.; Thomas, H.E.; Lew, A.M.; Kay, T.W.H. Pathogenic Mechanisms in Type 1 Diabetes: The Islet is Both Target and Driver of Disease. Rev. Diabet. Stud. 2012, 9, 148–168. [Google Scholar] [CrossRef]
- Boldison, J.; Wong, F.S. Immune and Pancreatic β Cell Interactions in Type 1 Diabetes. Trends Endocrinol. Metab. 2016, 27, 856–867. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Neiman, D.; Gillis, D.; Piyanzin, S.; Cohen, D.; Fridlich, O.; Moss, J.; Zick, A.; Oron, T.; Sundberg, F.; Forsander, G.; et al. Multiplexing DNA methylation markers to detect circulating cell-free DNA derived from human pancreatic β cells. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet Beta-Cell Endoplasmic Reticulum Stress Precedes the Onset of Type 1 Diabetes in the Nonobese Diabetic Mouse Model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef] [PubMed]
- Marhfour, I.; Lopez, X.M.; Lefkaditis, D.; Salmon, I.; Allagnat, F.; Richardson, S.J.; Morgan, N.G.; Eizirik, D.L. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012, 55, 2417–2420. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-Interacting Protein Mediates ER Stress-Induced β Cell Death through Initiation of the Inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Villalta, S.A.; Feldman, H.C.; Register, A.C.; Rosenthal, W.; Hoffmann-Petersen, I.T.; Mehdizadeh, M.; Ghosh, R.; Wang, L.; Colon-Negron, K.; et al. Targeting ABL-IRE1α Signaling Spares ER-Stressed Pancreatic β Cells to Reverse Autoimmune Diabetes. Cell Metab. 2017, 25, 883–897.e8. [Google Scholar] [CrossRef] [PubMed]
- Louvet, C.; Szot, G.L.; Lang, J.; Lee, M.R.; Martinier, N.; Bollag, G.; Zhu, S.; Weiss, A.; Bluestone, J.A. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18895–18900. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.S.; Harenda, Q.; Pietrzak, S.; Oktay, H.Z.; Schreiber, S.; Liao, Y.; Sonthalia, S.; Ciecko, A.E.; Chen, Y.G.; et al. Beta Cell Dedifferentiation Induced by IRE1α Deletion Prevents Type 1 Diabetes. Cell Metab. 2020, 31, 822–836.e5. [Google Scholar] [CrossRef]
- Chen, J.; Fontes, G.; Saxena, G.; Poitout, V.; Shalev, A. Lack of TXNIP Protects Against Mitochondria-Mediated Apoptosis but Not Against Fatty Acid-Induced ER Stress-Mediated β-Cell Death. Diabetes 2010, 59, 440–447. [Google Scholar] [CrossRef]
- Ovalle, F.; Grimes, T.; Xu, G.; Patel, A.J.; Grayson, T.B.; Thielen, L.A.; Li, P.; Shalev, A. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat. Med. 2018, 24, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.Q.; Zhang, P.; Li, S.; Yuan, L.; Xia, T.; Xie, C.; Clare-Salzler, M.J. C-Abl Inhibitor Imatinib Enhances Insulin Production by β Cells: C-Abl Negatively Regulates Insulin Production via Interfering with the Expression of NKx2.2 and GLUT-2. PLoS ONE 2014, 9, e97694. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.S.; Spaeth, J.M.; Karp, J.; Stocks, B.T.; Hoopes, E.M.; Stein, R.W.; Moore, D.J. B lymphocytes protect islet β cells in diabetes-prone NOD mice treated with imatinib. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Kusaczuk, M. Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471. [Google Scholar] [CrossRef] [PubMed]
- Lebensztejn, D.M. Application of ursodeoxycholic acid (UDCA) in the therapy of liver and biliary duct diseases in children. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2000, 6, 632–636. [Google Scholar]
- Heubi, J.E.; Wiechmann, D.A.; Creutzinger, V.; Setchell, K.D.R.; Squires, R.J.; Couser, R.; Rhodes, P. Tauroursodeoxycholic acid (TUDCA) in the prevention of total parenteral nutrition-associated liver disease. J. Pediatr. 2002, 141, 237–242. [Google Scholar] [CrossRef]
- Pozzilli, P.; Bosi, E.; Cirkel, D.; Harris, J.; Leech, N.; Tinahones, F.J.; Vantyghem, M.C.; Vlasakakis, G.; Ziegler, A.G.; Janmohamed, S. Randomized 52-week Phase 2 Trial of Albiglutide Versus Placebo in Adult Patients with Newly Diagnosed Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2020, 105, dgaa149. [Google Scholar] [CrossRef]
- Griffin, K.J.; Thompson, P.A.; Gottschalk, M.; Kyllo, J.H.; Rabinovitch, A. Combination therapy with sitagliptin and lansoprazole in patients with recent-onset type 1 diabetes (REPAIR-T1D): 12-month results of a multicentre, randomised, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2014, 2, 710–718. [Google Scholar] [CrossRef]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; De Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Farr, J.N.; Tchkonia, T.; Kirkland, J.L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 2020, 16, 263–275. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Passos, J.F.; Khosla, S.; Tchkonia, T.; Kirkland, J.L. Reducing Senescent Cell Burden in Aging and Disease. Trends Mol. Med. 2020, 26, 630–638. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- DeMaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Bhatia-Dey, N.; Kanherkar, R.R.; Stair, S.E.; Makarev, E.O.; Csoka, A.B. Cellular Senescence as the Causal Nexus of Aging. Front. Genet. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Klughammer, J.; Farlik, M.; Penz, T.; Spittler, A.; Barbieux, C.; Berishvili, E.; Bock, C.; Kubicek, S. Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Rep. 2016, 17, 178–187. [Google Scholar] [CrossRef]
- Camunas-Soler, J.; Dai, X.-Q.; Hang, Y.; Bautista, A.; Lyon, J.; Suzuki, K.; Kim, S.K.; Quake, S.R.; Macdonald, P.E. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab. 2020, 31, 1017–1031.e4. [Google Scholar] [CrossRef]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Migliorini, A.; Gegg, M.; Lickert, H. Impact of islet architecture on β-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol. 2016, 12, 695. [Google Scholar] [CrossRef]
- Van Der Meulen, T.; Mawla, A.M.; DiGruccio, M.R.; Adams, M.W.; Nies, V.; Dólleman, S.; Liu, S.; Ackermann, A.M.; Cáceres, E.; Hunter, A.E.; et al. Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab. 2017, 25, 911–926.e6. [Google Scholar] [CrossRef]
- Enge, M.; Arda, H.E.; Mignardi, M.; Beausang, J.; Bottino, R.; Kim, S.K.; Quake, S.R. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 2017, 171, 321–330.e14. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Horwitz, E.; Krogvold, L.; Zhitomirsky, S.; Swisa, A.; Fischman, M.; Lax, T.; Dahan, T.; Hurvitz, N.; Weinberg-Corem, N.; Klochendler, A.; et al. Beta-Cell DNA Damage Response Promotes Islet Inflammation in Type 1 Diabetes. Diabetes 2018, 67, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16Ink4a-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; Van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; Van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; Van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. β Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef]
- Thompson, P.J.; Shah, A.; Apostolopolou, H.; Bhushan, A. BET Proteins Are Required for Transcriptional Activation of the Senescent Islet Cell Secretome in Type 1 Diabetes. Int. J. Mol. Sci. 2019, 20, 4776. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Braña, I.; Cassier, P.A.; Moreno, V.; De Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr. 2020, 4, 1–9. [Google Scholar] [CrossRef]
- Fu, W.; Farache, J.; Clardy, S.M.; Hattori, K.; Mander, P.; Lee, K.; Rioja, I.; Weissleder, R.; Prinjha, R.K.; Benoist, C.; et al. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and Beta cells. eLife 2014, 3, e04631. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Hickson, L.T.J.; Prata, L.G.P.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sims, E.K.; Syed, F.; Nyalwidhe, J.; Bahnson, H.T.; Haataja, L.; Speake, C.; Morris, M.A.; Balamurugan, A.N.; Mirmira, R.G.; Nadler, J.; et al. Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Transl. Res. 2019, 213, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Wasserfall, C.; Nick, H.S.; Campbell-Thompson, M.; Beachy, D.; Haataja, L.; Kusmartseva, I.; Posgai, A.; Beery, M.; Rhodes, C.; Bonifacio, E.; et al. Persistence of Pancreatic Insulin mRNA Expression and Proinsulin Protein in Type 1 Diabetes Pancreata. Cell Metab. 2017, 26, 568–575.e3. [Google Scholar] [CrossRef]
- Lam, C.J.; Chatterjee, A.; Shen, E.; Cox, A.R.; Kushner, J.A. Low-Level Insulin Content Within Abundant Non-β Islet Endocrine Cells in Long-standing Type 1 Diabetes. Diabetes 2019, 68, 598–608. [Google Scholar] [CrossRef]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting Adult Pancreatic Islet α Cells into β Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef]
- Vakilian, M.; Tahamtani, Y.; Ghaedi, K. A review on insulin trafficking and exocytosis. Gene 2019, 706, 52–61. [Google Scholar] [CrossRef]
- Chen, Y.C.; Taylor, A.J.; Verchere, C.B. Islet prohormone processing in health and disease. Diabetes Obes. Metab. 2018, 20, 64–76. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Dhawan, S.; Shieh, C.; Butler, P.C.; Cory, M.; Butler, A.E. Increased Hormone-Negative Endocrine Cells in the Pancreas in Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3487–3496. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Cory, M.; Ong, A.; Choi, J.; Dhawan, S.; Butler, P.C.; Butler, A.E. Pancreatic Nonhormone Expressing Endocrine Cells in Children with Type 1 Diabetes. J. Endocr. Soc. 2017, 1, 385–395. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Beeck, A.O.; Eizirik, D.L. Viral infections in type 1 diabetes mellitus—Why the β cells? Nat. Rev. Endocrinol. 2016, 12, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Størling, J.; Pociot, F. Type 1 Diabetes Candidate Genes Linked to Pancreatic Islet Cell Inflammation and Beta-Cell Apoptosis. Genes 2017, 8, 72. [Google Scholar] [CrossRef]
- Li, Q.; Xu, B.; Michie, S.A.; Rubins, K.H.; Schreriber, R.D.; McDevitt, H.O. Interferon-α initiates type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12439–12444. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Targeting of type I interferon in systemic autoimmune diseases. Transl. Res. 2015, 165, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Dos Santos, R.S.; Op De Beeck, A.; De Brachène, A.C.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.A.; Hundhausen, C.; Errett, J.S.; Stone, A.E.; Allenspach, E.J.; Ge, Y.; Arkatkar, T.; Clough, C.; Dai, X.; Khim, S.; et al. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat. Immunol. 2017, 18, 744–752. [Google Scholar] [CrossRef]
- Osum, K.C.; Burrack, A.L.; Martinov, T.; Sahli, N.L.; Mitchell, J.S.; Tucker, C.G.; Pauken, K.E.; Papas, K.; Appakalai, B.; Spanier, J.A.; et al. Interferon-gamma drives programmed death-ligand 1 expression on islet β cells to limit T cell function during autoimmune diabetes. Sci. Rep. 2018, 8, 8295. [Google Scholar] [CrossRef]
- Colli, M.L.; Hill, J.L.E.; Marroquí, L.; Chaffey, J.; Dos Santos, R.S.; Leete, P.; De Brachène, A.C.; Paula, F.M.M.; Op De Beeck, A.; Castela, A.; et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine 2018, 36, 367–375. [Google Scholar] [CrossRef]
- Paterson, A.M.; Brown, K.E.; Keir, M.E.; Vanguri, V.K.; Riella, L.V.; Chandraker, A.; Sayegh, M.H.; Blazar, B.R.; Freeman, G.J.; Sharpe, A.H. The Programmed Death-1 Ligand 1:B7-1 Pathway Restrains Diabetogenic Effector T Cells In Vivo. J. Immunol. 2011, 187, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of Beta Cell Mass in Response to High-Fat Diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.W.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of Autophagy Diminishes Pancreatic Beta Cell Mass and Function with Resultant Hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Bugliani, M.; Mossuto, S.; Grano, F.; Suleiman, M.; Marselli, L.; Boggi, U.; De Simone, P.; Eizirik, D.L.; Cnop, M.; Marchetti, P.; et al. Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells Under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front. Endocrinol. 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, C.; Conteh, A.M.; Marasco, M.R.; Crowder, J.J. Pancreatic Beta Cell Autophagy is Impaired in Type 1 Diabetes. BioRXiv 2020. [Google Scholar] [CrossRef]
- Chen, J.; Stimpson, S.E.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372. [Google Scholar] [CrossRef]
- Masini, M.; Martino, L.; Marselli, L.; Bugliani, M.; Boggi, U.; Filipponi, F.; Marchetti, P.; De Tata, V. Ultrastructural alterations of pancreatic beta cells in human diabetes mellitus. Diabetes/Metab. Res. Rev. 2017, 33, e2894. [Google Scholar] [CrossRef]
- De Boer, P.; Pirozzi, N.M.; Wolters, A.H.G.; Kuipers, J.; Kusmartseva, I.; Atkinson, M.A.; Campbell-Thompson, M.; Giepmans, B.N.G. Large-scale electron microscopy database for human type 1 diabetes. Nat. Commun. 2020, 11, 2475. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Colli, M.L. Revisiting the role of inflammation in the loss of pancreatic β-cells in T1DM. Nat. Rev. Endocrinol. 2020, 16, 611–612. [Google Scholar] [CrossRef]
- Carré, A.; Richardson, S.J.; Larger, E.; Mallone, R. Presumption of guilt for T cells in type 1 diabetes: Lead culprits or partners in crime depending on age of onset? Diabetologia 2020, 64, 15–25. [Google Scholar] [CrossRef]
- Battaglia, M.; Ahmed, S.; Anderson, M.S.; Atkinson, M.A.; Becker, D.; Bingley, P.J.; Bosi, E.; Brusko, T.M.; DiMeglio, L.A.; Evans-Molina, C.; et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care 2020, 43, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Ma, X.; Li, X.; Xie, Z.; Zhou, Z. Fulminant type 1 diabetes: A comprehensive review of an autoimmune condition. Diabetes/Metab. Res. Rev. 2020, 36, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Lundgren, V.; Turunen, J.A.; Forsblom, C.; Isomaa, B.; Groop, P.H.; Groop, L.; Tuomi, T. Latent Autoimmune Diabetes in Adults Differs Genetically from Classical Type 1 Diabetes Diagnosed After the Age of 35 Years. Diabetes Care 2010, 33, 2062–2064. [Google Scholar] [CrossRef] [PubMed]
- Jörns, A.; Wedekind, D.; Jähne, J.; Lenzen, S. Pancreas Pathology of Latent Autoimmune Diabetes in Adults (LADA) in Patients and in a LADA Rat Model Compared with Type 1 Diabetes. Diabetes 2020, 69, 624–633. [Google Scholar] [CrossRef]
- Buzzetti, R.; Tuomi, T.; Mauricio, D.; Pietropaolo, M.; Zhou, Z.; Pozzilli, P.; Leslie, R.D. Management of Latent Autoimmune Diabetes in Adults: A Consensus Statement from an International Expert Panel. Diabetes 2020, 69, 2037–2047. [Google Scholar] [CrossRef]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2017, 66, 241–255. [Google Scholar] [CrossRef]
- Pozzilli, P.; Maddaloni, E.; Buzzetti, R. Combination immunotherapies for type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2015, 11, 289–297. [Google Scholar] [CrossRef]
- Brissova, M.; Haliyur, R.; Saunders, D.; Shrestha, S.; Dai, C.; Blodgett, D.M.; Bottino, R.; Campbell-Thompson, M.; Aramandla, R.; Poffenberger, G.; et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep. 2018, 22, 2667–2676. [Google Scholar] [CrossRef]
- Vecchio, F.; Messina, G.; Giovenzana, A.; Petrelli, A. New Evidence of Exocrine Pancreatopathy in Pre-symptomatic and Symptomatic Type 1 Diabetes. Curr. Diabetes Rep. 2019, 19, 92. [Google Scholar] [CrossRef]
- Alexandre-Heymann, L.; Mallone, R.; Boitard, C.; Scharfmann, R.; Larger, E. Structure and function of the exocrine pancreas in patients with type 1 diabetes. Rev. Endocr. Metab. Disord. 2019, 20, 129–149. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brawerman, G.; Thompson, P.J. Beta Cell Therapies for Preventing Type 1 Diabetes: From Bench to Bedside. Biomolecules 2020, 10, 1681. https://doi.org/10.3390/biom10121681
Brawerman G, Thompson PJ. Beta Cell Therapies for Preventing Type 1 Diabetes: From Bench to Bedside. Biomolecules. 2020; 10(12):1681. https://doi.org/10.3390/biom10121681
Chicago/Turabian StyleBrawerman, Gabriel, and Peter J. Thompson. 2020. "Beta Cell Therapies for Preventing Type 1 Diabetes: From Bench to Bedside" Biomolecules 10, no. 12: 1681. https://doi.org/10.3390/biom10121681
APA StyleBrawerman, G., & Thompson, P. J. (2020). Beta Cell Therapies for Preventing Type 1 Diabetes: From Bench to Bedside. Biomolecules, 10(12), 1681. https://doi.org/10.3390/biom10121681