New Trends for Antimalarial Drugs: Synergism between Antineoplastics and Antimalarials on Breast Cancer Cells



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Line and Cell Culture
2.3. Drug Treatment
2.4. Cell Viability Assay
2.5. Data Analysis
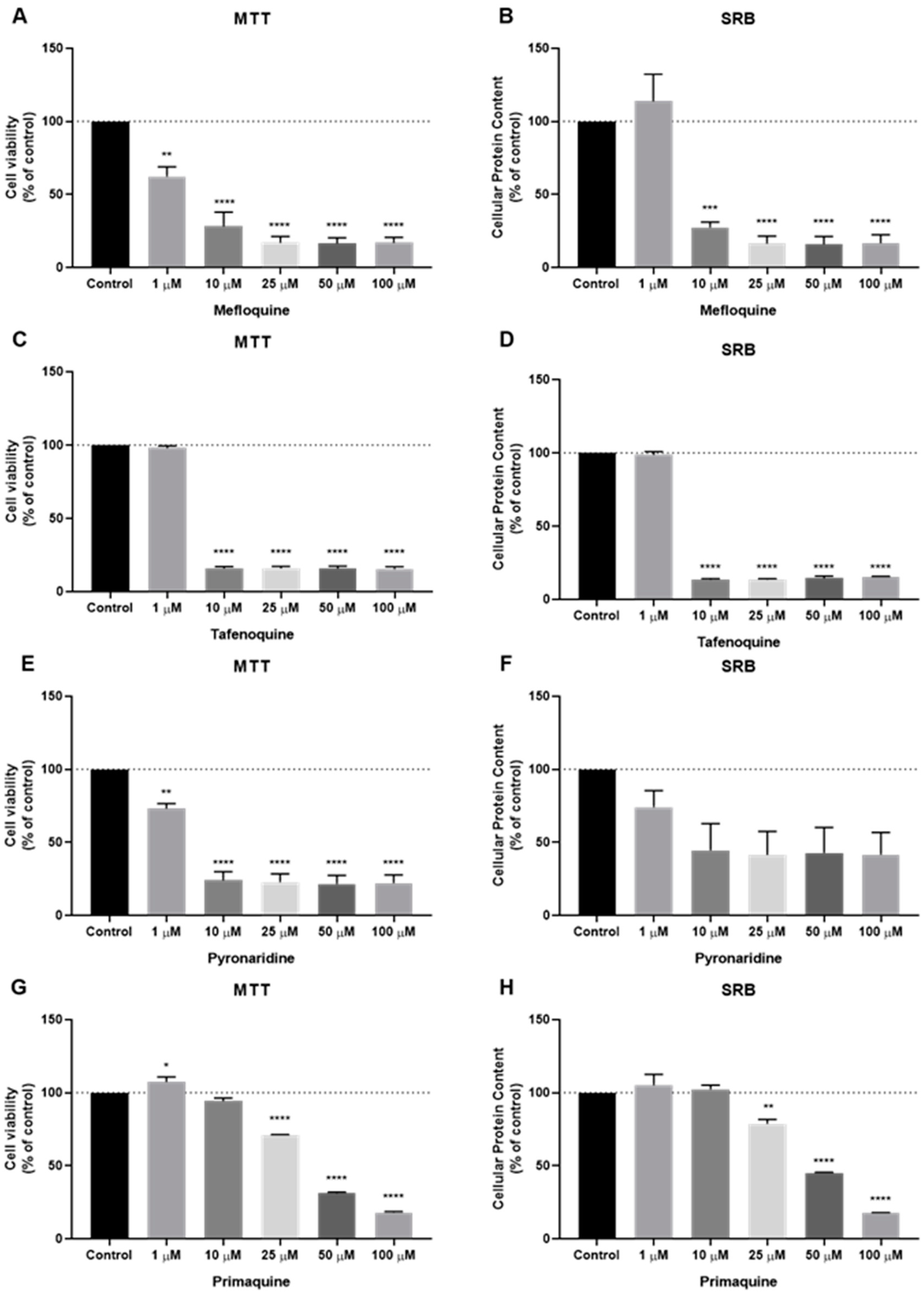
2.6. Analysis of Drug Interactions
2.7. Statistical Analysis
3. Results
3.1. Effect of DOX and PTX as Single Agents on MCF-7 Cellular Viability
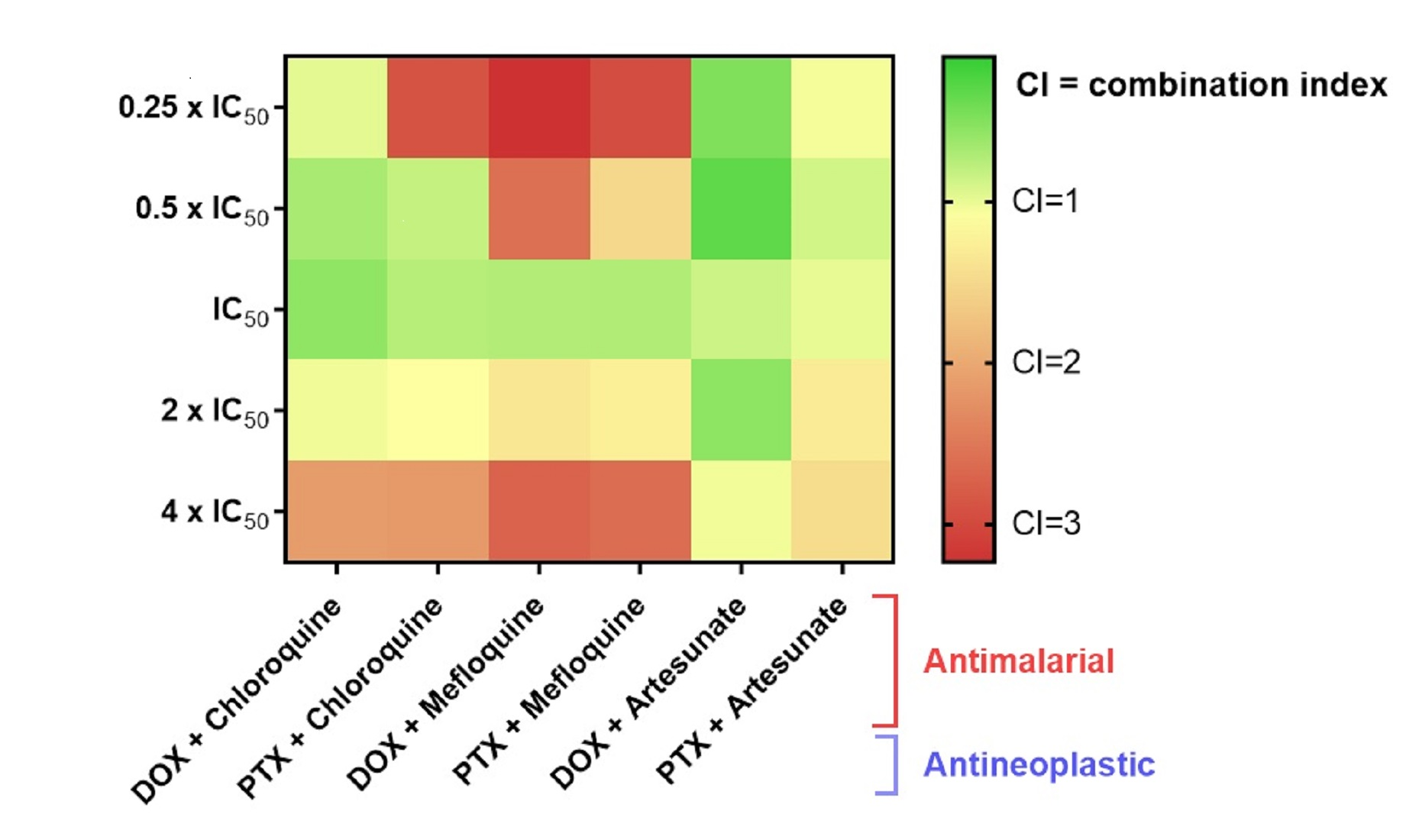
3.2. Effect of Antimalarial Quinolinic Derivates as Single Agents on MCF-7 Cellular Viability
3.3. Effect of Antimalarial Non-Quinolinic Derivates as Single Agents on MCF-7 Cellular Viability
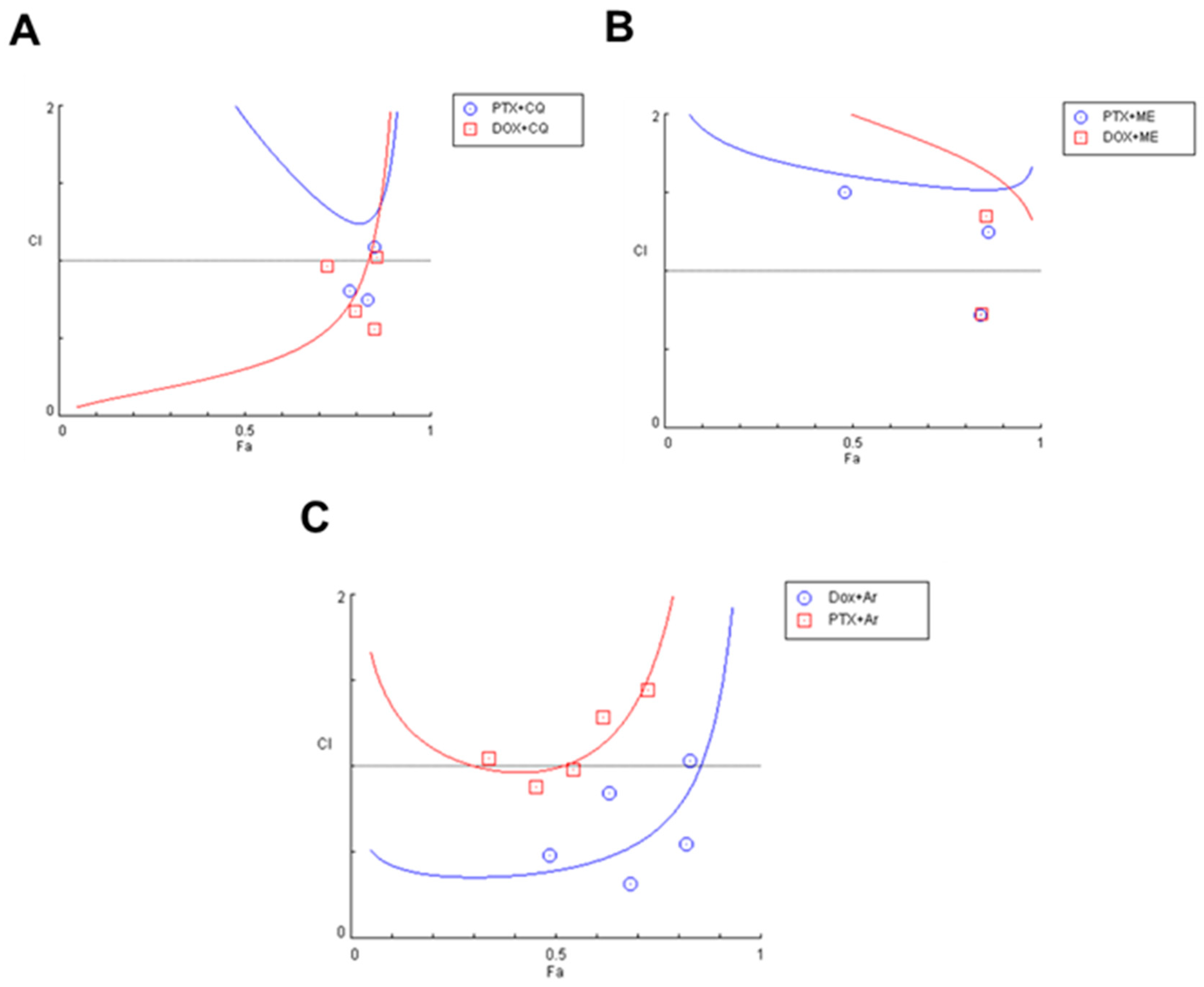
3.4. Effect of Various Combinations of DOX and Different Antimalarials in MCF-7 Cells
3.5. Effect of Various Combinations of PTX and Different Antimalarials in MCF-7 Cells
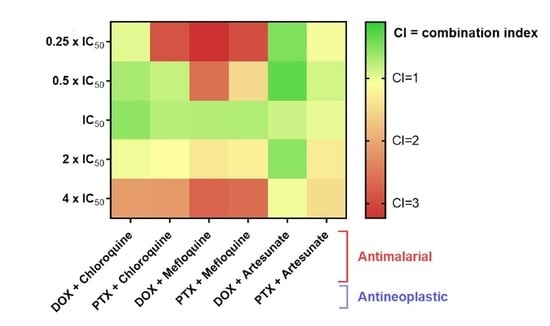
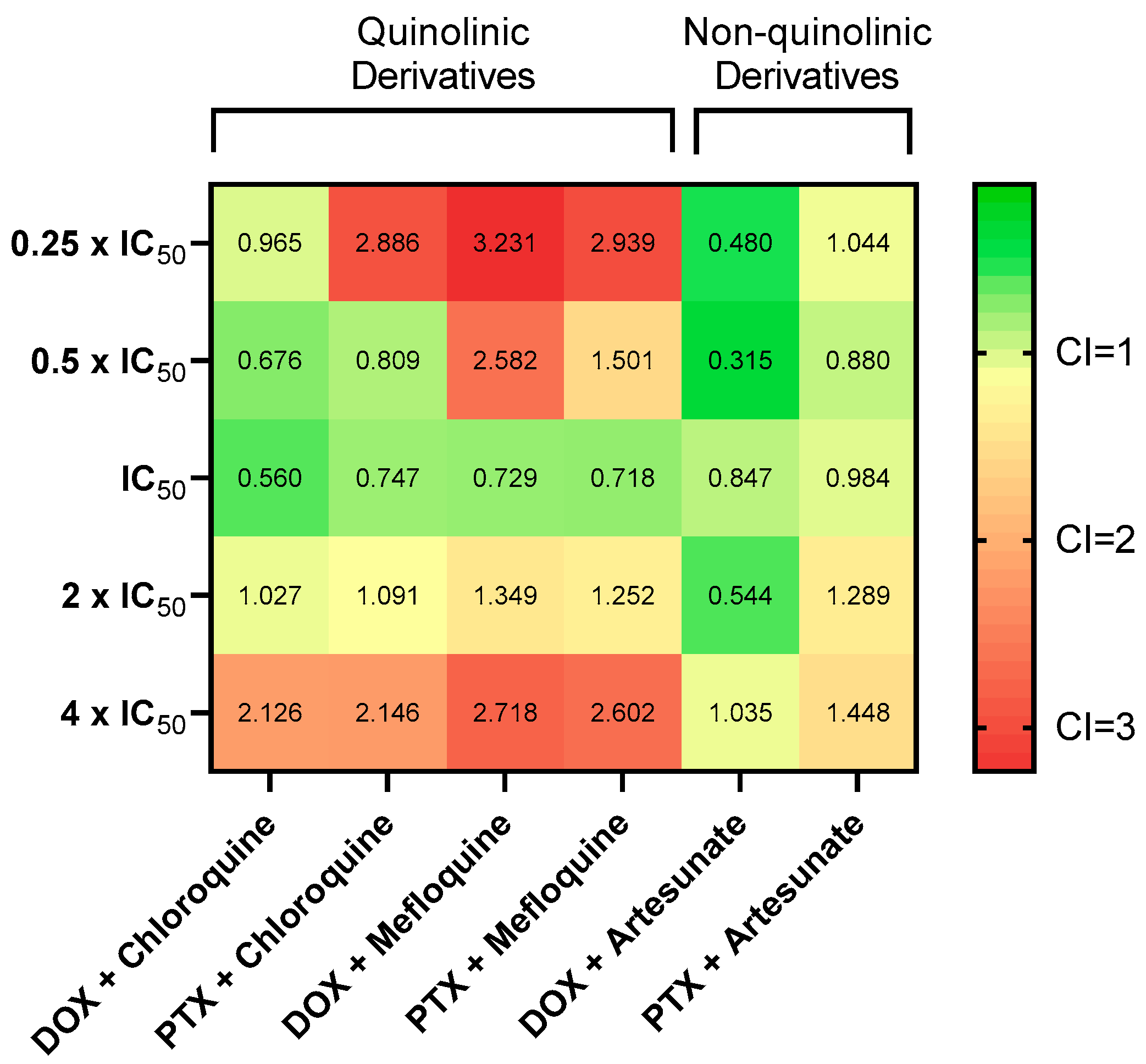
3.6. Synergistic Combinations of Antineoplastic and Antimalarial Drugs in MCF-7 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Breast Cancer Facts & Figures 2019–2020; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Hu, L.X.; Du, Y.Y.; Zhang, Y.; Pan, Y.Y. Synergistic effects of exemestane and aspirin on MCF-7 human breast cancer cells. Asian Pac. J. Cancer Prev. 2012, 13, 5903–5908. [Google Scholar] [CrossRef] [PubMed]
- Pagani, O.; Senkus, E.; Wood, W.; Colleoni, M.; Cufer, T.; Kyriakides, S.; Costa, A.; Winer, E.P.; Cardoso, F. International Guidelines for Management of Metastatic Breast Cancer: Can Metastatic Breast Cancer Be Cured? JNCI J. Natl. Cancer Inst. 2010, 102, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Coley, H.M. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev. 2008, 34, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Stegmeier, F.; Warmuth, M.; Sellers, W.R.; Dorsch, M. Targeted Cancer Therapies in the Twenty-First Century: Lessons from Imatinib. Clin. Pharmacol. Ther. 2010, 87, 543–552. [Google Scholar] [CrossRef]
- Wu, G.-S.; Lu, J.-J.; Guo, J.-J.; Huang, M.-Q.; Gan, L.; Chen, X.-P.; Wang, Y.-T. Synergistic anti-cancer activity of the combination of dihydroartemisinin and doxorubicin in breast cancer cells. Pharmacol. Rep. 2013, 65, 453–459. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Wonders, K.Y.; Reigle, B.S. Trastuzumab and Doxorubicin-Related Cardiotoxicity and the Cardioprotective Role of Exercise. Integr. Cancer Ther. 2009, 8, 17–21. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, D.; Yang, R.; Wang, S. Paclitaxel: New uses for an old drug. Drug Des. Dev. Ther. 2014, 8, 279. [Google Scholar] [CrossRef]
- Markman, M.; Mekhail, T.M. Paclitaxel in cancer therapy. Expert Opin. Pharmacother. 2002, 3, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Chen, S.; Wang, Y.; Watahiki, A.; Bohrer, L.; Sun, Z.; Wang, Y.; Huang, H. Inhibition of the Androgen Receptor as a Novel Mechanism of Taxol Chemotherapy in Prostate Cancer. Cancer Res. 2009, 69, 8386–8394. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel Action of Paclitaxel against Cancer Cells: Bystander Effect Mediated by Reactive Oxygen Species. Cancer Res. 2007, 67, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Taxol. Available online: https://www.breastcancer.org/treatment/druglist/taxol (accessed on 11 November 2020).
- Atkins, J.H.; Gershell, L.J. Selective anticancer drugs. Nat. Rev. Drug Discov. 2002, 1, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Vicent, M.J.; Ringsdorf, H.; Duncan, R. Polymer therapeutics: Clinical applications and challenges for development. Adv. Drug Deliv. Rev. 2009, 61, 1117–1120. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Chidley, C.; Sorger, P.K. A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Cokol, M. Drugs and their Interactions. Curr. Drug Discov. Technol. 2013, 10, 106–113. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, D.; Wu, B.; Quan, Y.; Liu, D.; Li, Y.; Zhang, X. Synergistic activity of an antimetabolite drug and tyrosine kinase inhibitors against breast cancer cells. Chem. Pharm. Bull. 2017, 65, 768–775. [Google Scholar] [CrossRef][Green Version]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.-S.; Choi, J.Y. Synergistic Anti-Cancer Effect of Phenformin and Oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef]
- Mei, L.; Chen, Y.; Wang, Z.; Wang, J.; Wan, J.; Yu, C.; Liu, X.; Li, W. Synergistic anti-tumour effects of tetrandrine and chloroquine combination therapy in human cancer: A potential antagonistic role for p21. Br. J. Pharmacol. 2015, 172, 2232–2245. [Google Scholar] [CrossRef]
- Gao, Y.; Xiao, X.; Zhang, C.; Yu, W.; Guo, W.; Zhang, Z.; Li, Z.; Feng, X.; Hao, J.; Zhang, K.; et al. Melatonin synergizes the chemotherapeutic effect of 5-fluorouracil in colon cancer by suppressing PI3K/AKT and NF-κB/iNOS signaling pathways. J. Pineal Res. 2017, 62, e12380. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.M.; Haass, N.K.; Brafford, P.A.; Lioni, M.; Flaherty, K.T.; Herlyn, M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol. Cancer Ther. 2006, 5, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- McMeekin, S.; Dizon, D.; Barter, J.; Scambia, G.; Manzyuk, L.; Lisyanskaya, A.; Oaknin, A.; Ringuette, S.; Mukhopadhyay, P.; Rosenberg, J.; et al. Phase III randomized trial of second-line ixabepilone versus paclitaxel or doxorubicin in women with advanced endometrial cancer. Gynecol. Oncol. 2015, 138, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.A.; Alonso, C.; Pérez, J.M. Biochemical Modulation of Cisplatin Mechanisms of Action: Enhancement of Antitumor Activity and Circumvention of Drug Resistance. Chem. Rev. 2003, 103, 645–662. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Zheng, W.; Sun, W.; Simeonov, A. Drug repurposing screens and synergistic drug-combinations for infectious diseases. Br. J. Pharmacol. 2018, 175, 181–191. [Google Scholar] [CrossRef]
- Grammer, A.C.; Lipsky, P.E. Drug Repositioning Strategies for the Identification of Novel Therapies for Rheumatic Autoimmune Inflammatory Diseases. Rheum. Dis. Clin. N. Am. 2017, 43, 467–480. [Google Scholar] [CrossRef]
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.; Capistrano, R.; Crispino, S.; Gyawali, B.; Rooman, I.; Van Nuffel, A.M.T.; Meheus, L.; Sukhatme, V.P.; et al. ReDO_DB: The repurposing drugs in oncology database. Ecancermedicalscience 2018, 12, 886. [Google Scholar] [CrossRef]
- Madden, R.M.; Pui, C.-H.; Hughes, W.T.; Flynn, P.M.; Leung, W. Prophylaxis ofPneumocystis carinii pneumonia with atovaquone in children with leukemia. Cancer 2007, 109, 1654–1658. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Stein, H.A.; Turschner, S.; Toegel, I.; Mora, R.; Jennewein, N.; Efferth, T.; Eils, R.; Brady, N.R. Artesunate Activates Mitochondrial Apoptosis in Breast Cancer Cells via Iron-catalyzed Lysosomal Reactive Oxygen Species Production. J. Biol. Chem. 2011, 286, 6587–6601. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Hooft van Huijsduijnen, R.; Guy, R.K.; Chibale, K.; Haynes, R.K.; Peitz, I.; Kelter, G.; Phillips, M.A.; Vennerstrom, J.L.; Yuthavong, Y.; Wells, T.N.C.C.; et al. Anticancer Properties of Distinct Antimalarial Drug Classes. PLoS ONE 2013, 8, e82962. [Google Scholar] [CrossRef]
- Laudisi, F.; Marônek, M.; Di Grazia, A.; Monteleone, G.; Stolfi, C. Repositioning of Anthelmintic Drugs for the Treatment of Cancers of the Digestive System. Int. J. Mol. Sci. 2020, 21, 4957. [Google Scholar] [CrossRef]
- Lamoureux, F.; Zoubeidi, A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy 2013, 9, 1119–1120. [Google Scholar] [CrossRef]
- Tang, H.; Sampath, P.; Yan, X.; Thorne, S.H. Potential for enhanced therapeutic activity of biological cancer therapies with doxycycline combination. Gene Ther. 2013, 20, 770–778. [Google Scholar] [CrossRef]
- Kim, J.-H.H.; Choi, A.-R.R.; Kim, Y.K.; Yoon, S. Co-treatment with the anti-malarial drugs mefloquine and primaquine highly sensitizes drug-resistant cancer cells by increasing P-gp inhibition. Biochem. Biophys. Res. Commun. 2013, 441, 655–660. [Google Scholar] [CrossRef]
- Wong, W.; Bai, X.-C.; Sleebs, B.E.; Triglia, T.; Brown, A.; Thompson, J.K.; Jackson, K.E.; Hanssen, E.; Marapana, D.S.; Fernandez, I.S.; et al. Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat. Microbiol. 2017, 2, 17031. [Google Scholar] [CrossRef]
- Yan, K.-H.; Lin, Y.-W.; Hsiao, C.-H.; Wen, Y.-C.; Lin, K.-H.; Liu, C.-C.; Hsieh, M.-C.; Yao, C.-J.; Yan, M.-D.; Lai, G.-M.; et al. Mefloquine induces cell death in prostate cancer cells and provides a potential novel treatment strategy in vivo. Oncol. Lett. 2013, 5, 1567–1571. [Google Scholar] [CrossRef]
- Sharma, N.; Thomas, S.; Golden, E.B.; Hofman, F.M.; Chen, T.C.; Petasis, N.A.; Schönthal, A.H.; Louie, S.G. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012, 326, 143–154. [Google Scholar] [CrossRef]
- Hounkpatin, A.B.; Kreidenweiss, A.; Held, J. Clinical utility of tafenoquine in the prevention of relapse of Plasmodium vivax malaria: A review on the mode of action and emerging trial data. Infect. Drug Resist. 2019, 12, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.V.V.P.; Pantziarka, P.; Sukhatme, V.P.V.V.P.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Thomé, R.; Lopes, S.C.P.; Costa, F.T.M.; Verinaud, L. Chloroquine: Modes of action of an undervalued drug. Immunol. Lett. 2013, 153, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.H.; Yoon, J.S.; Won, Y.-W.W.; Park, B.-B.B.; Lee, Y.Y. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. Apmis 2012, 120, 597–604. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef]
- Fan, C.; Wang, W.; Zhao, B.; Zhang, S.; Miao, J. Chloroquine inhibits cell growth and induces cell death in A549 lung cancer cells. Bioorg. Med. Chem. 2006, 14, 3218–3222. [Google Scholar] [CrossRef]
- Rahim, R.; Strobl, J.S. Hydroxychloroquine, chloroquine, and all-trans retinoic acid regulate growth, survival, and histone acetylation in breast cancer cells. Anticancer Drugs 2009, 20, 736–745. [Google Scholar] [CrossRef]
- Jiang, P.; Zhao, Y.; Shi, W.; Deng, X.; Xie, G.; Mao, Y.; Li, Z.; Zheng, Y.; Yang, S.; Wei, Y. Cell Growth Inhibition, G2/M Cell Cycle Arrest, and Apoptosis Induced by Chloroquine in Human Breast Cancer Cell Line Bcap-37. Cell. Physiol. Biochem. 2008, 22, 431–440. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhao, Y.-L.; Deng, X.; Yang, S.; Mao, Y.; Li, Z.; Jiang, P.; Zhao, X.; Wei, Y. Chloroquine Inhibits Colon Cancer Cell Growth In Vitro and Tumor Growth In Vivo via Induction of Apoptosis. Cancer Investig. 2009, 27, 286–292. [Google Scholar] [CrossRef]
- Villanueva, P.J.; Martinez, A.; Baca, S.T.; DeJesus, R.E.; Larragoity, M.; Contreras, L.; Gutierrez, D.A.; Varela-Ramirez, A.; Aguilera, R.J. Pyronaridine exerts potent cytotoxicity on human breast and hematological cancer cells through induction of apoptosis. PLoS ONE 2018, 13, 1–18. [Google Scholar] [CrossRef]
- Baird, J.K.; Hoffman, S.L. Primaquine Therapy for Malaria. Clin. Infect. Dis. 2004, 39, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.K.; Fryauff, D.J.; Hoffman, S.L.; Kevin Baird, J.; Fryauff, D.J.; Hoffman, S.L. Primaquine for Prevention of Malaria in Travelers. Clin. Infect. Dis. 2003, 37, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Camarda, G.; Jirawatcharadech, P.; Priestley, R.S.; Saif, A.; March, S.; Wong, M.H.L.; Leung, S.; Miller, A.B.; Baker, D.A.; Alano, P.; et al. Antimalarial activity of primaquine operates via a two-step biochemical relay. Nat. Commun. 2019, 10, 3226. [Google Scholar] [CrossRef] [PubMed]
- Jha, T.K.; Sundar, S.; Thakur, C.P.; Felton, J.M.; Sabin, A.J.; Horton, J. A phase II dose-ranging study of sitamaquine for the treatment of visceral leishmaniasis in India. Am. J. Trop. Med. Hyg. 2005, 73, 1005–1011. [Google Scholar] [CrossRef]
- Wasunna, M.K.; Rashid, J.R.; Mbui, J.; Kirigi, G.; Kinoti, D.; Lodenyo, H.; Felton, J.M.; Sabin, A.J.; Albert, M.J.; Horton, J. A phase II dose-increasing study of sitamaquine for the treatment of visceral leishmaniasis in Kenya. Am. J. Trop. Med. Hyg. 2005, 73, 871–876. [Google Scholar] [CrossRef]
- Carvalho, L.; Luque-Ortega, J.R.; López-Martín, C.; Castanys, S.; Rivas, L.; Gamarro, F. The 8-Aminoquinoline Analogue Sitamaquine Causes Oxidative Stress in Leishmania donovani Promastigotes by Targeting Succinate Dehydrogenase. Antimicrob. Agents Chemother. 2011, 55, 4204–4210. [Google Scholar] [CrossRef]
- Proguanil | DrugBank Online. Available online: https://go.drugbank.com/drugs/DB01131 (accessed on 3 November 2020).
- Hughes, W.; Leoung, G.; Kramer, F.; Bozzette, S.A.; Safrin, S.; Frame, P.; Clumeck, N.; Masur, H.; Lancaster, D.; Chan, C.; et al. Comparison of Atovaquone (566C80) with Trimethoprim-Sulfamethoxazole to Treat Pneumocystis carinii Pneumonia in Patients with AIDS. N. Engl. J. Med. 1993, 328, 1521–1527. [Google Scholar] [CrossRef]
- Radloff, P.; Philips, J.; Nkeyi, M.; Kremsner, P.; Radloff, P.; Hutchinson, D.; Kremsner, P. Atovaquone and proguanil for Plasmodium falciparum malaria. Lancet 1996, 347, 1511–1514. [Google Scholar] [CrossRef]
- Mather, M.W.; Darrouzet, E.; Valkova-Valchanova, M.; Cooley, J.W.; McIntosh, M.T.; Daldal, F.; Vaidya, A.B. Uncovering the Molecular Mode of Action of the Antimalarial Drug Atovaquone Using a Bacterial System. J. Biol. Chem. 2005, 280, 27458–27465. [Google Scholar] [CrossRef]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef]
- Gao, X.; Liu, X.; Shan, W.; Liu, Q.; Wang, C.; Zheng, J.J.; Yao, H.; Tang, R.; Zheng, J.J. Anti-malarial atovaquone exhibits anti-tumor effects by inducing DNA damage in hepatocellular carcinoma. Am. J. Cancer Res. 2018, 8, 1697–1711. [Google Scholar]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef]
- Yang, N.-D.; Tan, S.-H.; Ng, S.; Shi, Y.; Zhou, J.; Tan, K.S.W.; Wong, W.-S.F.; Shen, H.-M. Artesunate Induces Cell Death in Human Cancer Cells via Enhancing Lysosomal Function and Lysosomal Degradation of Ferritin. J. Biol. Chem. 2014, 289, 33425–33441. [Google Scholar] [CrossRef]
- Zhang, P.; Luo, H.-S.; Li, M.; Tan, S. Artesunate inhibits the growth and induces apoptosis of human gastric cancer cells by downregulating COX-2. OncoTargets Ther. 2015, 8, 845. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Fernando, W.; Hoskin, D.W. The anti-malarial drug artesunate causes cell cycle arrest and apoptosis of triple-negative MDA-MB-468 and HER2-enriched SK-BR-3 breast cancer cells. Exp. Mol. Pathol. 2019, 107, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Meyler’s Side Effects of Drugs; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 9780444537164.
- Lumefantrine | DrugBank Online. Available online: https://go.drugbank.com/drugs/DB06708 (accessed on 3 November 2020).
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Roell, K.R.; Reif, D.M.; Motsinger-Reif, A.A. An Introduction to Terminology and Methodology of Chemical Synergy—Perspectives from Across Disciplines. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef]
- Greish, K.; Fateel, M.; Abdelghany, S.; Rachel, N.; Alimoradi, H.; Bakhiet, M.; Alsaie, A. Sildenafil citrate improves the delivery and anticancer activity of doxorubicin formulations in a mouse model of breast cancer. J. Drug Target. 2018, 26, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Vogus, D.R.; Pusuluri, A.; Chen, R.; Mitragotri, S. Schedule dependent synergy of gemcitabine and doxorubicin: Improvement of in vitro efficacy and lack of in vitro-in vivo correlation. Bioeng. Transl. Med. 2018, 3, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Buranrat, B.; Suwannaloet, W.; Naowaboot, J. Simvastatin potentiates doxorubicin activity against MCF-7 breast cancer cells. Oncol. Lett. 2017, 14, 6243–6250. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Warri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Chloroquine Inhibits Autophagy to Potentiate Antiestrogen Responsiveness in ER+ Breast Cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Das, A. Anticancer effect of antimalarial artemisinin compounds. Ann. Med. Health Sci. Res. 2015, 5, 93. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | IC50/µM |
|---|---|
| DOX | 0.1699 ± 0.0924 |
| PTX | 0.003 ± 0.0001 |
| Mefloquine | 1.241 ± 0.064 |
| Tafenoquine | 2.595 ± 0.060 |
| Primaquine | 29.90 ± 0.02 |
| Pyronaridine | 1.388 ± 0.216 |
| Artesunate | 11.60 ± 0.04 |
| Cycloguanil | 20.30 ± 0.08 |
| Combination (Drug A + Drug B) | Concentration (Drug A + Drug B) | Fractional Effect(Fa) | CI |
|---|---|---|---|
| DOX + chloroquine | 0.05 µM + 16 µM 0.1 µM + 32 µM 0.2 µM + 64 µM 0.4 µM + 128 µM 0.8 µM + 256 µM | 0.724 0.797 0.851 0.856 0.854 | 0.965 0.676 0.560 1.027 2.126 |
| PTX + chloroquine | 0.75 nM + 16 µM 1.5 nM + 32 µM 3 nM + 64 µM 6 nM + 128 µM 12 nM + 256 µM | 0.636 0.782 0.831 0.850 0.851 | 2.886 0.809 0.747 1.091 2.146 |
| DOX + mefloquine | 0.05 µM + 2 µM 0.1 µM + 4 µM 0.2 µM + 8 µM 0.4 µM + 16 µM 0.8 µM + 32 µM | 0.176 0.375 0.845 0.856 0.855 | 3.231 2.582 0.729 1.349 2.718 |
| PTX + mefloquine | 0.75 nM + 2 µM 1.5 nM + 4 µM 3 nM + 8 µM 6 nM + 16 µM 12 nM + 32 µM | 0.158 0.479 0.841 0.863 0.857 | 2.939 1.501 0.718 1.252 2.602 |
| DOX + artesunate | 0.05 µM + 3 µM 0.1 µM + 6 µM 0.2 µM + 12 µM 0.4 µM + 24 µM 0.8 µM + 48 µM | 0.487 0.684 0.631 0.821 0.828 | 0.480 0.315 0.847 0.544 1.035 |
| PTX + artesunate | 0.75 nM + 3 µM 1.5 nM + 6 µM 3 nM + 12 µM 6 nM + 24 µM 12 nM + 48 µM | 0.337 0.453 0.544 0.617 0.726 | 1.044 0.880 0.984 1.289 1.448 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, D.; Vale, N. New Trends for Antimalarial Drugs: Synergism between Antineoplastics and Antimalarials on Breast Cancer Cells. Biomolecules 2020, 10, 1623. https://doi.org/10.3390/biom10121623
Duarte D, Vale N. New Trends for Antimalarial Drugs: Synergism between Antineoplastics and Antimalarials on Breast Cancer Cells. Biomolecules. 2020; 10(12):1623. https://doi.org/10.3390/biom10121623
Chicago/Turabian StyleDuarte, Diana, and Nuno Vale. 2020. "New Trends for Antimalarial Drugs: Synergism between Antineoplastics and Antimalarials on Breast Cancer Cells" Biomolecules 10, no. 12: 1623. https://doi.org/10.3390/biom10121623
APA StyleDuarte, D., & Vale, N. (2020). New Trends for Antimalarial Drugs: Synergism between Antineoplastics and Antimalarials on Breast Cancer Cells. Biomolecules, 10(12), 1623. https://doi.org/10.3390/biom10121623