Aberrant Non-Coding RNA Expression in Patients with Systemic Lupus Erythematosus: Consequences for Immune Dysfunctions and Tissue Damage

 , , , ,
, , , ,
Abstract
1. Introduction
2. Understanding the Genetics/Epigenetics of Patients with SLE in the Post-GWAS Era
2.1. Genetic Loci Susceptible to Lupus Pathogenesis
2.2. Epigenetic Regulation in Patients with SLE
2.2.1. DNA Hypomethylation in SLE-CD4+ T Cells
2.2.2. DNA Hydroxymethylation by miRNA Interference in Patients with SLE
2.2.3. Alternation of Histone Modifications in Immune-Related Cells in SLE
2.2.4. Post-Translational Non-Histone Protein Modifications in SLE
2.3. Aberrant Expression of ncRNAs in Patients with SLE
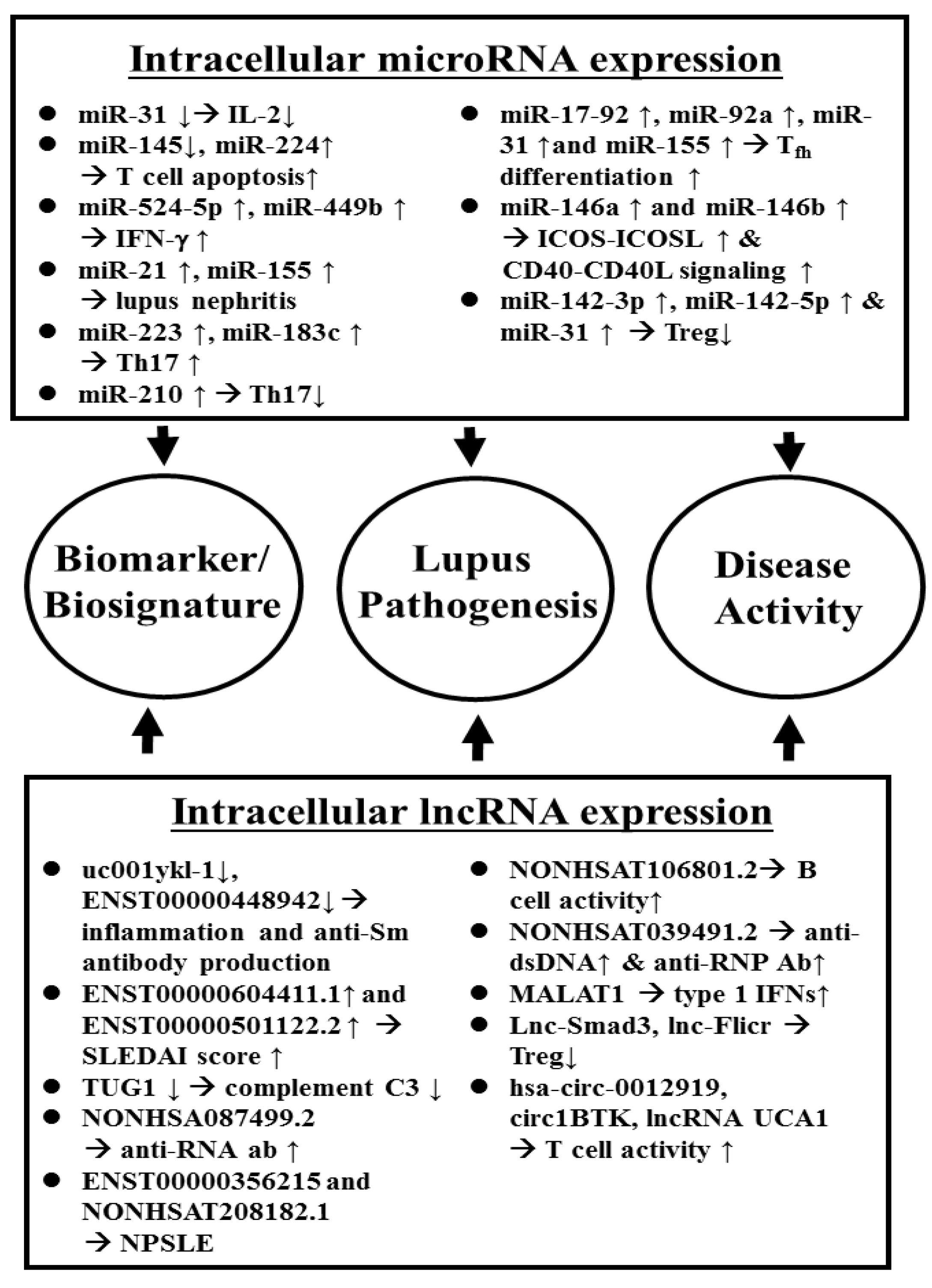
2.3.1. Aberrant Intracellular ncRNA Expression Associated with Pathogenesis and as Biomarkers/Biosignatures in SLE Patients
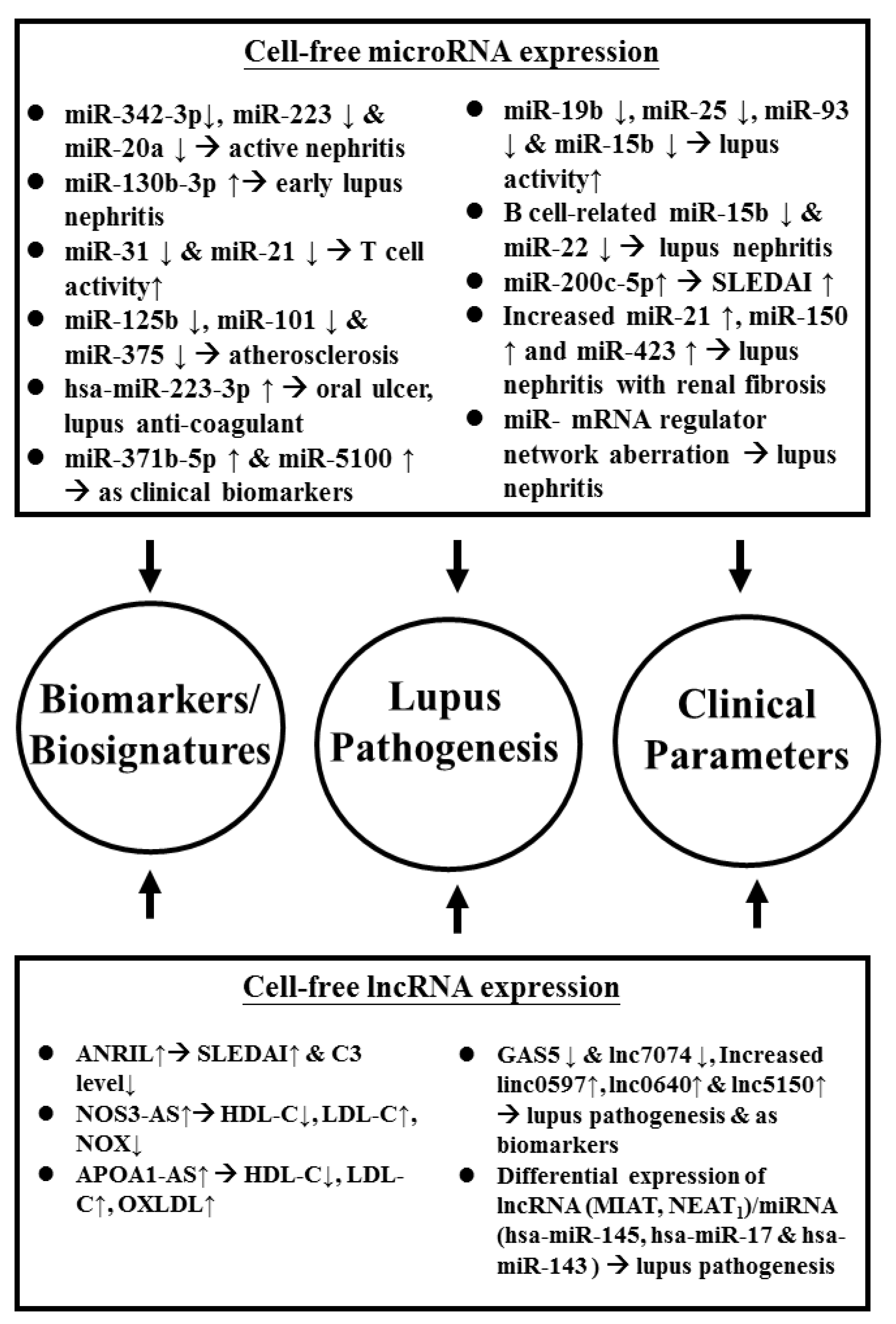
2.3.2. Abnormal Cell-Free ncRNA Expression as Biomarkers/Biosignatures and in the Pathogenesis of SLE
2.3.3. An Abnormal Cell-Free ncRNA Expression in SLE
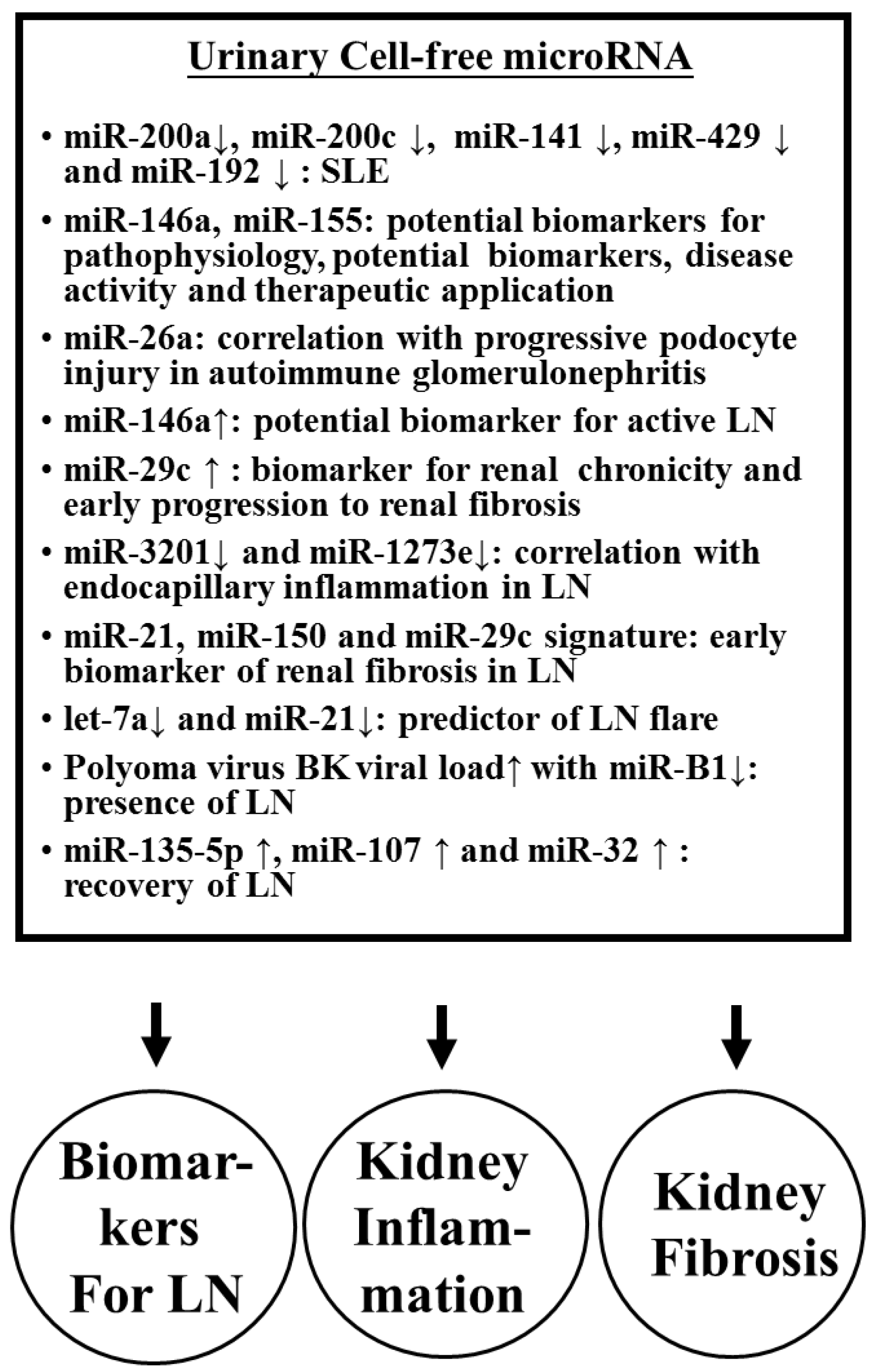
2.3.4. Imbalanced Urinary Cell-Free ncRNA Expression in SLE Patients with Nephritis
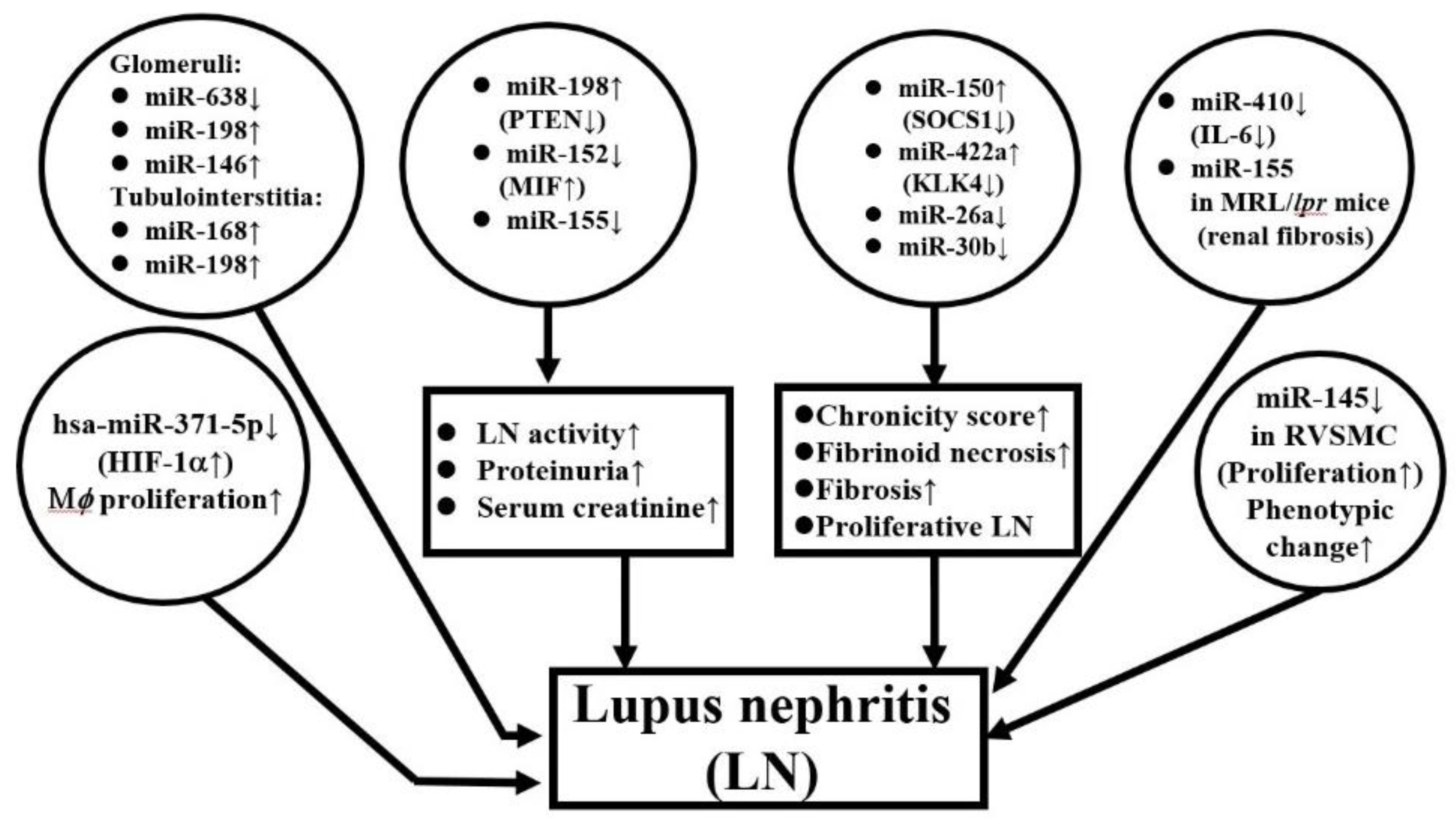
2.3.5. Aberrant ncRNA Expression in the Kidney Tissues of Patients with LN
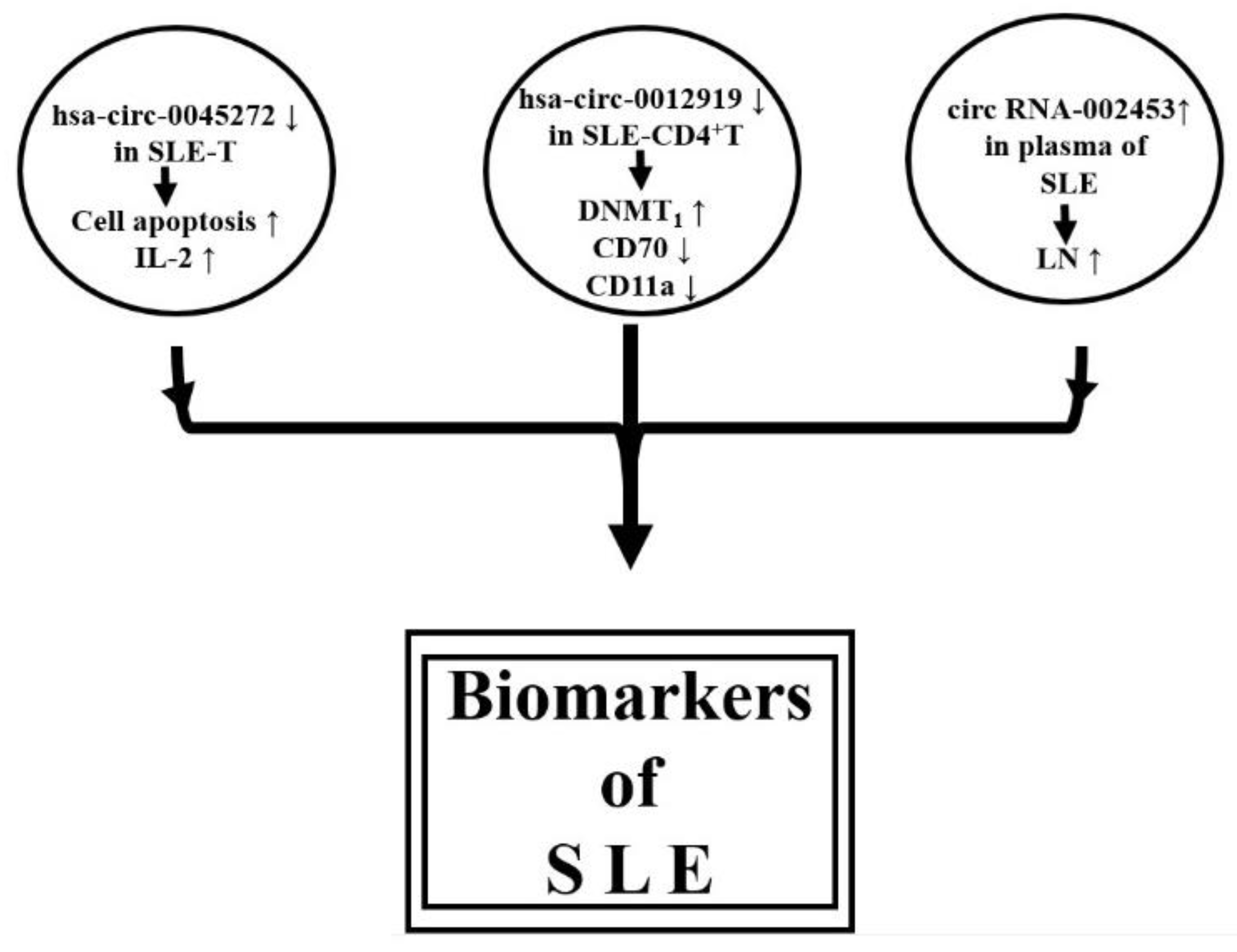
2.3.6. Circular RNAs Expression Profile in SLE
3. Conclusions
4. Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rönnblom, L.; Pascual, V. The innate immune system in SLE: Type I interferons and dendritic cells. Lupus 2008, 17, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Katsiari, C.G.; Liossis, S.-N.C.; Sfikakis, P.P. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: A reappraisal. Semin. Arthritis Rheum. 2010, 39, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Park, S.-H.; Kim, H.-Y.; Kwok, S.-K. A plasmacytoid dendritic cells-type I interferon axis is critically implicated in the pathogenesis of systemic lupus erythematosus. Int. J. Mol. Sci. 2015, 16, 14158–14170. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-Y.; Li, K.-J.; Hsieh, S.-C.; Liao, H.-T.; Yu, C.-L. What’s wrong with neutrophils in lupus? Clin. Exp. Rheumatol. 2019, 37, 684–693. [Google Scholar] [PubMed]
- Spada, R.; Rojas, J.M.; Barber, D.F. Recent findings on the role of natural killer cells in the pathogenesis of systemic lupus erythematosus. J. Leukoc. Biol. 2015, 98, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Cruz-González, D.d.J.; Gómez-Martin, D.; Layseca-Espinosa, E.; Baranda, L.; Abud-Mendoza, C.; Alcocer-Varela, J.; González-Amaro, R.; Monsiváis-Urenda, A.E. Analysis of the regulatory function of natural killer cells from patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2018, 191, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Weidenbusch, M.; Kulkarni, O.P.; Anders, H.-J. The innate immune system in human systemic lupus erythematosus. Clin. Sci. 2017, 131, 625–634. [Google Scholar] [CrossRef]
- Leffler, J.; Bengtsson, A.A.; Blom, A.M. The complement system in systemic lupus erythematosus: An update. Ann. Rheum. Dis. 2014, 73, 1601–1606. [Google Scholar] [CrossRef]
- Liu, T.; Son, M.; Diamond, B. HMGB1 in systemic lupus erythematosus. Front. Immunol. 2020, 11, 1057. [Google Scholar] [CrossRef]
- Liu, Y.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Moore, E.; Goel, R.; O’Neil, L.; Mistry, P.; Hoffmann, V.; Mondal, S.; et al. Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7-dependent lupus. JCI Insight 2018, 3, e124729. [Google Scholar] [CrossRef]
- Moulton, V.R.; Tsokos, G.C. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J. Clin. Investig. 2015, 125, 2220–2227. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, T.; Tsokos, G.C.; Moulton, V.R. Aberrant T cell signaling and subsets in systemic lupus erythematosus. Front. Immunol. 2018, 9, 1088. [Google Scholar] [CrossRef] [PubMed]
- Morawski, P.A.; Bolland, S. Expanding the B cell centric view of systemic lupus erythematosus. Trends Immunol. 2017, 38, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.H.; Chan, T.M. B cell abnormalities in systemic lupus erythematosus and lupus nephritis—Role in pathogenesis and effect of immunosuppressive treatments. Int. J. Mol. Sci. 2019, 20, 6231. [Google Scholar] [CrossRef]
- Wang, T.; Marken, J.; Chen, J.; Tran, V.B.; Li, Q.-Z.; Li, M.; Cerosaletti, K.; Elkon, K.B.; Zeng, X.; Giltiay, N.V. High TCR7 expression drives the expansion of CD19+CD24hiCD38hi transitional B cells and autoantibody production in SLE patients. Front. Immunol. 2019, 10, 1243. [Google Scholar] [CrossRef]
- O’Neil, L.J.; Kaplan, M.J.; Carmona-Rivera, C. The role of neutrophils and neutrophil extracellular traps in vascular damage in systemic lupus erythematosus. J. Clin. Med. 2019, 8, 1325. [Google Scholar] [CrossRef]
- Mackern-Oberti, J.P.; Llanos, C.; Riedel, C.A.; Bueno, S.M.; Kalergis, A.M. Contribution of dendritic cells to the autoimmune pathology of systemic lupus erythematosus. Immunology 2015, 146, 497–507. [Google Scholar] [CrossRef]
- Hervier, B.; Beziat, V.; Haroche, J.; Mathian, A.; Lebon, P.; Ghillani-Dalbin, P.; Musset, L.; Debré, P.; Amoura, Z.; Vieillard, V. Phenotype and function of natural killer cells in systemic lupus erythematosus: Excess interferon-γ production in patients with active disease. Arthritis Rheum. 2011, 63, 1698–1706. [Google Scholar] [CrossRef]
- Henriques, A.; Teixeira, L.; Inês, L.; Carvalheiro, T.; Gonçalves, A.; Martinho, A.; Pais, M.L.; da Silva, J.A.P.; Paiva, A. NK cells dysfunction in systemic lupus erythematosus: Relation to disease activity. Clin. Rheumatol. 2013, 32, 805–813. [Google Scholar] [CrossRef]
- Rose, T.; Dörner, T. Drivers of the immunopathogenesis in systemic lupus erythematosus. Best Pract. Res. Clin. Rheumatol. 2017, 31, 321–333. [Google Scholar] [CrossRef]
- Kubo, S.; Nakayamada, S.; Yoshikawa, M.; Miyazaki, Y.; Sakata, K.; Nakano, K.; Hanami, K.; Iwata, S.; Miyagawa, I.; Saito, K.; et al. Peripheral immunophenotyping identifies three subgroups based on T cell heterogeneity in lupus patients. Arthritis Rheumatol. 2017, 69, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Rodríguez, L.; Martínez-Taboada, V.; Calvo-Alén, J.; Beares, I.; Villa, I.; López-Hoyos, M. Altered Th17/Treg ratio in peripheral blood of systemic lupus erythematosus but not primary antiphospholipid syndrome. Front. Immunol. 2019, 10, 391. [Google Scholar] [CrossRef]
- Yang, S.; Hu, C.; Huang, B.; Zhang, L.; Li, Q.; Jiang, S.; Zhang, Q.; Liu, J.; Zhang, X.; Tan, J. Aberration of CCR7CD8 memory T cells from patients with systemic lupus erythematosus: An inducer of T helper type 2 bias of CD4 T cells. Immunology 2004, 112, 274–289. [Google Scholar]
- Blanco, P.; Ueno, H.; Schmitt, N. T follicular helper (Tfh) cells in lupus: Activation and involvement in SLE pathogenesis. Eur. J. Immunol. 2016, 46, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.; Weber, C.; Storch, S.; Federlin, K. Relationship between CD5+ B lymphocytes and the activity of systemic autoimmunity. Clin. Immunol. Immnopathol. 1990, 56, 219–225. [Google Scholar] [CrossRef]
- Omar, H.H.; Nasef, S.I.; Omar, H.H.; Ghaly, M.S. CD5+ B lymphocytes in systemic lupus erythematosus patients: Relation to disease activity. Clin. Rheumatol. 2017, 36, 2719–2726. [Google Scholar] [CrossRef]
- Wang, T.; Mei, Y.; Li, Z. Research progress on regulatory B cells in systemic lupus erythematosus. BioMed Res. Int. 2019, 2019, 7948687. [Google Scholar] [CrossRef]
- Pan, L.; Lu, M.-P.; Wang, J.-H.; Xu, M.; Yang, S.-R. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J. Pediatr. 2020, 16, 19–30. [Google Scholar] [CrossRef]
- Qiu, L.-J.; Xu, K.; Liang, Y.; Cen, H.; Zhang, M.; Wen, P.-F.; Ni, J.; Xu, W.-D.; Leng, R.-X.; Pan, H.-F.; et al. Decreased SOCS1 mRNA expression levels in peripheral blood mononuclear cells from patients with systemic lupus erythematosus in a Chinese population. Clin. Exp. Med. 2015, 15, 261–267. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Xia, Y. Defective suppressor of cytokine signaling 1 signaling contributes to the pathogenesis of systemic lupus erythematosus. Front. Immunol. 2017, 8, 1292. [Google Scholar] [CrossRef]
- Goropevšek, A.; Holcar, M.; Avčin, T. The role of STAT signaling pathways in the pathogenesis of systemic lupus erythematosus. Clin. Rev. Allergy Immunol. 2017, 52, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Vélez, G.; Medina, F.; Ramírez-Montaño, L.; Zarazúa-Lozada, A.; Hernández, R.; Llorente, L.; Moreno, J. Constitutive phosphorylation of interferon receptor A-associated signaling proteins in systemic lupus erythematosus. PLoS ONE 2012, 7, e41414. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Qin, Y.; Xia, S.; Dai, M.; Han, X.; Wu, Y.; Zhang, X.; Ma, J.; Wang, Y.; Tang, Y.; et al. Identification of cyclin-dependent kinase 1 as a novel regulator of type I interferon signaling in systemic lupus erythematosus. Arthritis Rheumatol. 2016, 68, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Su, Z.; Wang, Z.; Xu, H. USP7 is associated with greater disease activity in systemic lupus erythematosus via stabilization of the IFNα receptor. Mol. Med. Rep. 2017, 16, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Li, K.-J.; Wu, C.-H.; Hsieh, S.-C.; Lu, M.-C.; Tsai, C.-Y.; Yu, C.-L. Deranged bioenergetics and defective redox capacity in T lymphocytes and neutrophils are related to cellular dysfunction and increased oxidative stress in patients with systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 2012, 548516. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-T.; Lin, C.-S.; Chen, W.-S.; Liao, H.-T.; Tsai, C.-Y.; Wei, Y.-H. Leukocyte mitochondrial DNA alteration in systemic lupus erythematosus and its relevance to the susceptibility to lupus nephritis. Int. J. Mol. Sci. 2012, 13, 8853–8868. [Google Scholar] [CrossRef]
- Lee, H.-T.; Wu, T.-H.; Lin, C.-S.; Lee, C.-S.; Wei, Y.-H.; Tsai, C.-Y.; Chang, D.-M. The pathogenesis of systemic lupus erythematosus-from the viewpoint of oxidative stress and mitochondrial dysfunction. Mitochondrion 2016, 30, 1–7. [Google Scholar] [CrossRef]
- Lee, H.-T.; Wu, T.-H.; Lin, C.-S.; Lee, C.-S.; Pan, S.-C.; Chang, D.-M.; Wei, Y.-H.; Tsai, C.-Y. Oxidative DNA and mitochondrial DNA change in patients with SLE. Front. Biosci. 2017, 22, 493–503. [Google Scholar]
- Lee, H.-T.; Lin, C.-S.; Pan, S.-C.; Wu, T.-H.; Lee, C.-S.; Chang, D.-M.; Tsai, C.-Y.; Wei, Y.-H. Alterations of oxygen consumption and extracellular acidification rates by glutamine in PBMCs of SLE patients. Mitochondrion 2019, 44, 65–74. [Google Scholar] [CrossRef]
- Tsai, C.-Y.; Shen, C.-Y.; Liao, H.-T.; Li, K.-J.; Lee, H.-T.; Lu, C.-S.; Wu, C.-H.; Kuo, Y.-M.; Hsieh, S.-C.; Yu, C.-L. Molecular and cellular bases of immunosenescence, inflammation, and cardiovascular complications mimicking “inflammaging” in patients with systemic lupus erythematosus. Int. J. Mol. Sci. 2019, 20, 3878. [Google Scholar] [CrossRef]
- Almlöf, J.C.; Alexsson, A.; Imgenberg-Kreuz, J.; Sylwan, L.; Bäcklin, C.; Leonard, D.; Nordmark, G.; Tandre, K.; Eloranta, M.L.; Padyukov, L.; et al. Novel risk genes for systemic lupus erythematosus predicted by random forest classification. Sci. Rep. 2017, 7, 6236. [Google Scholar] [CrossRef] [PubMed]
- Julià, A.; López-Longo, F.J.; Venegas, J.J.P.; Bonàs-Guarch, S.; Olivé, A.; Andreu, J.L.; Aquirre-Zamorano, M.Á.; Vela, P.; Nolla, J.M.; da la Fuente, J.L.M.; et al. Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Res. Ther. 2018, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Regunathan-Shenk, R.; Radhakrishnan, J. Pathogenesis of SLE nephritis in the era of precision medicine. Curr. Rheumatol. Rev. 2018, 14, 140–144. [Google Scholar] [PubMed]
- Goulielmos, G.N.; Zervou, M.I.; Vazgiourakis, V.M.; Ghodke-Puranik, Y.; Garyfallos, A.; Niewold, T.B. The genetics and molecular pathogenesis of systemic lupus erythematosus (SLE) in populations of different ancestry. Gene 2018, 668, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-C.; Chun, S.; Kim, K.; Mak, A. Update on the genetics of systemic lupus erythematosus: Genome-wide association studies and beyond. Cells 2019, 8, 1180. [Google Scholar] [CrossRef]
- Fike, A.J.; Elcheva, I.; Rahman, Z.S.M. The post-GWAS era: How to validate the contribution of gene variants in lupus. Curr. Rheumatol. Rep. 2019, 21, 3. [Google Scholar] [CrossRef]
- Deng, Y.; Tsao, B.P. Advances in lupus genetics and epigenetics. Curr. Opin. Rheumatol. 2014, 26, 482–492. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Metzler, G.; Wray-Dutra, M.; Jackson, S.W. Altered B cell signalling in autoimmunity. Nat. Rev. Immunol. 2017, 17, 421–436. [Google Scholar] [CrossRef]
- Gorradin, O.; Cohen, A.J.; Luppino, J.M.; Bayles, I.M.; Schumacher, F.R.; Scacheri, P.C. Modeling disease risk through analysis of physical interactions between genetic variants within chromatin regulatory circuitry. Nat. Genet. 2016, 48, 1313–1320. [Google Scholar] [CrossRef]
- Jackson, S.W.; Kolhatkar, N.S.; Rawlings, D.J. B cells take the front seat: Dysregulated B cell signals orchestrate loss of tolerance and autoantibody production. Curr. Opin. Immunol. 2015, 33, 70–77. [Google Scholar] [CrossRef]
- Bronson, P.G.; Chaivorapol, C.; Ortmann, W.; Behrens, T.W.; Graham, R.R. The genetics of type I interferon in systemic lupus erythematosus. Curr. Opin. Immunol. 2012, 24, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Catalina, M.D.; Owen, K.A.; Labonte, A.C.; Grammer, A.C.; Lipsky, P.E. The pathogenesis of systemic lupus erythematosus: Harnessing big data to understand the molecular basis of lupus. J. Autoimmun. 2020, 110, 102359. [Google Scholar] [CrossRef] [PubMed]
- Lever, E.; Alves, M.R.; Isenberg, D.A. Towards precision medicine in systemic lupus erythematosus. Pharm. Pers. Med. 2020, 13, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Fielder, A.H.L.; Walport, M.J.; Batchelor, J.R.; Rynes, R.I.; Black, C.M.; Dodi, I.A.; Hughes, G.R.V. Family study of the major histocompatibility complex in patients with systemic lupus erythematosus: Importance of null alleles of C4A and C4B in determining disease susceptibility. Br. Med. J. (Clin. Res. Ed.) 1983, 286, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Davidson, A. Taming lupus—A new understanding of pathogenesis is leading to clinical advances. Nat. Med. 2012, 18, 871–882. [Google Scholar] [CrossRef]
- Alarcón, G.S.; McGwin, G., Jr.; Petri, M.; Ramsey-Goldman, R.; Fessler, B.J.; Vilá, L.M.; Edberg, J.C.; Reveille, J.D.; Kimberly, R.P.; PROFILE study group. Time to renal disease and end-stage renal disease in PROFILE: A multiethnic lupus cohort. PLoS. Med. 2006, 3, e396. [Google Scholar] [CrossRef]
- Brown, E.E.; Edberg, J.C.; Kimberly, R.P. Fc receptor genes and the systemic lupus erythematosus diathesis. Autoimmunity 2007, 40, 567–581. [Google Scholar] [CrossRef]
- Morris, D.L.; Roberts, A.L.; Witherden, A.S.; Tarzi, R.; Barros, P.; Whittaker, J.C.; Cook, T.H.; Aitman, T.J.; Vyse, T.J. Evidence for both copy number and allelic (NA1/NA2) risk at the FCGR3B locus in systemic lupus erythematosus. Eur. J. Hum. Genet. 2010, 18, 1027–1031. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Wang, C.-M.; Chang, S.-W.; Cheng, C.-H.; Wu-Jan, Y.-J.; Lin, J.-C.; Yang, B.; Ho, H.-H.; Wu, J. Association of FCGR3A and FCGR3B copy number variations with systemic lupus erythematosus and rheumatoid arthritis in Taiwanese patients. Arthritis Rheumatol. 2014, 66, 3113–3121. [Google Scholar] [CrossRef]
- Fernando, M.M.A.; Stevens, C.R.; Walsh, E.C.; De Jager, P.L.; Goyette, P.; Plenge, R.M.; Vyse, T.J.; Rioux, J.D. Defining the role of the MHC in autoimmunity: A review and pooled analysis. PLoS Genet. 2008, 4, e1000024. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Niewold, T.B. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J. Autoimmun. 2015, 64, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-Y.; Hsieh, S.-C.; Lu, C.-S.; Wu, T.-H.; Liao, H.-T.; Wu, C.-H.; Li, K.-J.; Kuo, Y.-M.; Lee, H.-T.; Shen, C.-Y.; et al. Cross-talk between mitochondrial dysfunction-provoked oxidative stress and aberrant noncoding RNA expression in the pathogenesis and pathophysiology of SLE. Int. J. Mol. Sci. 2019, 20, 5183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, M.; Sawalha, A.H.; Richardson, B.; Lu, Q. Impaired DNA methylation and its mechanisms in CD4(+) T cells of systemic lupus erythematosus. J. Autoimmun. 2013, 41, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Coit, P.; Renauer, P.; Jeffries, M.A.; Merrill, J.T.; McCune, W.J.; Maksimowicz-McKinnon, K.; Sawalha, A.H. Renal involvement in lupus is characterized by unique DNA methylation changes in naïve CD4+ T cells. J. Autoimmun. 2015, 61, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Almlöf, J.C.; Leonard, D.; Alexsson, A.; Nordmark, G.; Eloranta, M.-L.; Rantapää-Dahlqvist, S.; Bengtsson, A.A.; Jönsen, A.; Padyukov, L.; et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 736–743. [Google Scholar] [CrossRef]
- Yeung, K.S.; Chung, B.H.-Y.; Choufani, S.; Mok, M.Y.; Wong, W.L.; Mak, C.C.Y.; Yang, W.; Lee, P.P.W.; Wong, W.H.S.; Chen, Y.; et al. Genome-wide DNA methylation analysis of Chinese patients with systemic lupus erythematosus identified hypomethylation in genes related to the type I interferon pathway. PLoS ONE 2017, 12, e0169553. [Google Scholar] [CrossRef]
- Teruel, M.; Sawalha, A.H. Epigenetic variability in systemic lupus erythematosus: What we learned from genome-wide DNA methylation studies. Curr. Rheumatol. Rep. 2017, 19, 32. [Google Scholar] [CrossRef]
- Weeding, E.; Sawalha, A.H. Deoxyribonucleic acid methylation in systemic lupus erythematosus: Implications for future clinical practice. Front. Immunol. 2018, 9, 875. [Google Scholar] [CrossRef]
- Joseph, S.; George, N.I.; Green-knox, B.; Treadwell, E.L.; Word, B.; Yim, S.; Lyn-Cook, B. Epigenome-wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon-related genes based on ethnicity and SLEDAI. J. Autoimmun. 2019, 96, 147–157. [Google Scholar] [CrossRef]
- de la Calle-Fabregat, C.; Morante-Palacios, O.; Ballestar, E. Understanding the relevance of DNA methylation changes in immune differentiation and disease. Genes 2020, 11, 110. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, H.; Li, Y.; Dai, X.; Zhou, B.; Li, Q.; Zuo, X.; Luo, H. The role of IF135 in lupus nephritis and related mechanisms. Mod. Rheumatol. 2017, 27, 1010–1018. [Google Scholar] [CrossRef]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.-S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Sui, W.; Tan, Q.; Yang, M.; Yang, Q.; Lin, H.; Ou, M.; Xue, W.; Chen, J.; Zou, T.; Jing, H.; et al. Genome-wide analysis of 5-hmC in the peripheral blood of systemic lupus erythematosus patients using an hMeDIP-chip. Int. J. Mol. Med. 2015, 35, 1467–1479. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, M.; Wang, J.; Liao, W.; Li, D.; Li, M.; Wu, H.; Zhang, Y.; Gershwin, M.E.; Lu, Q. Increased 5-hydroxymethylcytosine in CD4(+) T cells in systemic lupus erythematosus. J. Autoimmun. 2016, 69, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Lei, W.; Zhang, G.; Zhou, Y.; Su, Y.; Lu, Q. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 2008, 35, 804–810. [Google Scholar] [PubMed]
- Dai, Y.; Zhang, L.; Hu, C.; Zhang, Y. Genome-wide analysis of histone H3 lysine 4 trimethylation by ChIP-chip in peripheral blood mononuclear cells of systemic lupus erythematosus patients. Clin. Exp. Rheumatol. 2010, 28, 158–168. [Google Scholar]
- Zhang, Z.; Song, L.; Maurer, K.; Petri, M.A.; Sullivan, K.E. Global H4 acetylation analysis by ChIP-chip in SLE monocytes. Genes Immun. 2010, 11, 124–133. [Google Scholar] [CrossRef]
- Zhou, Y.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Zhong, Q.; Zhao, M.; Lu, Q. Histone modifications and methyl-CpG-binding domain protein levels at the TNFSF7 (CD70) promoter in SLE CD4+ T cells. Lupus 2011, 20, 1365–1371. [Google Scholar] [CrossRef]
- Hedrick, C.M. Epigenetics in SLE. Curr. Rheumatol. Rep. 2017, 19, 58. [Google Scholar] [CrossRef]
- Hedrich, C.M. Mechanistic aspects of epigenetic dysregulation in SLE. Clin. Immunol. 2018, 196, 3–11. [Google Scholar] [CrossRef]
- Farivar, S.; Aghamaleki, F.S. Effects of major epigenetic factors on systemic lupus erythematosus. Iran. Biomed. J. 2018, 22, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, P.; Tzioufas, A.G. Epigenetic perspectives on systemic autoimmune disease. J. Autoimmun. 2019, 104, 102315. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Arito, M.; Takakuwa, Y.; Ooka, S.; Sato, T.; Kurokawa, M.S.; Okamoto, K.; Uchida, T.; Suematsu, N.; Kato, T. Altered posttranslational modification on U1 small nuclear ribonucleoprotein 68k in systemic autoimmune diseases detected by 2D Western blot. Electrophoresis 2012, 33, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Routsias, J.G.; Tzioufas, A.G. B-cell epitopes of the intracellular autoantigens Ro/SSA and La/SSB: Tools to study the regulation of the autoimmune response. J. Autoimmun. 2010, 35, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Terzoglou, A.G.; Routsias, J.G.; Avrameas, S.; Moutsopoulos, H.M.; Tzioufas, A.G. Preferential recognition of the phosphorylated major linear B-cell epitope of La/SSB 349-368 aa by anti-La/SSB autoantibodies from patients with systemic autoimmune diseases. Clin. Exp. Immunol. 2006, 144, 432–439. [Google Scholar] [CrossRef]
- Brahms, H.; Raymackers, J.; Union, A.; de Keyser, F.; Meheus, L.; Lührmann, R. The C-terminal RG dipeptide repeats of the spliceosomal Sm proteins D1 and D3 contain symmetrical dimethylarginines, which form a major B-cell epitope for anti-Sm autoantibodies. J. Biol. Chem. 2000, 275, 17122–17129. [Google Scholar] [CrossRef] [PubMed]
- Zavala-Cerna, M.G.; Martínez-García, E.A.; Torres-Bugarin, O.; Rubio-Jurado, B.; Riebeling, C.; Nava, A. The clinical significance of posttranslational modification of autoantigens. Clin. Rev. Allergy Immunol. 2014, 47, 73–90. [Google Scholar] [CrossRef]
- Moulton, V.R.; Gillooly, A.R.; Tsokos, G.C. Ubiquitination regulates expression of the serine/arginine-rich splicing factor 1 (SRSF1) in normal and systemic lupus erythematosus (SLE) T cells. J. Biol. Chem. 2014, 289, 4126–4134. [Google Scholar] [CrossRef]
- Barrera-Vargas, A.; Gómez-Martín, D.; Carmona-Rivera, C.; Merayo-Chalico, J.; Torres-Ruiz, J.; Manna, Z.; Hasni, S.; Alcocer-Varela, J. Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 944–950. [Google Scholar] [CrossRef]
- Quiroz, E.N.; Chavez-Estrada, V.; Macias-Ochoa, K.; Ayala-Navarro, M.F.; Flores-Aguilar, A.S.; Morales-Navarrete, F.; de la Cruz Lopez, F.; Escorcia, L.G.; Musso, C.G.; Martinez, G.A.; et al. Epigenetic mechanisms and posttranslational modifications in systemic lupus erythematosus. Int. J. Mol. Sci. 2019, 20, 5679. [Google Scholar] [CrossRef]
- Wei, J.-W.; Huang, K.; Yang, C.; Kang, C.-S. Non-coding RNAs as regulators in epigenetics (review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lui, C. Coding or non-coding, the converging concepts of RNAs. Front. Genet. 2019, 10, 496. [Google Scholar] [CrossRef] [PubMed]
- Turpin, D.; Truchetet, M.-E.; Faustin, B.; Augusto, J.-F.; Contin-Bordes, C.; Brisson, A.; Blanco, P.; Duffau, P. Role of extracellular vesicles in autoimmune diseases. Autoimmun. Rev. 2016, 15, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Wu, H.; Liu, Y.; Zhao, M.; Li, D.; Lu, Q. Recent advances of exosomes in immune modulation and autoimmune diseases. Autoimmunity 2016, 49, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Liang, D.; Tang, Y.; Qu, B.; Cui, H.; Luo, X.; Huang, X.; Chen, S.; Higgs, B.W.; Jallal, B.; et al. Identification of microRNA-31 as a novel regulator contributing to impaired inerleukin-2 production in T cells from patients with systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 3715–3725. [Google Scholar] [CrossRef]
- Lu, M.-C.; Lai, N.-S.; Chen, H.-C.; Yu, H.-C.; Huang, K.-Y.; Tung, C.-H.; Huang, H.-B.; Yu, C.-L. Decreased microRNA (miR)-145 and increased miR-224 expression in T cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin. Exp. Immunol. 2013, 171, 91–99. [Google Scholar] [CrossRef]
- Lu, M.-C.; Yu, C.-L.; Chen, H.-C.; Yu, H.-C.; Huang, H.-B.; Lai, N.-S. Aberrant T cell expression of Ca2+ influx-regulated miRNAs in patients with systemic lupus erythematosus promotes lupus pathogenesis. Rheumatology 2015, 54, 343–348. [Google Scholar] [CrossRef]
- Khoshmirsafa, M.; Kianmehr, N.; Falak, R.; Mowla, S.J.; Seif, F.; Mirzaei, B.; Valizadeh, M.; Shekarabi, M. Elevated expression of miR-21 and miR-155 in peripheral blood mononuclear cells as potential biomarkers for lupus nephritis. Int. J. Rheum. Dis. 2019, 22, 458–467. [Google Scholar] [CrossRef]
- Li, L.-J.; Zhao, W.; Tao, S.-S.; Li, J.; Xu, S.-Z.; Wang, J.-B.; Leng, R.-X.; Fan, Y.-G.; Pan, H.-F.; Ye, D.-Q. Comprehensive long non-coding RNA expression profiling reveals their potential roles in systemic lupus erythematosus. Cell. Immunol. 2017, 319, 17–27. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Chen, S.; Du, J.; Lin, J.; Qin, H.; Wang, J.; Liang, J.; Xu, J. Long noncoding RNA expression profile and association with SLEDAI score in monocyte-derived dendritic cells from patients with systemic lupus erythematosus. Arthritis Res. Ther. 2018, 20, 138. [Google Scholar] [CrossRef]
- Cao, H.-Y.; Li, D.; Wang, Y.-P.; Lu, H.-X.; Sun, J.; Li, H.-B. Clinical significance of reduced expression of lncRNA TUG1 in the peripheral blood of systemic lupus erythematosus patients. Int. J. Rheum. Dis. 2020, 23, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Xu, X.; Zhang, H.; Chen, C.; Hou, Y.; Yao, G.; Wang, S.; Wang, D.; Feng, X.; Sun, L.; et al. Comprehensive expression profile of long non-coding RNAs in peripheral blood mononuclear cells from patients with neuropsychiatric systemic lupus erythematosus. Ann. Transl. Med. 2020, 8, 349. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Tan, Y.; Luo, H. MALAT1 is involved in type I IFNs-mediated systemic lupus erythematosus by up-regulating OAS2, OAS3, and OASL. Braz. J. Med. Biol. Res. 2020, 53, e9292. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Wang, X.; Wang, L.; Chu, X.; Hu, X.; Sun, L.; Jiang, M.; Wang, H.; Wang, Z.; Zhao, H.; et al. Full high-throughput sequencing analysis of differences in expression profiles of long noncoding RNAs and their mechanisms of action in systemic lupus erythematosus. Arthritis Res. Ther. 2019, 21, 70. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liu, L.; Min, X.; Jia, S.; Zhao, M. Non-coding RNAs in CD4+ T cells: New insights into the pathogenesis of systemic lupus erythematosus. Front. Immunol. 2020, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Perez-Hernandez, J.; Cortes, R. Extracellular vesicles as biomarkers of systemic lupus erythematosus. Dis. Markers 2015, 2015, 613536. [Google Scholar] [CrossRef]
- Yáñez-Mό, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extraceull. Vesicles 2015, 4. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Perez-Hernandez, J.; Redon, J.; Cortes, R. Extracellular vesicles as therapeutic agents in systemic lupus erythematosus. Int. J. Mol. Sci. 2017, 18, 717. [Google Scholar] [CrossRef]
- Wang, G.; Tam, L.-S.; Li, E.K.-M.; Kwan, B.C.-H.; Chow, K.-M.; Luk, C.C.-W.; Li, P.K.-T.; Szeto, C.-C. Serum and urinary cell-free miR-146a and miR-155 in patients with systemic lupus erythematosus. J. Rheumatol. 2010, 37, 2516–2522. [Google Scholar] [CrossRef]
- Carlsen, A.L.; Schetter, A.J.; Nielsen, C.T.; Lood, C.; Knudsen, S.; Voss, A.; Harris, C.C.; Hellmark, T.; Segelmark, M.; Jacobsen, S.; et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mou, S.; Wang, L.; Zhang, M.; Shao, X.; Fang, W.; Lu, R.; Qi, C.; Fan, Z.; Cao, Q.; et al. Up-regulation of serum miR-130b-3p level is associated with renal damage in early lupus nephritis. Sci. Rep. 2015, 5, 12644. [Google Scholar] [CrossRef]
- Amr, K.S.; Bayoumi, F.S.; Elgengehy, F.T.; Abdallah, S.O.; Ahmed, H.H.; Eissa, E. The role of microRNA-31 and microRNA-21 as regulatory biomarkers in the activation of T lymphocytes of Egyptian lupus patients. Rheumatol. Int. 2016, 36, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Kay, S.D.; Carlsen, A.L.; Voss, A.; Burton, M.; Diederichsen, A.; Poulsen, M.K.; Heegaard, N. Associations of circulating cell-free mcrioRNA with vasculopathy and vascular events in systemic lupus erythematosus patients. Scand. J. Rheumatol. 2019, 48, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-S.; Jung, J.-Y.; Jeon, J.-Y.; Kim, H.-A.; Suh, C.-H. Circulating hsa-miR-30e-5p, hsa-miR-92a-3p, and hsa-miR-223-3p may be novel biomarkers in systemic lupus erythematosus. HLA 2016, 88, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Wu, J.-L.; Liu, L.-M.; Jiang, J.-Q.; Wu, H.-J.; Zhao, M.; Lu, Q.-J. Serum miRNA-371b-5p and miR-5100 act as biomarkers for systemic lupus erythematosus. Clin. Immunol. 2018, 196, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Quiroz, E.; Pacheco-Lugo, L.; Navarro-Quiroz, R.; Lorenzi, H.; España-Puccini, P.; Díaz-Olmos, Y.; Almendrales, L.; Olave, V.; Gonzalez-Torres, H.; Diaz-Perez, A.; et al. Profiling analysis of circulating microRNA in peripheral blood of patients with class IV lupus nephritis. PLoS ONE 2017, 12, e0187973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, X.; Ye, L.; Guo, G.; Li, X.; Chen, C.; Sun, L.; Li, B.; Chen, N.; Xue, X. B cell related circulating microRNAs with the potential value of biomarkers in the differential diagnosis, and distinguishment between the disease activity and lupus nephritis for systemic lupus erythematosus. Front. Immunol. 2018, 9, 1473. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y. The correlation of plasma microRNA-200 family expressions with risk and disease severity of lupus nephritis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3118–3125. [Google Scholar]
- Nakhjavani, M.; Etemadi, J.; Pourlak, T.; Mirhosaini, Z.; Vahed, S.Z.; Abediazar, S. Plasma levels of miR-21, miR-150, miR-423 in patients with lupus nephritis. Iran. J. Kidney Dis. 2019, 13, 198–206. [Google Scholar]
- Abd-Elmawla, M.A.; Fawzy, M.W.; Rizk, S.M.; Shaheen, A.A. Role of long non-coding RNAs expression (ANRIL, NOS3-AS, and APOA1-AS) in development of atherosclerosis in Egyptian systemic lupus erythematosus patients. Clin. Rheumatol. 2018, 37, 3319–3328. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-C.; Hu, Y.; Guan, S.-Y.; Ye, D.-Q.; Pan, H.-F. Differential plasma expression profiles of long non-coding RNAs reveal potential biomarkers for systemic lupus erythematosus. Biomolecules 2019, 9, 206. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, W.; Zheng, F.; Tang, D.; Liu, D.; Wang, G.; Xu, Y.; Yin, L.; Zhang, X.; Dai, Y. Reconstruction and analysis of the aberrant lncRNA-miRNA-mRNA network in systemic lupus erythematosus. Lupus 2020, 29, 398–406. [Google Scholar] [CrossRef]
- Lucafò, M.; Di Silvestre, A.; Romano, M.; Avian, A.; Antonelli, R.; Martelossi, S.; Naviglio, S.; Tommasini, A.; Stocco, G.; Ventura, A.; et al. Role of the long non-coding RNA growth arrest-specific 5 in glucocorticoid response in children with inflammatory bowel disease. Basic Clin. Pharmacol. Toxicol. 2018, 122, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Lucafò, M.; Pugnetti, L.; Bramuzzo, M.; Curci, D.; Di Silvestre, A.; Marcuzzi, A.; Bergamo, A.; Martelossi, S.; Villanacci, V.; Bozzola, A.; et al. Long non-coding RNA GAS5 and intestinal MMP2 and MMP9 expression: A translational study in pediatric patients with IBD. Int. J. Mol. Sci. 2019, 20, 5280. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, T.-P.; Zhao, Y.-L.; Li, B.-Z.; Leng, R.-X.; Pan, H.-F.; Ye, D.-Q. Decreased H19, GAS5, and linc0597 expression and association analysis of related gene polymorphisms in rheumatoid arthritis. Biomolecules 2020, 10, 55. [Google Scholar] [CrossRef]
- Santoro, M.; Nociti, V.; Lucchini, M.; De Fino, C.; Losavio, F.A.; Mirabella, M. Expression profile of long non-coding RNAs in serum of patients with multiple sclerosis. J. Mol. Neurosci. 2016, 59, 18–23. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, N.; Du, L.; Wang, Y. Downregulation of lncRNA NEAT1 inhibits mouse mesangial cell proliferation, fibrosis, and inflammation but promotes apoptosis in diabetic nephropathy. Int. J. Clin. Exp. Pathol. 2019, 12, 1174–1183. [Google Scholar]
- Dong, G.; Yang, Y.; Li, X.; Yao, X.; Zhu, Y.; Zhang, H.; Wang, H.; Ma, Q.; Zhang, J.; Shi, H.; et al. Granulocytic myeloid-derived suppressor cells contribute to IFN-I signaling activation of B cells and disease progression through the lncRNA NEAT1-BAFF axis in systemic lupus erythematosus. Biochim. Biophys. Act. Mol. Bas. Dis. 2020, 1866, 165554. [Google Scholar] [CrossRef]
- Wang, G.; Tam, L.S.; Li, E.K.M.; Kwan, B.C.H.; Chow, K.M.; Luk, C.C.W.; Li, P.K.T.; Szeto, C.C. Serum and urinary free microRNA level in patients with systemic lupus erythematosus. Lupus 2011, 20, 493–500. [Google Scholar] [CrossRef]
- Wang, G.; Tam, L.-S.; Kwan, B.C.-H.; Li, E.K.-M.; Chow, K.-M.; Luk, C.C.-W.; Li, P.K.-T.; Szeto, C.-C. Expression of miR-146a and miR-155 in the urinary sediment of systemic lupus erythematosus. Clin. Rheumatol. 2012, 31, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Ichii, O.; Otsuka-Kanazawa, S.; Horino, T.; Kimura, J.; Nakamura, T.; Matsumoto, M.; Toi, M.; Kon, Y. Decreased miR-26a expression correlates with the progression of podocyte injury in autoimmune glomerulonephritis. PLoS ONE 2014, 9, e110383. [Google Scholar] [CrossRef] [PubMed]
- Perez-Hernandez, J.; Forner, M.J.; Pinto, C.; Chaves, F.J.; Cortes, R.; Redon, J. Increased urinary exosomal microRNAs in patients with systemic lupus erythematosus. PLoS ONE 2015, 10, e0138618. [Google Scholar] [CrossRef] [PubMed]
- Solé, C.; Cortés-Hernández, J.; Felip, M.L.; Vidal, M.; Ordi-Ros, J. miR-29c in urinary exosomes as predictor of early renal fibrosis in lupus nephritis. Nephrol. Dial. Transplant. 2015, 30, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Gonzalez, M.; Srivastava, A.; Pavkovic, M.; Bijol, V.; Rennke, H.G.; Stillman, I.E.; Zhang, X.; Parikh, S.; Rovin, B.H.; Afkarian, M.; et al. Identification, confirmation, and replication of novel urinary microRNA biomarkers in lupus nephritis and diabetic nephropathy. Clin. Chem. 2017, 63, 1515–1526. [Google Scholar] [CrossRef]
- Solé, C.; Moliné, T.; Vidal, M.; Ordi-Ros, J.; Cortés-Hernández, J. An exosomal urinary miRNA signature for early diagnosis of renal fibrosis in lupus nephritis. Cells 2019, 8, 773. [Google Scholar] [CrossRef] [PubMed]
- Tangtanatakul, P.; Klinchanhom, S.; Sodsai, P.; Sutichet, T.; Promjeen, C.; Avihingsanon, Y.; Hirankarn, N. Down-regulation of let-7a and miR-21 in urine exosomes from lupus nephritis patients during disease flare. Asian Pac. J. Allergy Immunol. 2019, 37, 189–197. [Google Scholar] [PubMed]
- Li, Y.-J.; Wu, H.-H.; Liu, S.-H.; Tu, K.-H.; Lee, C.-C.; Hsu, H.-H.; Chang, M.-Y.; Yu, K.-H.; Chen, W.; Tian, Y.-C. Polyomavirus BK, BKV microRNA, and urinary neutrophil gelatinase-associated lipocalin can be used as potential biomarkers of lupus nephritis. PLoS ONE 2019, 14, e0210633. [Google Scholar] [CrossRef]
- Cao, Y.; Cao, X.; Sun, L.; Li, Y. miR-206 inhibits cell proliferation and extracellular matrix accumulation by targeting hypoxia-inducible factor 1- alpha (HIF-1α) in mesangial cells treated with high glucose. Med. Sci. Monit. 2019, 25, 10036–10044. [Google Scholar] [CrossRef]
- Garcia-Vives, E.; Solé, C.; Moliné, T.; Vidal, M.; Agraz, I.; Ordi-Ros, J.; Cortés-Hernández, J. The urinary exosomal miRNA expression profile is predictive of clinical response in lupus nephritis. Int. J. Mol. Sci. 2020, 21, 1372. [Google Scholar] [CrossRef]
- Lu, J.; Kwan, B.C.-H.; Lai, F.M.-M.; Tam, L.-S.; Li, E.K.-M.; Chow, K.-M.; Wang, G.; Li, P.K.-T.; Szeto, C.-C. Glomerular and tubulointerstitial miR-638, miR-198 and miR-146a expression in lupus nephritis. Nephrology 2012, 17, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hasni, S.A.; Perez, P.; Tandon, M.; Jang, S.I.; Zheng, C.; Kopp, J.B.; Austin, H., 3rd; Balow, J.E.; Alevizos, I.; et al. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. J. Am. Soc. Nephrol. 2013, 24, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Krasoudaki, E.; Banos, A.; Stagakis, E.; Loupasakis, K.; Drakos, E.; Sinatkas, V.; Zampoulaki, A.; Papagianni, A.; Iliopoulos, D.; Boumpas, D.T.; et al. Micro-RNA analysis of renal biopsies in human lupus nephritis demonstrates up-regulated miR-422a driving reduction of akllikrein-related peptidase 4. Nephrol. Dial. Transplant. 2016, 31, 1076–1686. [Google Scholar] [CrossRef] [PubMed]
- Costa-Reis, P.; Russo, P.A.; Zhang, Z.; Colonna, L.; Maurer, K.; Gallucci, S.; Schulz, S.W.; Kiani, A.N.; Petri, M.; Sullivan, K.E. The role of microRNAs and human epidermal growth factor receptor 2 in proliferative lupus nephritis. Arthritis Rheumatol. 2015, 67, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Sun, L.; Fang, W.; Wang, H.; Yao, D.; Cui, R.; Xu, J.; Wang, L.; Wang, X. Hsa-miR-371-5p inhibits human mesangial cell proliferation and promotes apoptosis in lupus nephritis by directly targeting hypoxia-inducible factor 1α. Mol. Med. Rep. 2016, 14, 5693–5698. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, N.; Zhang, J.; Zhao, H.; Wang, X. miR-410 suppresses the expression of IL-6 as well as renal fibrosis in the pathogenesis of lupus nephritis. Clin. Exp. Pharmacol. Physiol. 2016, 43, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Leiss, H.; Salzberger, W.; Jacobs, B.; Gessl, I.; Kozakowski, N.; Blüml, S.; Puchner, A.; Kiss, A.; Podesser, B.K.; Smolen, J.S.; et al. microRNA 155-deficiency leads to decreased autoantibody levels and reduced severity of nephritis and pneumonitis in pristane-induced lupus. PLoS ONE 2017, 12, e0181015. [Google Scholar] [CrossRef]
- Kong, J.; Li, L.; Lu, Z.; Song, J.; Yan, J.; Yang, J.; Gu, Z.; Da, Z. microRNA-155 suppresses mesangial cell proliferation and TGF-β1 production via inhibiting CXCR5-ERK signaling pathway in lupus nephritis. Inflammation 2019, 42, 255–263. [Google Scholar] [CrossRef]
- Li, X.; Luo, F.; Li, J.; Luo, C. miR-183 delivery attenuates murine lupus nephritis-related injuries via targeting mTOR. Scand. J. Immunol. 2019, 90, e12810. [Google Scholar] [CrossRef]
- Cui, D.; Zhu, D.; Ren, H.; Lin, J.; Lai, W.; Huang, Q.; Zhao, J.; Yang, M. MciroRNA198 contributes to lupus nephritis progression by inhibition of phosphatase and tensin homology deleted on chromosome ten expression. Mol. Med. Rep. 2017, 7813–7820. [Google Scholar] [CrossRef]
- Zheng, J.; Guo, R.; Tang, Y.; Fu, Q.; Chen, J.; Wu, L.; Leng, L.; Bucala, R.; Song, Y.; Lu, L. miR-152 attenuates the severity of lupus nephritis through the downregulation of macrophage migration inhibitory factor (MIF)-induced expression of COL1A1. Front. Immunol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xiang, W.; Peng, X.; Ding, Y.; Liao, W.; He, X. MicroRNA-145 involves in the pathogenesis of renal vascular lesions and may become a potential therapeutic target in patients with juvenile lupus nephritis. Kidney Blood Press. Res. 2019, 44, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, S.; Chen, H.; Mo, X.; Li, T.; Shao, Y.; Xiao, B.; Guo, J. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin. Chim. Acta 2015, 444, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhu, H.; Shi, Y.; Wu, W.; Cai, H.; Chen, X. cir-ITCH plays an inhibitory role in colorectal cancer by regulating the Wnt/β–catenin pathway. PLoS ONE 2015, 10, e0131225. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Liu, G.; Huo, X.; Tao, X.; Sun, X.; Ge, Z.; Yang, J.; Fan, J.; Liu, L.; Qin, W. Has_circ_0001649: A circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark. 2016, 16, 161–169. [Google Scholar] [CrossRef]
- Su, H.; Lin, F.; Deng, X.; Shen, L.; Fang, Y.; Fei, Z.; Zhao, L.; Zhang, X.; Pan, H.; Xie, D.; et al. Profiling and bioinformatics analyses reveal differential circular RNA expression in radioresistant esophageal cancer cells. J. Trasl. Med. 2016, 14, 225. [Google Scholar] [CrossRef]
- Li, L.-J.; Huang, Q.; Pan, H.-F.; Ye, D.-Q. Circular RNAs and systemic lupus erythematosus. Exp. Cell Res. 2016, 346, 248–254. [Google Scholar] [CrossRef]
- Li, L.-J.; Zhu, Z.-W.; Zhao, W.; Tao, S.-S.; Li, B.-Z.; Xu, S.-Z.; Wang, J.-B.; Zhang, M.-Y.; Wu, J.; Leng, R.-X.; et al. Circular RNA expression profile and potential function of has_circ_0045272 in systemic lupus erythematosus. Immunology 2018, 155, 137–149. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.; Chen, Y.; Wu, Z.; Zhang, C.; Shi, W. The down-regulation of hsa_circ_0012919, the sponge for miR-125a-3p, contributes to DNA methylation of CD11a and CD70 in CD4+ T cells of systemic lupus erythematosus. Clin. Sci. 2018, 132, 2285–2298. [Google Scholar] [CrossRef]
- Quyang, Q.; Huang, Q.; Jiang, Z.; Zhao, J.; Shi, G.-P.; Yang, M. Using plasma circRNA_002453 as a novel biomarker in the diagnosis of lupus nephritis. Mol. Immunol. 2018, 101, 531–538. [Google Scholar]
- Li, H.; Li, K.; Lai, W.; Li, X.; Wang, H.; Yang, J.; Chu, S.; Wang, H.; Kang, C.; Qui, Y. Comprehensive circular RNA profiles in plasma reveals that circular RNAs can be used as novel biomarkers for systemic lupus erythematosus. Clin. Chim. Acta 2018, 480, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Cortes, R.; Forner, M.J. Circular RNAs: Novel biomarkers of disease activity in systemic lupus erythematosus? Clin. Sci. 2019, 133, 1049–1052. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Immunological Function | References |
|---|---|---|
| [I] Innate immune cells: | ||
|
| [1,2,3,17] |
| ||
| ||
| IL-1β, IL-6, TNF-α, IFN-α | ||
|
| [5,6,18,19] |
| ||
| ||
|
| [4,16] |
| ||
| ||
| ||
| [II] Adaptive immune cells: | ||
|
| [11,12,20,21,22,23,24] |
| ||
| ||
| ||
|
| [13,14,15,25,26,27] |
| ||
| ||
| [III] Intracellular signaling: | ||
|
↑ (?) | [29,30] [31] [32,33] [34] |
| [IV] Immunometabolism |
| [35] |
| [V] Redox capacity ● Oxidative stresses | ↓ ↑ | [36,37,38,39,40] |
| ● Mitochondrial DNA heteroplasmy | ↑ |
| [I] Advances in genome-wide association studies on SLE [43,44,45,46,47,48,49,50,51,52,53]. | ||
| ● Immune complex processing genes: | ||
| • | C1q, C2, C4 | |
| • | FcγR2A, FcγR3A | |
| • | CRP | |
| • | ITGAM | |
| ● Immune signal transduction genes: | ||
| • | STAT4 | • TNFSF4 |
| • | IRF5 | • BLK |
| • | BAN K1 | • MECP2 (?) |
| • | PTPN22 | • PXK (?) |
| • | PCDCD1 | • XKR6 (?) |
| ● TLR/IFN-1 pathway: | ||
| • | TREX1 | • IRAK1 (?) |
| • | TNFAIP3 | • STAT1 (?) |
| ● HLA-DR: | ||
| • | Disease-association: DR3, DR9, DR15, DQA1*0101 | |
| • | Disease-protection: DR4, DR11, DR14 | |
| ● Others: | ||
| • | PDHX/CD44 | • IFNK |
| • | ICAM1-ICAM4-ICAM5 | • UBE2 L3 |
| • | TRAF6 | • IRF8 |
| • | PPP2CA | • MAV5 |
| • | MYG9-APOL1 | |
| ● IFN-1 signaling: | ||
| • | IRF4 | • MYC |
| • | IRF5 | • IFIH1 (MDA5) |
| • | ERK1 | • TNFAIP3 |
| ● B cell receptor signaling: | ||
| • | FcγRIIb (FcγRIIB) | • PRDM1 (BLIMP1) |
| • | BANK1 (BANK1) | • CSK (C-Src tyrosine kinase, CSK) |
| • | BLK (BLK) | |
| [I] DNA hypomethylation in CD4+ T cells [63,64,65,66,67,68,69,70] | ||
| ● ITGAL (CD11a) | ● PT PRC (CD45) | |
| ● PRF1 (perforin) | ● UHRF1 BP1 | |
| ● TNF SF7 (CD70) | ● IRF5 | |
| ● TNF SF5 (CD40L) | ● IKZF3 | |
| ● PP2A cα | ● UBE2L3 | |
| ● IRF7 (IFN-1 master regulatory gene) | ● MHC-class III ● HLA-DQβ2 | |
| [II] DNA hypomethylation in lupus nephritis specimen [64,71] | ||
| ● IFNγR | ● IFI35 | |
| ● STAT1 | ● IRF7 | |
| [III] Increased DNA hydroxymethylation in CD4+ T cells [72,73,74] | ||
| ● TREX1 | ● SOCS1 | |
| ● CDKN1A | ● NR2F | |
| ● CDKN1B | ● IL15RA | |
| ● CTCF | ||
| [I] | Histone modifications in SLE |
| ● Global histone H3 and H4 hyperacetylation in active lupus CD4+ T cells [75,76] | |
| ● H3K4me variants in PBMC of SLE [76] | |
| ● Increased H4 acetylation in SLE-monocytes [77] | |
| ● Elevated histone H3 acetylation and dimethylated H3 lysine 4 (H3K4me2) levels in lupus CD4+ T cells [78] | |
| ● Increased H3K18ac and reduced H3K27me3 of IL-17 gene cluster in SLE-T cells [79] | |
| ● H3K18 deacetylation and H3K27 trimethylation of IL-2 in CD3+ T, CD4+ T and effector (CD4+) T cells in SLE [79,80,81] | |
| ● H3K18ac elevation for IL-10 in CD3+ and CD4+ T cells in SLE [79,80,81] | |
| ● H3ac elevation for TNF-α in SLE-monocytes [79,80,81] | |
| ● H3K4me elevation for IRF1 in SLE-monocytes [82] | |
| [II] | Non-histone protein modifications in patients with SLE |
| ● U1 small nuclear ribonucleoprotein 68 k [83] | |
| ● MAPK signaling molecules [84] | |
| ● Complex ribonuclear proteins SSA/Ro and SSB/La [84,85] | |
| ● Spliceosomal Sm protein [86,87] | |
| ● Serine/arginine rich splicing factor 1 (SRSF1) [88] | |
| ● Neutrophilic myeloperoxidase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-Y.; Shen, C.-Y.; Liu, C.-W.; Hsieh, S.-C.; Liao, H.-T.; Li, K.-J.; Lu, C.-S.; Lee, H.-T.; Lin, C.-S.; Wu, C.-H.; et al. Aberrant Non-Coding RNA Expression in Patients with Systemic Lupus Erythematosus: Consequences for Immune Dysfunctions and Tissue Damage. Biomolecules 2020, 10, 1641. https://doi.org/10.3390/biom10121641
Tsai C-Y, Shen C-Y, Liu C-W, Hsieh S-C, Liao H-T, Li K-J, Lu C-S, Lee H-T, Lin C-S, Wu C-H, et al. Aberrant Non-Coding RNA Expression in Patients with Systemic Lupus Erythematosus: Consequences for Immune Dysfunctions and Tissue Damage. Biomolecules. 2020; 10(12):1641. https://doi.org/10.3390/biom10121641
Chicago/Turabian StyleTsai, Chang-Youh, Chieh-Yu Shen, Chih-Wei Liu, Song-Chou Hsieh, Hsien-Tzung Liao, Ko-Jen Li, Cheng-Shiun Lu, Hui-Ting Lee, Cheng-Sung Lin, Cheng-Han Wu, and et al. 2020. "Aberrant Non-Coding RNA Expression in Patients with Systemic Lupus Erythematosus: Consequences for Immune Dysfunctions and Tissue Damage" Biomolecules 10, no. 12: 1641. https://doi.org/10.3390/biom10121641
APA StyleTsai, C.-Y., Shen, C.-Y., Liu, C.-W., Hsieh, S.-C., Liao, H.-T., Li, K.-J., Lu, C.-S., Lee, H.-T., Lin, C.-S., Wu, C.-H., Kuo, Y.-M., & Yu, C.-L. (2020). Aberrant Non-Coding RNA Expression in Patients with Systemic Lupus Erythematosus: Consequences for Immune Dysfunctions and Tissue Damage. Biomolecules, 10(12), 1641. https://doi.org/10.3390/biom10121641