Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts

Abstract
1. Introduction
2. TGF-β Signaling and Fibrotic Diseases
2.1. Introduction to Fibrotic Diseases
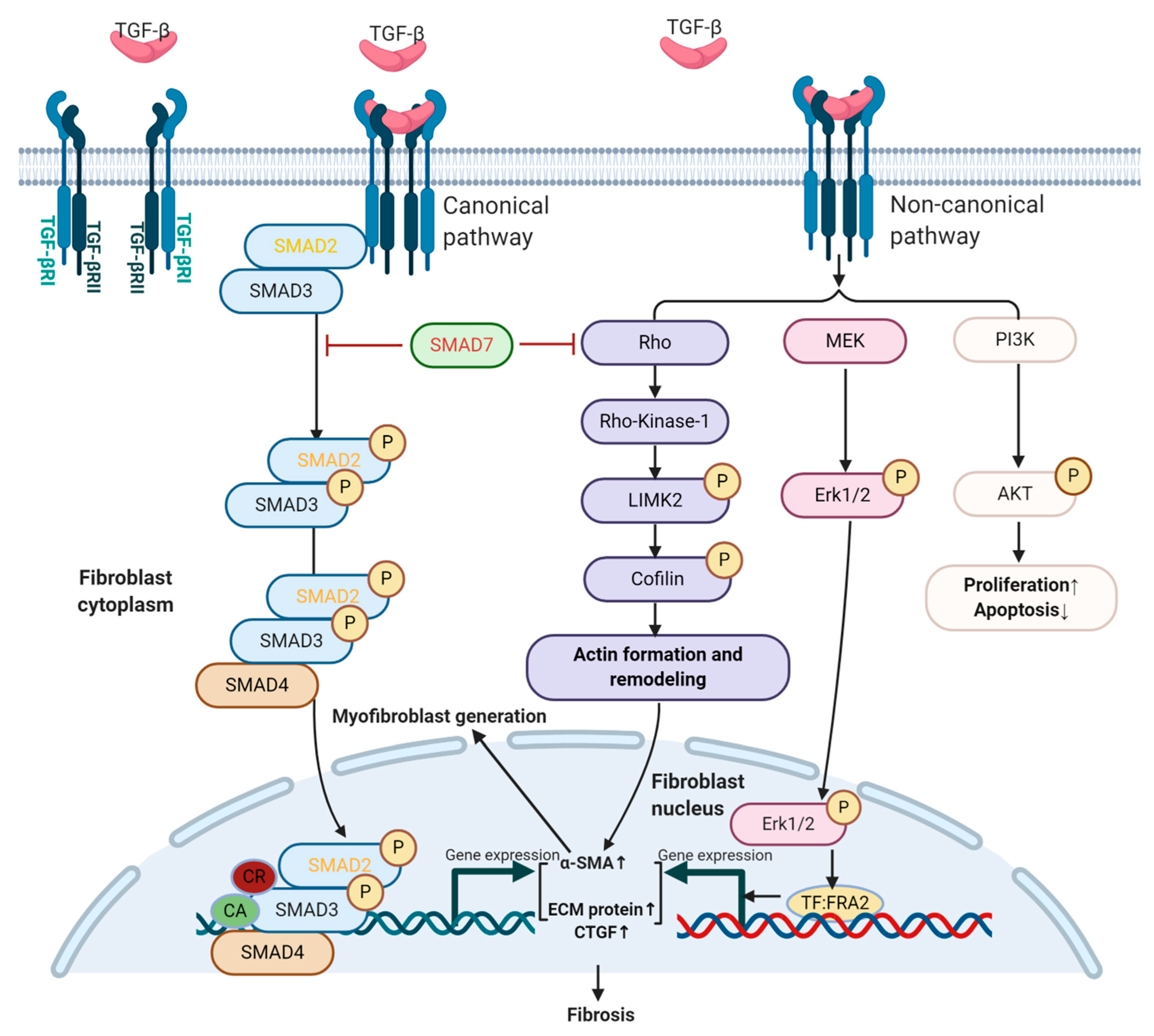
2.2. TGF-β Signaling in Fibroblasts and Fibrosis
2.2.1. TGF-β Signaling and Myofibroblast Generation
2.2.2. Role of TGF-β in Proliferation and Apoptosis of Fibroblasts
2.3. Paracrine TGF-β Signaling-Mediated Fibrosis
3. Tumor Promoting Effect of TGF-β Signaling in CAFs
3.1. TGF-β Signaling and Origin of CAFs
3.2. The Effect of TGF-β Signaling on the Biological Properties of CAFs
3.3. TGF-β Signaling and Metabolic Reprogramming of CAFs
3.4. Effect of CAF-Mediated TGF-β Signaling on Cancer Progression and Therapy Resistance
4. TGF-β Regulated Fibrotic Diseases and CAF-Mediated Cancer Progression
5. Therapeutic Targeting of TGF-β Signaling in Fibrosis and CAF-Mediated Cancer
5.1. TGF-β Inhibition and Fibrotic Diseases
5.2. TGF-β Targeted Therapy and Anti-CAF-Mediated Cancer Progression

{kind=link}
{kind=link}
{kind=link}
| Agent | Target | Status | Treatment Application | Efficacy |
|---|---|---|---|---|
| Terguride | TGF-β1 generation and activation | Phase II clinical trial | Systemic sclerosis (SSc) | Decreased dermal thickness with reduced myofibroblast numbers [139] |
| BG00011 | TGF-β activation | Phase II clinical trial | Idiopathic pulmonary fibrosis | Dose-dependent reductions in p-SMAD2 in bronchoalveolar lavage cells (NCT01371305) |
| Fresolimumab or GC1008) | TGF-β1, TGF-β2, and TGF-β3 | Phase I clinical trial | Resistant primary focal segmental glomerulosclerosis | Among 16 patients, one case of complete remission and two cases of partial remission of proteinuria [142] |
| Fresolimumab (GC1008) | TGF-β1, TGF-β2, and TGF-β3 | Phase I Clinical trial | SSc | Decreased skin fibrosis with reduced myofibroblast marker(α-SMA) expression [143] |
| IN-1130 | TGF-βRI | Preclinical rat model study | Renal fibrosis | Decreased renal fibrosis with reduced myofibroblast marker(α-SMA) expression [144] |
| GW788388 | TGF-βRI and TGF-βRII | Preclinical mouse model study | Renal fibrosis | Decreased renal fibrosis with reduced albuminuria [145] |
| LY2109761 | TGF-βRI and TGF-βRII | Preclinical chick embryo model study | Hepatocellular carcinoma | Suppressed tumor growth and progression by blocking TGF-β dependent production of CTGF and CAF proliferation [102] |
| LY2157299 or Galunisertib | TGF-βRI | Preclinical mouse model study | Ovarian cancer | Suppressed ovarian tumor growth partially via CAF inactivation [158] |
| Artemisinin derivatives | p-SMAD3 | Preclinical mouse model study | Breast cancer | Inhibited cancer growth and metastasis via CAFs inactivation [161] |
| M7824 or Bintrafusp alfa | TGF-β1, TGF-β2, TGF-β3, PD-L1 | Preclinical mouse model study | Breast and colorectal cancer | Suppressed tumor growth and metastasis partially by CAF inactivation [166] |
| Relaxin-2 | p-SMAD2 | Preclinical mouse model study | Pancreatic ductal adenocarcinoma | Retarded tumor growth and improved chemotherapy efficacy by impairing fibrosis [167] |
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, P.M.; Massagué, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.H.; Li, M.O.; Jenison, M.C.; Shlomchik, W.D.; Flavell, R.A.; Shlomchik, M.J. Autocrine/paracrine TGFbeta1 is required for the development of epidermal Langerhans cells. J. Exp. Med. 2007, 204, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Yan, X.; Chen, Y.-G. Smad7: Not only a regulator, but also a cross-talk mediator of TGF-β signalling. Biochem. J. 2011, 434. [Google Scholar] [CrossRef]
- Finnson, K.W.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: Mechanisms and targets. Semin. Cell Dev. Biol. 2020, 101, 115–122. [Google Scholar] [CrossRef]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.-P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrogenesis. Cell Signal. 2018, 43. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L.; Chen, A.; Xiang, G.; Wang, Y.; Wu, J.; Mitchelson, K.; Cheng, J.; Zhou, Y. Identification of the gene transcription and apoptosis mediated by TGF-beta-Smad2/3-Smad4 signaling. J. Cell. Physiol. 2008, 215, 422–433. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Wilkes, M.C.; Penheiter, S.G.; Gupta, S.K.; Edens, M.; Leof, E.B. Non-Smad transforming growth factor-β signaling regulated by focal adhesion kinase binding the p85 subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 2011, 286, 17841–17850. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Overstreet, J.M.; Higgins, S.P.; Higgins, P.J. TGF-β1 → SMAD/p53/USF2 → PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell Tissue Res. 2012, 347, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, J.M.; Samarakoon, R.; Meldrum, K.K.; Higgins, P.J. Redox control of p53 in the transcriptional regulation of TGF-β1 target genes through SMAD cooperativity. Cell Signal. 2014, 26, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M. TGFβ signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J. Cell Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Li, A.G.; Wang, D.; Feng, X.-H.; Wang, X.-J. Latent TGFbeta1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J. 2004, 23, 1770–1781. [Google Scholar] [CrossRef]
- Liu, X.; Alexander, V.; Vijayachandra, K.; Bhogte, E.; Diamond, I.; Glick, A. Conditional epidermal expression of TGFbeta 1 blocks neonatal lethality but causes a reversible hyperplasia and alopecia. Proc Natl. Acad. Sci. USA 2001, 98, 9139–9144. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Wu, F.; Weigel, K.J.; Zhou, H.; Wang, X.-J. Paradoxical roles of TGF-β signaling in suppressing and promoting squamous cell carcinoma. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Rosenberg, S.A. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J. Immunol. 2005, 174, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis--A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 373, 96. [Google Scholar] [CrossRef]
- Rosenbloom, J.; Castro, S.V.; Jimenez, S.A. Narrative review: Fibrotic diseases: Cellular and molecular mechanisms and novel therapies. Ann. Intern. Med. 2010, 152, 159–166. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Maier, C.; Ramming, A.; Bergmann, C.; Weinkam, R.; Kittan, N.; Schett, G.; Distler, J.H.W.; Beyer, C. Inhibition of phosphodiesterase 4 (PDE4) reduces dermal fibrosis by interfering with the release of interleukin-6 from M2 macrophages. Ann. Rheum. Dis. 2017, 76, 1133–1141. [Google Scholar] [CrossRef]
- Attari, H.; Cao, Y.; Elmholdt, T.R.; Zhao, Y.; Prince, M.R. A Systematic Review of 639 Patients with Biopsy-confirmed Nephrogenic Systemic Fibrosis. Radiology 2019, 292, 376–386. [Google Scholar] [CrossRef]
- Rosenbloom, J.; Macarak, E.; Piera-Velazquez, S.; Jimenez, S.A. Human Fibrotic Diseases: Current Challenges in Fibrosis Research. Methods Mol. Biol. 2017, 1627. [Google Scholar] [CrossRef]
- Belmares, R.; Raychaudhuri, U.; Maansson, S.; Clark, A.F. Histological investigation of human glaucomatous eyes: Extracellular fibrotic changes and galectin 3 expression in the trabecular meshwork and optic nerve head. Clin. Anat. 2018, 31, 1031–1049. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.B.; Hansen, N.-U.B.; Bay-Jensen, A.-C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol. Gastrointest Liver Physiol. 2015, 308, G807–G830. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Henke, C.A.; Horowitz, J.C.; Noble, P.W.; Roman, J.; Sime, P.J.; Zhou, Y.; Wells, R.G.; White, E.S.; Tschumperlin, D.J. Matrix biology of idiopathic pulmonary fibrosis: A workshop report of the national heart, lung, and blood institute. Am. J. Pathol. 2014, 184, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Castelino, F.V.; Varga, J. Emerging cellular and molecular targets in fibrosis: Implications for scleroderma pathogenesis and targeted therapy. Curr. Opin. Rheumatol. 2014, 26, 607–614. [Google Scholar] [CrossRef]
- Rosenbloom, J.; Mendoza, F.A.; Jimenez, S.A. Strategies for anti-fibrotic therapies. Biochim. Biophys. Acta 2013, 1832, 1088–1103. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Hu, B.; Phan, S.H. Myofibroblasts. Curr. Opin. Rheumatol. 2013, 25, 71–77. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 2007, 39, 666–671. [Google Scholar] [CrossRef]
- Murray, L.A.; Chen, Q.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Peng, X.; Gulati, M.; Homer, R.J.; Russell, T.; van Rooijen, N.; et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int. J. Biochem. Cell Biol. 2011, 43, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Distler, J.H.W. Tyrosine kinase signaling in fibrotic disorders: Translation of basic research to human disease. Biochim. Biophys. Acta 2013, 1832, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-H.; Cao, G.; Wu, X.-Q.; Vaziri, N.D.; Zhao, Y.-Y. Wnt signaling pathway in aging-related tissue fibrosis and therapies. Ageing Res. Rev. 2020, 60, 101063. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Varelas, X.; Guan, K.-L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Lafyatis, R. Transforming growth factor β--at the centre of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 706–719. [Google Scholar] [CrossRef]
- Li, R.K.; Li, G.; Mickle, D.A.; Weisel, R.D.; Merante, F.; Luss, H.; Rao, V.; Christakis, G.T.; Williams, W.G. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation 1997, 96, 874–881. [Google Scholar] [CrossRef]
- Schieren, G.; Gambichler, T.; Skrygan, M.; Burkert, B.; Altmeyer, P.; Rump, L.C.; Kreuter, A. Balance of profibrotic and antifibrotic [corrected] signaling in nephrogenic systemic fibrosis skin lesions. Am. J. Kidney Dis. 2010, 55, 1040–1049. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Li, F.; Bian, L.; Iriyama, S.; Jian, Z.; Fan, B.; Luo, J.; Wang, D.D.; Young, C.D.; Han, G.; Wang, X.-J. Smad7 Ameliorates TGF-β-Mediated Skin Inflammation and Associated Wound Healing Defects but Not Susceptibility to Experimental Skin Carcinogenesis. J. Invest. Dermatol. 2019, 139, 940–950. [Google Scholar] [CrossRef]
- Penke, L.R.; Peters-Golden, M. Molecular determinants of mesenchymal cell activation in fibroproliferative diseases. Cell Mol. Life Sci. 2019, 76, 4179–4201. [Google Scholar] [CrossRef]
- Jelaska, A.; Korn, J.H. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 2000, 43, 2230–2239. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M.; Sundqvist, A.; Heldin, A.; van Dam, H.; Kletsas, D.; Heldin, C.-H.; Moustakas, A. Tamoxifen Inhibits TGF-β-Mediated Activation of Myofibroblasts by Blocking Non-Smad Signaling Through ERK1/2. J. Cell Physiol. 2015, 230, 3084–3092. [Google Scholar] [CrossRef]
- Samarakoon, R.; Dobberfuhl, A.D.; Cooley, C.; Overstreet, J.M.; Patel, S.; Goldschmeding, R.; Meldrum, K.K.; Higgins, P.J. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013, 25, 2198–2209. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF--Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Reich, N.; Maurer, B.; Akhmetshina, A.; Venalis, P.; Dees, C.; Zerr, P.; Palumbo, K.; Zwerina, J.; Nevskaya, T.; Gay, S.; et al. The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum. 2010, 62, 280–290. [Google Scholar] [CrossRef]
- Eferl, R.; Hasselblatt, P.; Rath, M.; Popper, H.; Zenz, R.; Komnenovic, V.; Idarraga, M.-H.; Kenner, L.; Wagner, E.F. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc. Natl. Acad. Sci. USA 2008, 105, 10525–10530. [Google Scholar] [CrossRef]
- Ni, J.; Dong, Z.; Han, W.; Kondrikov, D.; Su, Y. The role of RhoA and cytoskeleton in myofibroblast transformation in hyperoxic lung fibrosis. Free Radic. Biol. Med. 2013, 61, 26–39. [Google Scholar] [CrossRef]
- Vardouli, L.; Moustakas, A.; Stournaras, C. LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J. Biol. Chem. 2005, 280, 11448–11457. [Google Scholar] [CrossRef]
- Katzenstein, A.L.; Myers, J.L. Idiopathic pulmonary fibrosis: Clinical relevance of pathologic classification. Am. J Respir Crit. Care Med. 1998, 157, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Meran, S.; Thomas, D.W.; Stephens, P.; Enoch, S.; Martin, J.; Steadman, R.; Phillips, A.O. Hyaluronan facilitates transforming growth factor-beta1-mediated fibroblast proliferation. J. Biol. Chem. 2008, 283, 6530–6545. [Google Scholar] [CrossRef] [PubMed]
- Lakos, G.; Takagawa, S.; Chen, S.-J.; Ferreira, A.M.; Han, G.; Masuda, K.; Wang, X.-J.; DiPietro, L.A.; Varga, J. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am. J. Pathol. 2004, 165, 203–217. [Google Scholar] [CrossRef]
- Wilkes, M.C.; Mitchell, H.; Penheiter, S.G.; Doré, J.J.; Suzuki, K.; Edens, M.; Sharma, D.K.; Pagano, R.E.; Leof, E.B. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 2005, 65, 10431–10440. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Horowitz, J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef]
- Yoshida, K.; Kuwano, K.; Hagimoto, N.; Watanabe, K.; Matsuba, T.; Fujita, M.; Inoshima, I.; Hara, N. MAP kinase activation and apoptosis in lung tissues from patients with idiopathic pulmonary fibrosis. J. Pathol. 2002, 198, 388–396. [Google Scholar] [CrossRef]
- Kulasekaran, P.; Scavone, C.A.; Rogers, D.S.; Arenberg, D.A.; Thannickal, V.J.; Horowitz, J.C. Endothelin-1 and transforming growth factor-beta1 independently induce fibroblast resistance to apoptosis via AKT activation. Am. J. Respir. Cell Mol. Biol. 2009, 41, 484–493. [Google Scholar] [CrossRef]
- Kis, K.; Liu, X.; Hagood, J.S. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med. 2011, 13, e27. [Google Scholar] [CrossRef]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 2010, 207, 535–552. [Google Scholar] [CrossRef]
- Fielding, C.A.; Jones, G.W.; McLoughlin, R.M.; McLeod, L.; Hammond, V.J.; Uceda, J.; Williams, A.S.; Lambie, M.; Foster, T.L.; Liao, C.-T.; et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 2014, 40, 40–50. [Google Scholar] [CrossRef]
- Ramalingam, T.R.; Gieseck, R.L.; Acciani, T.H.; Hart, K.M.; Cheever, A.W.; Mentink-Kane, M.M.; Vannella, K.M.; Wynn, T.A. Enhanced protection from fibrosis and inflammation in the combined absence of IL-13 and IFN-γ. J. Pathol. 2016, 239, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Lu, X.; Ren, J.; Privratsky, J.R.; Yang, B.; Rudemiller, N.P.; Zhang, J.; Griffiths, R.; Jain, M.K.; Nedospasov, S.A.; et al. KLF4 in Macrophages Attenuates TNF-Mediated Kidney Injury and Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 1925–1938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Meng, X.-M.; Huang, X.-R.; Wang, X.-J.; Yang, L.; Lan, H.Y. Transforming growth factor-β1 mediates psoriasis-like lesions via a Smad3-dependent mechanism in mice. Clin. Exp. Pharmacol. Physiol. 2014, 41, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Huang, X.R.; Xiao, J.; Chung, A.C.K.; Qin, W.; Chen, H.-y.; Lan, H.Y. Disruption of Smad4 impairs TGF-β/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012, 81, 266–279. [Google Scholar] [CrossRef]
- Li, J.H.; Zhu, H.-J.; Huang, X.R.; Lai, K.N.; Johnson, R.J.; Lan, H.Y. Smad7 inhibits fibrotic effect of TGF-Beta on renal tubular epithelial cells by blocking Smad2 activation. J. Am. Soc. Nephrol. 2002, 13, 1464–1472. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Wu, G.R.; Zhou, Q.; Yue, H.; Rao, L.Z.; Yuan, T.; Mo, B.; Wang, F.X.; Chen, L.M.; et al. MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci. Adv. 2020. [Google Scholar] [CrossRef]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef]
- Chung, S.; Overstreet, J.M.; Li, Y.; Wang, Y.; Niu, A.; Wang, S.; Fan, X.; Sasaki, K.; Jin, G.-N.; Khodo, S.N.; et al. TGF-β promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Loktev, A.; Lindner, T.; Mier, W.; Debus, J.; Altmann, A.; Jäger, D.; Giesel, F.; Kratochwil, C.; Barthe, P.; Roumestand, C.; et al. A Tumor-Imaging Method Targeting Cancer-Associated Fibroblasts. J. Nucl. Med. 2018, 59, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10GPR77 Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172. [Google Scholar] [CrossRef] [PubMed]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef]
- Leung, C.S.; Yeung, T.-L.; Yip, K.-P.; Wong, K.-K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2018, 128, 589–606. [Google Scholar] [CrossRef]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar] [CrossRef]
- Meng, W.; Wu, Y.; He, X.; Liu, C.; Gao, Q.; Ge, L.; Wu, L.; Liu, Y.; Guo, Y.; Li, X.; et al. A systems biology approach identifies effective tumor-stroma common targets for oral squamous cell carcinoma. Cancer Res. 2014, 74, 2306–2315. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Dumont, N.; Liu, B.; Defilippis, R.A.; Chang, H.; Rabban, J.T.; Karnezis, A.N.; Tjoe, J.A.; Marx, J.; Parvin, B.; Tlsty, T.D. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 2013, 15, 249–262. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, G.; Gander, R.; Lilg, C.; Lepperdinger, G.; Plas, E.; Berger, P. Profiling molecular targets of TGF-beta1 in prostate fibroblast-to-myofibroblast transdifferentiation. Mech. Ageing Dev. 2005, 126, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Ringuette Goulet, C.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes Induce Fibroblast Differentiation into Cancer-Associated Fibroblasts through TGFβ Signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.A.C.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.N.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene 2014, 33. [Google Scholar] [CrossRef]
- Lamprecht, S.; Sigal-Batikoff, I.; Shany, S.; Abu-Freha, N.; Ling, E.; Delinasios, G.J.; Moyal-Atias, K.; Delinasios, J.G.; Fich, A. Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-β1 Signaling and the Epigenetic Corruption of Stromal Naïve Fibroblasts. Cancers (Basel) 2018, 10, 61. [Google Scholar] [CrossRef]
- Barcellos-de-Souza, P.; Comito, G.; Pons-Segura, C.; Taddei, M.L.; Gori, V.; Becherucci, V.; Bambi, F.; Margheri, F.; Laurenzana, A.; Del Rosso, M.; et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-β1. Stem Cells 2016, 34, 2536–2547. [Google Scholar] [CrossRef]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.C.; Franzen, C.A.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015, 34, 4821–4833. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-X.; Cao, Z.-B.; He, T.-T.; Huang, T.; Xiang, C.-L.; Liu, Y. TGFβ1 is essential for MSCs-CAFs differentiation and promotes HCT116 cells migration and invasion via JAK/STAT3 signaling. Onco. Targets Ther. 2019, 12, 5323–5334. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, L.; Ti, X.; Krause, U.; Hai, B.; Zhao, Y.; Yang, Z.; Liu, F. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells 2012, 30, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A.; Fransvea, E.; Dituri, F.; Lupo, L.; Antonaci, S.; Giannelli, G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010, 51, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Schaeffer, D.F.; Cho, C.-K.J.; Musrap, N.; Saraon, P.; Batruch, I.; Grin, A.; Mitrovic, B.; Kirsch, R.; Riddell, R.H.; et al. Collective migration of cancer-associated fibroblasts is enhanced by overexpression of tight junction-associated proteins claudin-11 and occludin. Mol. Oncol. 2014, 8, 178–195. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, T.; Shen, J.; Li, S.F.; Lin, J.W.; Zheng, X.H.; Gao, Q.H.; Zhou, H.M. Separation, cultivation and biological characteristics of oral carcinoma-associated fibroblasts. Oral. Dis. 2006, 12, 375–380. [Google Scholar] [CrossRef]
- Sloan, E.K.; Ciocca, D.R.; Pouliot, N.; Natoli, A.; Restall, C.; Henderson, M.A.; Fanelli, M.A.; Cuello-Carrión, F.D.; Gago, F.E.; Anderson, R.L. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am. J. Pathol. 2009, 174, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with "Warburg-like" cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Yashiro, M.; Nishii, T.; Matsuoka, J.; Fuyuhiro, Y.; Morisaki, T.; Fukuoka, T.; Shimizu, K.; Shimizu, T.; Miwa, A.; et al. Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-β signaling. Int. J. Cancer 2014, 134, 1785–1795. [Google Scholar] [CrossRef]
- Fuyuhiro, Y.; Yashiro, M.; Noda, S.; Matsuoka, J.; Hasegawa, T.; Kato, Y.; Sawada, T.; Hirakawa, K. Cancer-associated orthotopic myofibroblasts stimulates the motility of gastric carcinoma cells. Cancer Sci. 2012, 103, 797–805. [Google Scholar] [CrossRef]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.F.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Saccà, M.; Vigneri, P.; De Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Clarke, M.F. Cancer stem cells and radiotherapy: New insights into tumor radioresistance. J. Natl. Cancer Inst. 2006, 98, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Rosell, R. Cancer stem cells and immunoresistance: Clinical implications and solutions. Transl. Lung Cancer Res. 2015, 4, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-β-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem. Cell 2017, 21. [Google Scholar] [CrossRef]
- Tang, Y.-A.; Chen, Y.-F.; Bao, Y.; Mahara, S.; Yatim, S.M.J.M.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef]
- Shyamsundar, M.; McAuley, D.F.; Ingram, R.J.; Gibson, D.S.; O’Kane, D.; McKeown, S.T.; Edwards, A.; Taggart, C.; Elborn, J.S.; Calfee, C.S.; et al. Keratinocyte growth factor promotes epithelial survival and resolution in a human model of lung injury. Am. J. Respir. Crit. Care Med. 2014, 189, 1520–1529. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Bajpai, S.; Chaturvedi, P.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J. Biol. Chem. 2013, 288, 10819–10829. [Google Scholar] [CrossRef]
- Nandi, T.; Pradyuth, S.; Singh, A.K.; Chitkara, D.; Mittal, A. Therapeutic agents for targeting desmoplasia: Current status and emerging trends. Drug Discov. Today 2020. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Berhan, A.; Harris, T.; Jaffar, J.; Jativa, F.; Langenbach, S.; Lönnstedt, I.; Alhamdoosh, M.; Ng, M.; Lee, P.; Westall, G.; et al. Cellular Microenvironment Stiffness Regulates Eicosanoid Production and Signaling Pathways. Am. J. Respir. Cell Mol. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Najafi, M.; Farhood, B.; Mortezaee, K. Transforming growth factor-β signaling: Tumorigenesis and targeting for cancer therapy. J. Cell Physiol. 2019, 234, 12173–12187. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Ghahremanifard, P.; Chanda, A.; Bonni, S.; Bose, P. TGF-β Mediated Immune Evasion in Cancer-Spotlight on Cancer-Associated Fibroblasts. Cancers (Basel) 2020, 12, 3650. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug. Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Dees, C.; Akhmetshina, A.; Zerr, P.; Reich, N.; Palumbo, K.; Horn, A.; Jüngel, A.; Beyer, C.; Krönke, G.; Zwerina, J.; et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. J. Exp. Med. 2011, 208, 961–972. [Google Scholar] [CrossRef]
- Distler, O.; Maurer, B.; Vettori, S.; Blumhardt, S.; Frey, D.; Distler, A.; Beyer, C.; Distler, J.H. OP0034 The Serotonin Receptor 2 Inhibitor Terguride Has Beneficial Effects on Skin Fibrosis: Results from A Phase 2 Proof of Concept Study. J. Ann. Rheum. Dis. 2016, 75, 66. [Google Scholar] [CrossRef]
- Puthawala, K.; Hadjiangelis, N.; Jacoby, S.C.; Bayongan, E.; Zhao, Z.; Yang, Z.; Devitt, M.L.; Horan, G.S.; Weinreb, P.H.; Lukashev, M.E.; et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 82–90. [Google Scholar] [CrossRef]
- Hahm, K.; Lukashev, M.E.; Luo, Y.; Yang, W.J.; Dolinski, B.M.; Weinreb, P.H.; Simon, K.J.; Chun Wang, L.; Leone, D.R.; Lobb, R.R.; et al. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am. J. Pathol. 2007, 170, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Trachtman, H.; Fervenza, F.C.; Gipson, D.S.; Heering, P.; Jayne, D.R.W.; Peters, H.; Rota, S.; Remuzzi, G.; Rump, L.C.; Sellin, L.K.; et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-β antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011, 79, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.A.; Kim, H.T.; Cho, I.S.; Sheen, Y.Y.; Kim, D.K. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006, 70, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; ten Dijke, P.; Laping, N.J. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Berzofsky, J.A.; Hsu, F.J.; Guitart, J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother 2015, 64, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Alvira, C.M.; Guignabert, C.; Kim, Y.-M.; Chen, C.; Wang, L.; Duong, T.T.; Yeung, R.S.M.; Li, D.Y.; Rabinovitch, M. Inhibition of transforming growth factor β worsens elastin degradation in a murine model of Kawasaki disease. Am. J. Pathol. 2011, 178, 1210–1220. [Google Scholar] [CrossRef]
- Chen, X.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Cassis, L.A.; Daugherty, A. TGF-β Neutralization Enhances AngII-Induced Aortic Rupture and Aneurysm in Both Thoracic and Abdominal Regions. PLoS ONE 2016, 11, e0153811. [Google Scholar] [CrossRef]
- Wang, Y.; Ait-Oufella, H.; Herbin, O.; Bonnin, P.; Ramkhelawon, B.; Taleb, S.; Huang, J.; Offenstadt, G.; Combadière, C.; Rénia, L.; et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J. Clin. Investig. 2010, 120, 422–432. [Google Scholar] [CrossRef]
- Lutgens, E.; Gijbels, M.; Smook, M.; Heeringa, P.; Gotwals, P.; Koteliansky, V.E.; Daemen, M.J.A.P. Transforming growth factor-beta mediates balance between inflammation and fibrosis during plaque progression. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 975–982. [Google Scholar] [CrossRef]
- Mallat, Z.; Gojova, A.; Marchiol-Fournigault, C.; Esposito, B.; Kamaté, C.; Merval, R.; Fradelizi, D.; Tedgui, A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 2001, 89, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFβ: Current knowledge and future perspectives. Ann. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, T.; Karasawa, H.; Funayama, R.; Shirota, M.; Suzuki, T.; Maeda, S.; Suzuki, H.; Yamamura, A.; Naitoh, T.; Nakayama, K.; et al. Cancer-associated fibroblasts secrete Wnt2 to promote cancer progression in colorectal cancer. Cancer Med. 2019, 8, 6370–6382. [Google Scholar] [CrossRef]
- Sun, D.-Y.; Wu, J.-Q.; He, Z.-H.; He, M.-F.; Sun, H.-B. Cancer-associated fibroblast regulate proliferation and migration of prostate cancer cells through TGF-β signaling pathway. Life Sci. 2019, 235, 116791. [Google Scholar] [CrossRef]
- Yegodayev, K.M.; Novoplansky, O.; Golden, A.; Prasad, M.; Levin, L.; Jagadeeshan, S.; Zorea, J.; Dimitstein, O.; Joshua, B.-Z.; Cohen, L.; et al. TGF-Beta-Activated Cancer-Associated Fibroblasts Limit Cetuximab Efficacy in Preclinical Models of Head and Neck Cancer. Cancers (Basel) 2020, 12, 339. [Google Scholar] [CrossRef]
- Zhang, Q.; Hou, X.; Evans, B.J.; VanBlaricom, J.L.; Weroha, S.J.; Cliby, W.A. LY2157299 Monohydrate, a TGF-βR1 Inhibitor, Suppresses Tumor Growth and Ascites Development in Ovarian Cancer. Cancers (Basel) 2018, 10, 260. [Google Scholar] [CrossRef]
- Li, Y. Qinghaosu (artemisinin): Chemistry and pharmacology. Acta Pharmacol. Sin. 2012, 33, 1141–1146. [Google Scholar] [CrossRef]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef]
- Yao, Y.; Guo, Q.; Cao, Y.; Qiu, Y.; Tan, R.; Yu, Z.; Zhou, Y.; Lu, N. Artemisinin derivatives inactivate cancer-associated fibroblasts through suppressing TGF-β signaling in breast cancer. J. Exp. Clin. Cancer Res. 2018, 37, 282. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Evans, K.; Xiao, C.; DeVito, N.; Theivanthiran, B.; Holtzhausen, A.; Siska, P.J.; Blobe, G.C.; Hanks, B.A. Stromal Fibroblasts Mediate Anti-PD-1 Resistance via MMP-9 and Dictate TGFβ Inhibitor Sequencing in Melanoma. Cancer Immunol. Res. 2018, 6, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massagué, J. TGF-β Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef]
- Knudson, K.M.; Hicks, K.C.; Luo, X.; Chen, J.-Q.; Schlom, J.; Gameiro, S.R. M7824, a novel bifunctional anti-PD-L1/TGFβ Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology 2018, 7, e1426519. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Mardhian, D.F.; Storm, G.; Bansal, R.; Prakash, J. Nano-targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo. J. Control. Release 2018, 290. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, X.; Young, C.D.; Zhou, H.; Wang, X.-J. Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules 2020, 10, 1666. https://doi.org/10.3390/biom10121666
Shi X, Young CD, Zhou H, Wang X-J. Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules. 2020; 10(12):1666. https://doi.org/10.3390/biom10121666
Chicago/Turabian StyleShi, Xueke, Christian D. Young, Hongmei Zhou, and Xiao-Jing Wang. 2020. "Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts" Biomolecules 10, no. 12: 1666. https://doi.org/10.3390/biom10121666
APA StyleShi, X., Young, C. D., Zhou, H., & Wang, X.-J. (2020). Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules, 10(12), 1666. https://doi.org/10.3390/biom10121666