Phosphorylation of Ykt6 SNARE Domain Regulates Its Membrane Recruitment and Activity

 ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Antibodies
2.2. Drosophila Stocks and Genetics
2.3. Kinase Screen
2.4. Cell Culture and Transfection
2.5. Blue Sepharose Precipitation
2.6. Extracellular Vesicle purification
2.7. Immunostainings, Microscopy, and Image Analysis
2.8. Rab5QL Assay and Quantification
2.9. Membrane Fractionation
2.10. BioID Pull Down and Mass Spectrometry
2.11. Mass Spectrometry Data Processing
2.12. Statistics
3. Results
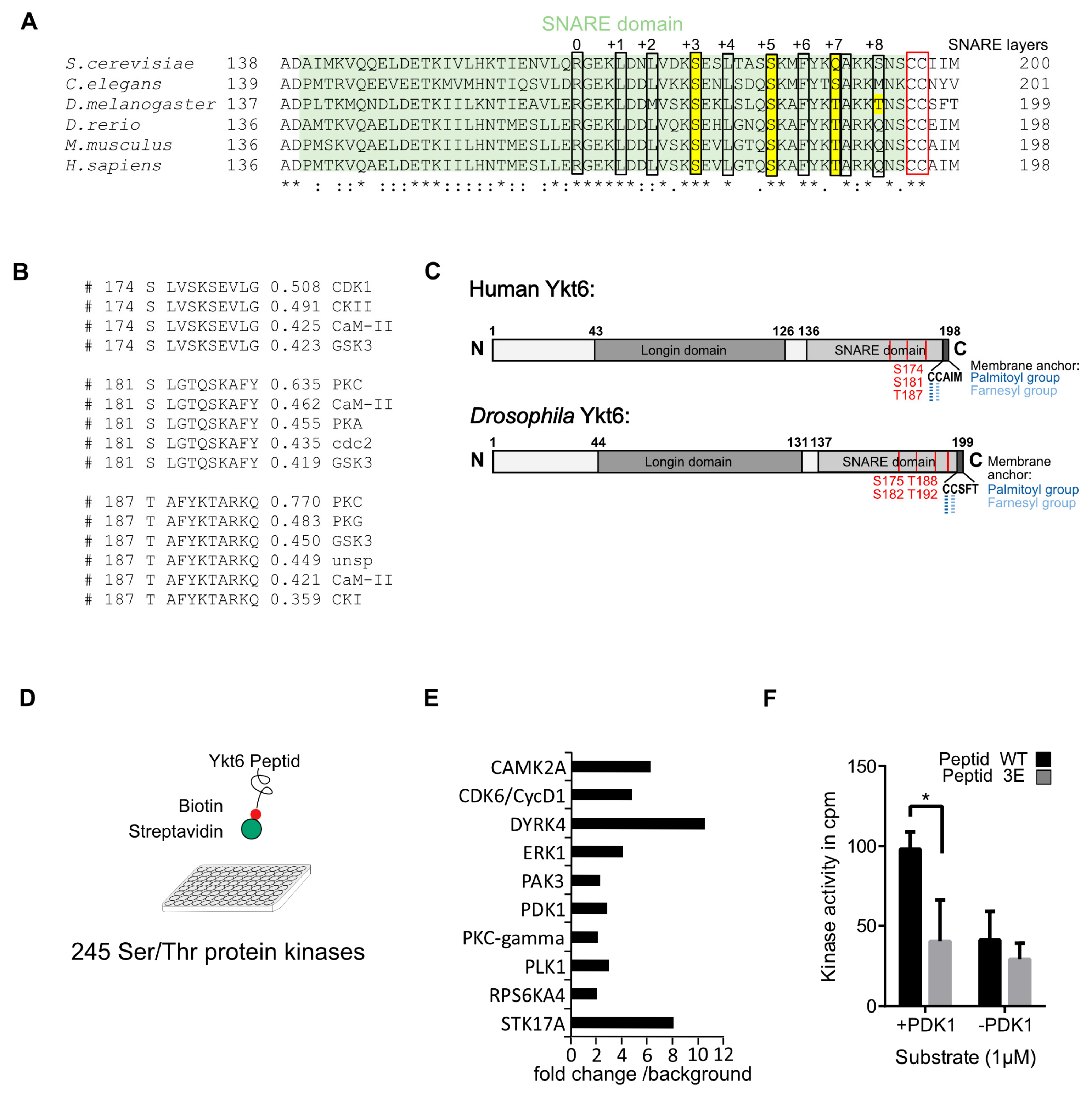
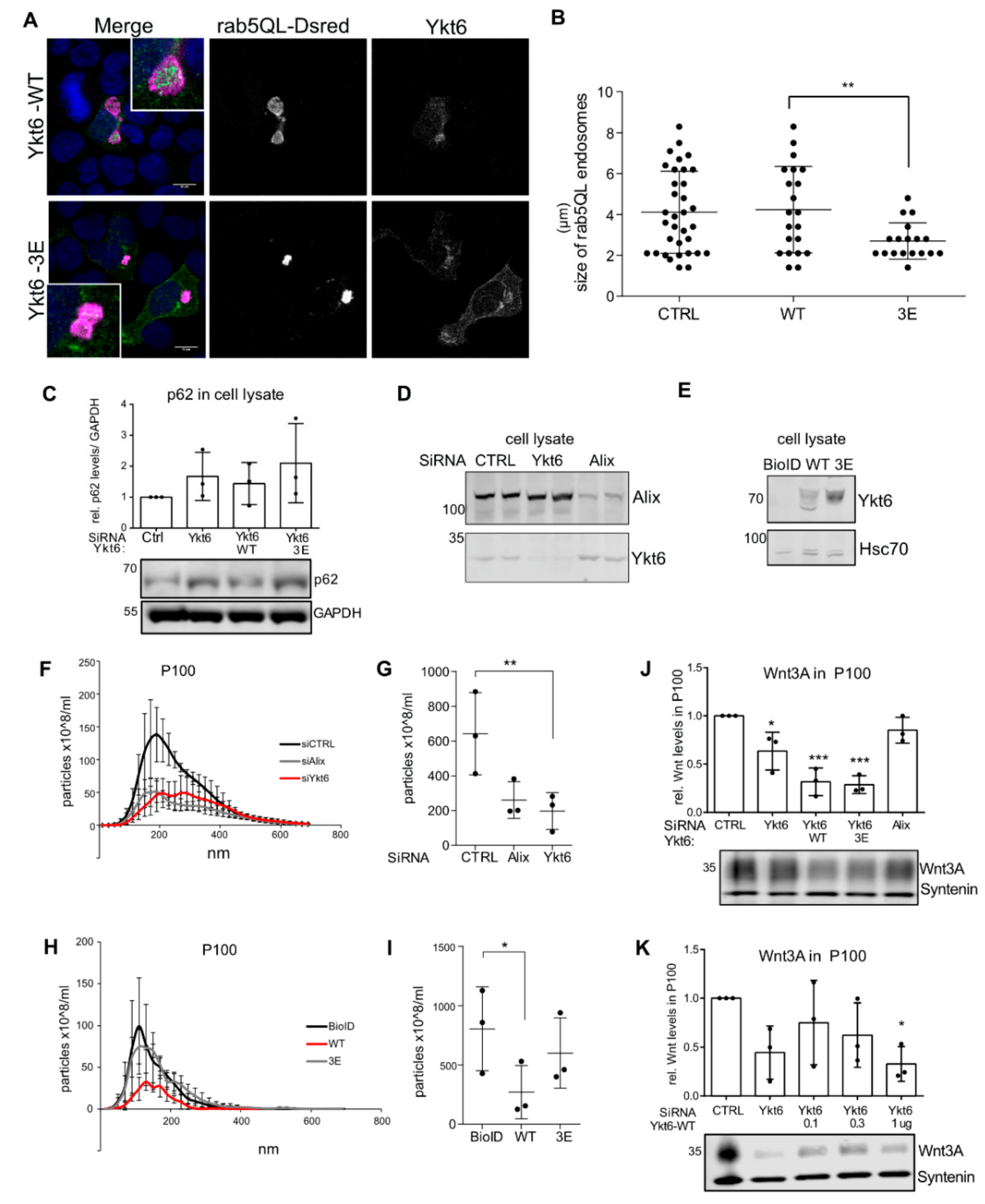
3.1. Several Phosphorylation Sites in the Ykt6 SNARE Domain Are Evolutionarily Conserved
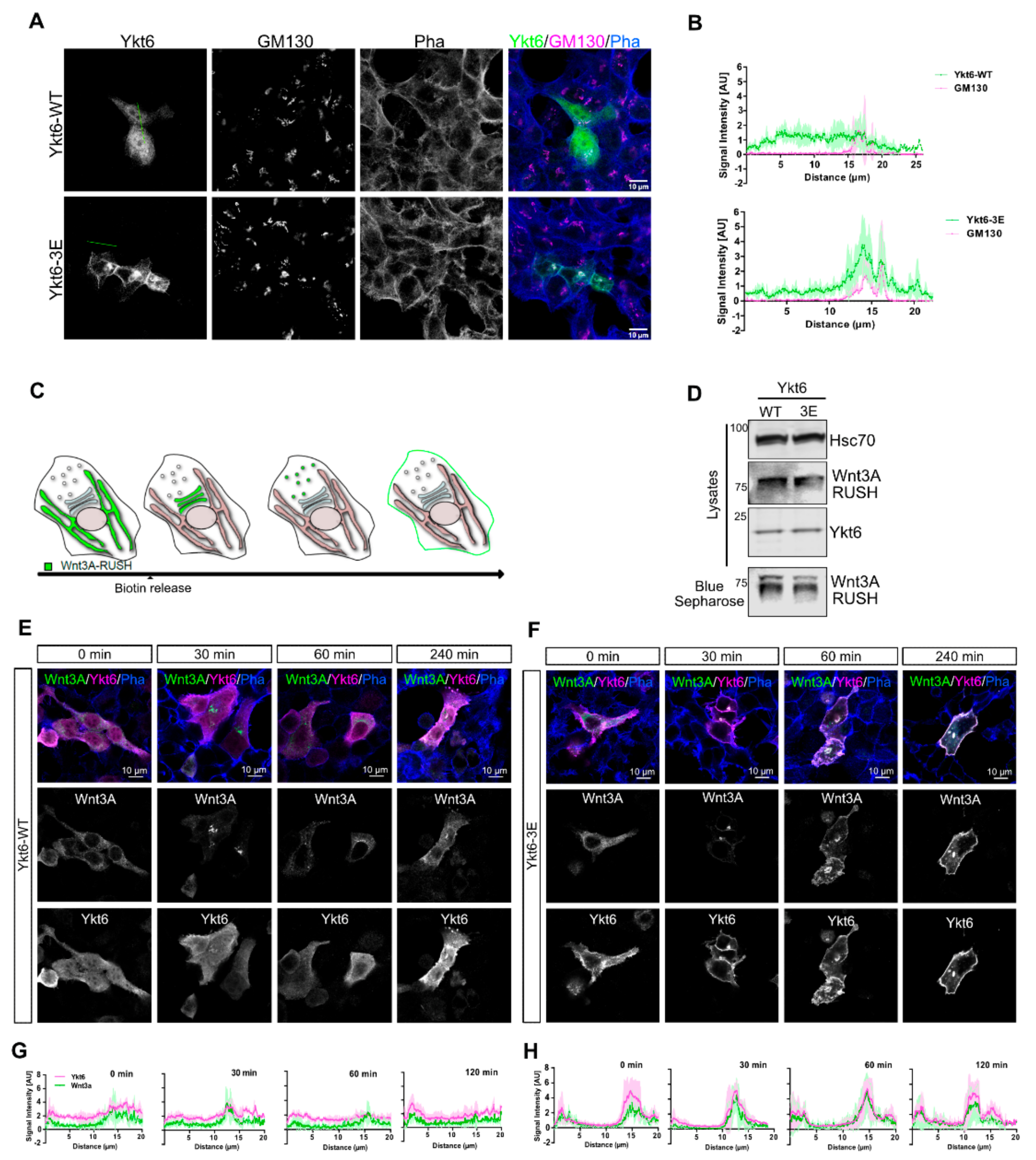
3.2. Phosphomimicking Mutations Accumulate Ykt6 at Membranes in the Secretory Pathway
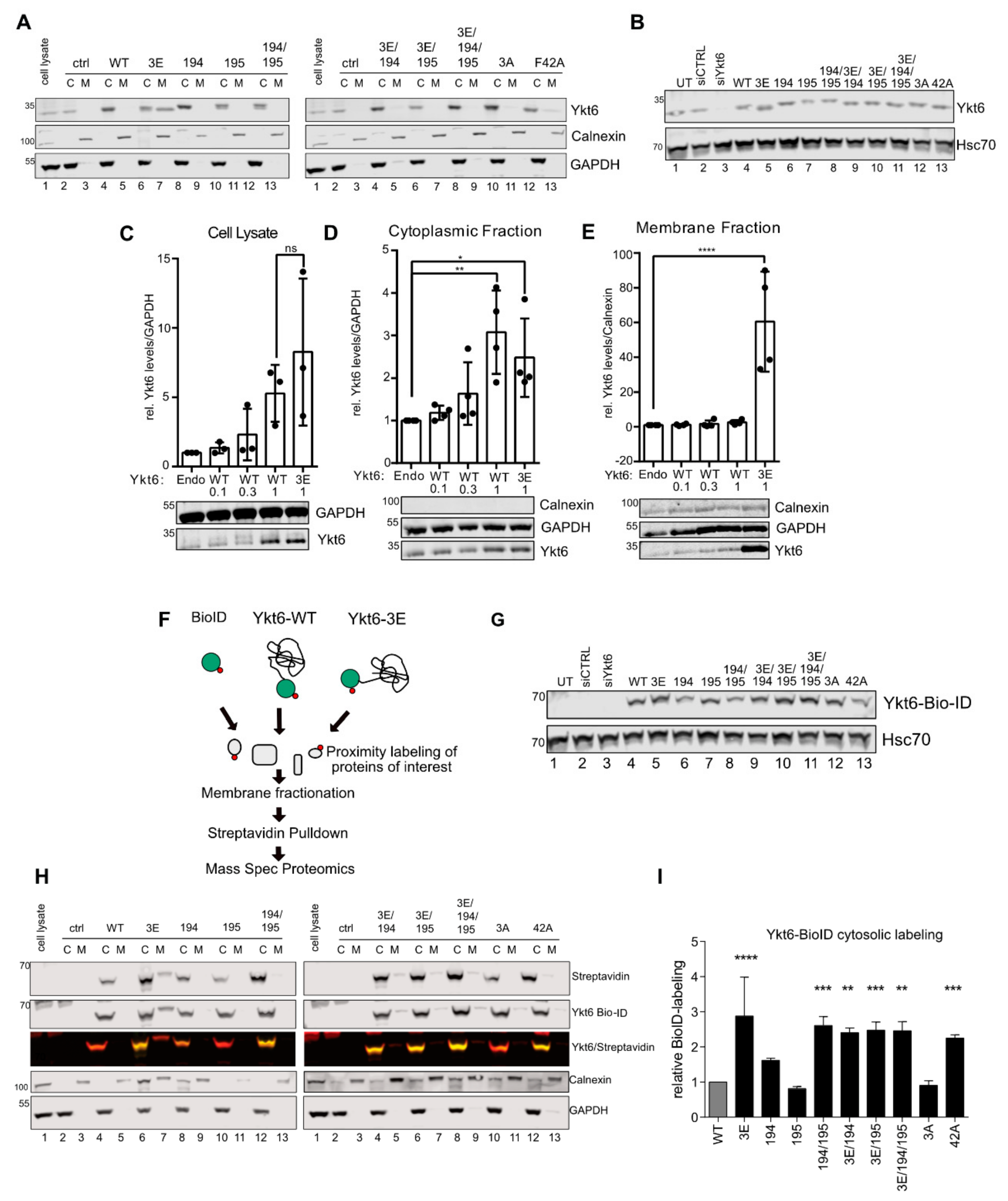
3.3. SNARE Phosphorylation Sites Determine Membrane Attachment and Autoinhibited Conformation
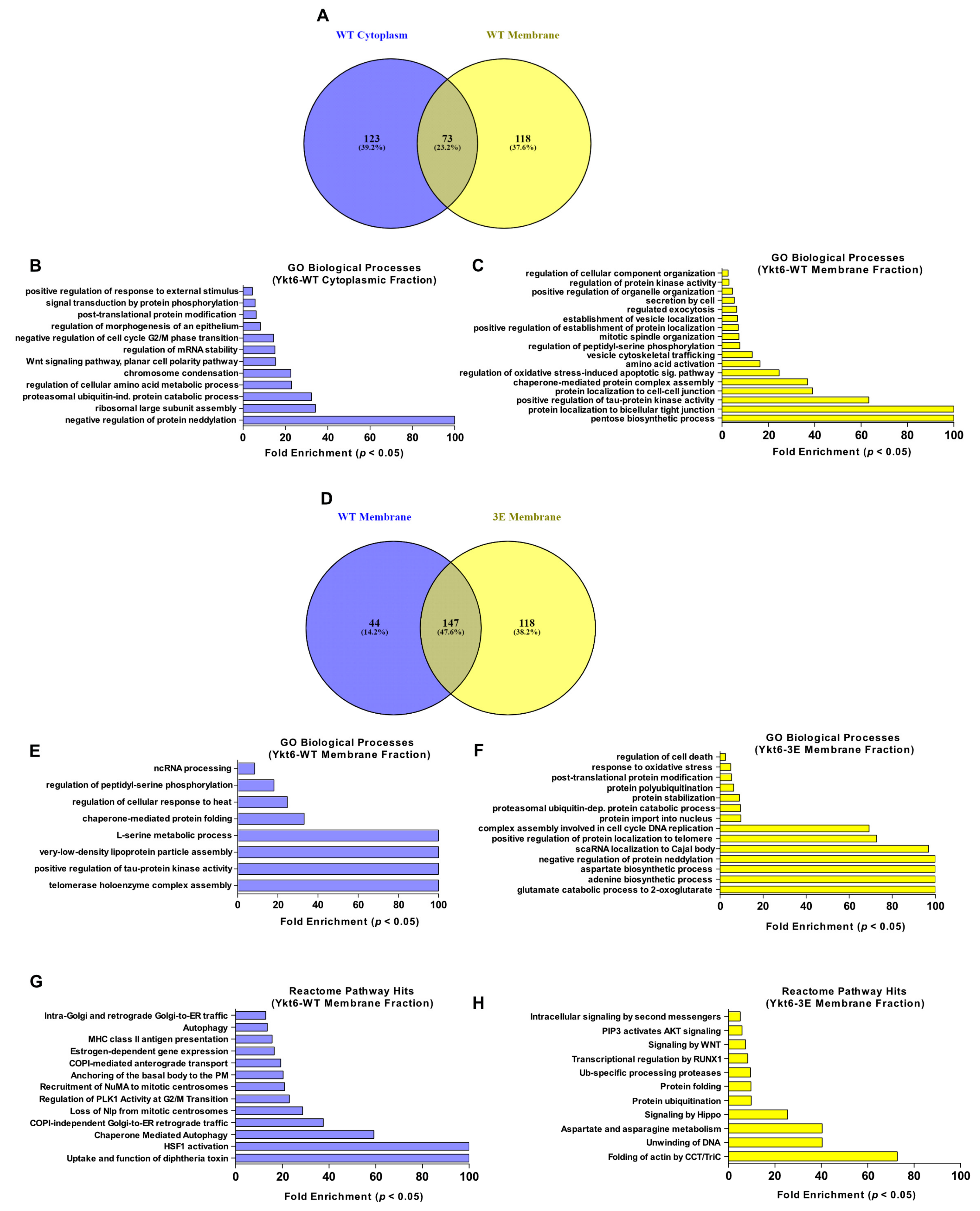
3.4. Ykt6 SNARE Domain-Dependent Proximity Proteome
3.5. Ykt6 Regulates EV Secretion in a Concentration-Dependent Manner
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weimbs, T.; Low, S.H.; Chapin, S.J.; Mostov, K.E.; Bucher, P.; Hofmann, K. A conserved domain is present in different families of vesicular fusion proteins: A new superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 3046–3051. [Google Scholar] [CrossRef] [PubMed]
- Antonin, W.; Fasshauer, D.; Becker, S.; Jahn, R.; Schneider, T.R. Crystal structure of the endosomal SNARE complex reveals common structural principles of all SNAREs. Nat. Genet. 2002, 9, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.B.; Fasshauer, D.; Jahn, R.; Brunger, A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature 1998, 395, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Ungar, D.; Hughson, F.M. SNARE Protein Structure and Function. Annu. Rev. Cell Dev. Biol. 2003, 19, 493–517. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, M.; Varlamov, O.; Eng, W.S.; Söllner, T.H.; Rothman, J.E. Localization and activity of the SNARE Ykt6 determined by its regulatory domain and palmitoylation. Proc. Natl. Acad. Sci. USA 2004, 101, 4815–4820. [Google Scholar] [CrossRef]
- Tochio, H. An Autoinhibitory Mechanism for Nonsyntaxin SNARE Proteins Revealed by the Structure of Ykt6p. Science 2001, 293, 698–702. [Google Scholar] [CrossRef]
- Shirakawa, R.; Goto-Ito, S.; Goto, K.; Wakayama, S.; Kubo, H.; Sakata, N.; Trinh, D.A.; Yamagata, A.; Sato, Y.; Masumoto, H.; et al. A SNARE geranylgeranyltransferase essential for the organization of the Golgi apparatus. EMBO J. 2020, 39, e104120. [Google Scholar] [CrossRef]
- Dingjan, I.; Linders, P.T.A.; Verboogen, D.R.J.; Revelo, N.H.; Ter Beest, M.; Bogaart, G.V.D. Endosomal and Phagosomal SNAREs. Physiol. Rev. 2018, 98, 1465–1492. [Google Scholar] [CrossRef]
- Tsui, M.M.; Banfield, D.K. Yeast Golgi SNARE interactions are promiscuous. J. Cell Sci. 2000, 113, 145–152. [Google Scholar]
- Dietrich, L.E.P.; Gurezka, R.; Veit, M.; Ungermann, C. The SNARE Ykt6 mediates protein palmitoylation during an early stage of homotypic vacuole fusion. EMBO J. 2003, 23, 45–53. [Google Scholar] [CrossRef]
- Ungermann, C.; Nichols, B.J.; Pelham, H.R.B.; Wickner, W. A Vacuolar v–t-SNARE Complex, the Predominant Form In Vivo and on Isolated Vacuoles, Is Disassembled and Activated for Docking and Fusion. J. Cell Biol. 1998, 140, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ungermann, C.; Von Mollard, G.F.; Jensen, O.N.; Margolis, N.; Stevens, T.H.; Wickner, W. Three v-SNAREs and Two t-SNAREs, Present in a Pentameric cis-SNARE Complex on Isolated Vacuoles, Are Essential for Homotypic Fusion. J. Cell Biol. 1999, 145, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- McNew, J.A.; Søgaard, M.; Lampen, N.M.; Machida, S.; Ye, R.R.; Lacomis, L.; Tempst, P.; Rothman, J.E.; Söllner, T.H. Ykt6p, a Prenylated SNARE Essential for Endoplasmic Reticulum-Golgi Transport. J. Biol. Chem. 1997, 272, 17776–17783. [Google Scholar] [CrossRef] [PubMed]
- Kweon, Y.; Rothe, A.; Conibear, E.; Stevens, T.H. Ykt6p is a multifunctional yeast R-SNARE that is required for multiple membrane transport pathways to the vacuole. Mol. Biol. Cell 2003, 14, 1868–1881. [Google Scholar] [CrossRef] [PubMed]
- Bas, L.; Papinski, D.; Licheva, M.; Torggler, R.; Rohringer, S.; Schuschnig, M.; Kraft, C. Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome–vacuole fusion. J. Cell Biol. 2018, 217, 3656–3669. [Google Scholar] [CrossRef]
- Nair, U.; Klionsky, D.J. Autophagosome biogenesis requires SNAREs. Autophagy 2011, 7, 1570–1572. [Google Scholar] [CrossRef]
- Cuddy, L.K.; Wani, W.Y.; Morella, M.L.; Pitcairn, C.; Tsutsumi, K.; Fredriksen, K.; Justman, C.J.; Grammatopoulos, T.N.; Belur, N.R.; Zunke, F.; et al. Stress-Induced Cellular Clearance Is Mediated by the SNARE Protein ykt6 and Disrupted by α-Synuclein. Neuron 2019, 104, 869–884. [Google Scholar] [CrossRef]
- Matsui, T.; Jiang, P.; Nakano, S.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 2018, 217, 2633–2645. [Google Scholar] [CrossRef]
- Takáts, S.; Glatz, G.; Szenci, G.; Boda, A.; Horváth, G.V.; Hegedűs, K.; Kovács, A.L.; Juhász, G. Non-canonical role of the SNARE protein Ykt6 in autophagosome-lysosome fusion. PLoS Genet. 2018, 14, e1007359. [Google Scholar] [CrossRef]
- Gao, J.; Reggiori, F.; Ungermann, C. A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J. Cell Biol. 2018, 217, 3670–3682. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Boncompain, G.; Divoux, S.; Gareil, N.; De Forges, H.; Lescure, A.; Latreche, L.; Mercanti, V.; Jollivet, F.; Raposo, G.; Perez, F. Synchronization of secretory protein traffic in populations of cells. Nat. Methods 2012, 9, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.C.; Chaudhary, V.; Bartscherer, K.; Boutros, M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 2012, 14, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.; Maeda, R.K.; Hediger, M.; Karch, F.; Basler, K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. USA 2007, 104, 3312–3317. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, K.; Boutros, M.; Gross, J.C. Biochemical Methods to Analyze Wnt Protein Secretion. Breast Cancer 2016, 1481, 17–28. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Menck, K.; Sönmezer, C.; Worst, T.S.; Schulz, M.; Dihazi, G.H.; Streit, F.; Erdmann, G.; Kling, S.; Boutros, M.; Binder, C.; et al. Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J. Extracell. Vesicles 2017, 6, 1378056. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.E.; Hiner, M.C.; Dezonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 1–26. [Google Scholar] [CrossRef]
- Baghirova, S.; Hughes, B.G.; Hendzel, M.J.; Schulz, R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2015, 2, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Atanassov, I.; Urlaub, H. Increased proteome coverage by combining PAGE and peptide isoelectric focusing: Comparative study of gel-based separation approaches. Proteomics 2013, 13, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.; Hopfgartner, G.; Varesio, E. The Use of Variable Q1 Isolation Windows Improves Selectivity in LC–SWATH–MS Acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-P.; Ivosev, G.; Couzens, A.L.; Larsen, B.; Taipale, M.; Lin, Z.-Y.; Zhong, Q.; Lindquist, S.; Vidal, M.; Aebersold, R.; et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 2013, 10, 1239–1245. [Google Scholar] [CrossRef]
- Gobert, C.; Bracco, L.; Rossi, F.; Olivier, M.; Tazi, J.; Lavelle, F.; Larsen, A.K.; Riou, J.-F. Modulation of DNA Topoisomerase I Activity byp53. Biochemistry 1996, 35, 5778–5786. [Google Scholar] [CrossRef]
- Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
- Malmersjö, S.; Di Palma, S.; Diao, J.; Lai, Y.; A Pfuetzner, R.; Wang, A.L.; A McMahon, M.; Hayer, A.; Porteus, M.; Bodenmiller, B.; et al. Phosphorylation of residues inside the SNARE complex suppresses secretory vesicle fusion. EMBO J. 2016, 35, 1810–1821. [Google Scholar] [CrossRef]
- Stuart, S.A.; Houel, S.; Lee, T.; Wang, N.; Old, W.M.; Ahn, N.G. A phosphoproteomic comparison of B-RAFV600E and MKK1/2 inhibitors in melanoma cells. Mol. Cell. Proteom. 2015, 14, 1599–1615. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar]
- Minakhina, S.; Steward, R. Melanotic Mutants in Drosophila: Pathways and Phenotypes. Genet 2006, 174, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Parchure, A.; Vyas, N.; Mayor, S. Wnt and Hedgehog: Secretion of Lipid-Modified Morphogens. Trends Cell Biol. 2017, 28, 157–170. [Google Scholar] [CrossRef]
- Swarup, S.; Verheyen, E.M. Wnt/wingless signaling in Drosophila. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Linnemannstöns, K.; Witte, L.; Pradhipa, K.M.; Kittel, J.C.; Danieli, A.; Müller, D.; Nitsch, L.; Honemann-Capito, M.; Grawe, F.; Wodarz, A.; et al. Ykt6-dependent endosomal recycling is required for Wnt secretion in the Drosophila wing epithelium. Development 2020, 147, dev185421. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Burke, B. BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci. 2013, 74, 19.23.1–19.23.14. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, L.E.; Peplowska, K.; LaGrassa, T.J.; Hou, H.; Rohde, J.; Ungermann, C. The SNARE Ykt6 is released from yeast vacuoles during an early stage of fusion. EMBO Rep. 2005, 6, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; DeGeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan–syntenin–ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef]
- Stenmark, H.; Parton, R.; Steele-Mortimer, O.; Lütcke, A.; Gruenberg, J.; Zerial, M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994, 13, 1287–1296. [Google Scholar] [CrossRef]
- Saito, Y.; Li, L.; Coyaud, E.; Luna, A.; Sander, C.; Raught, B.; Asara, J.M.; Brown, M.; Muthuswamy, S.K. LLGL2 rescues nutrient stress by promoting leucine uptake in ER+ breast cancer. Nat. Cell Biol. 2019, 569, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Kurre, R.; Rose, J.; Walter, S.; Fröhlich, F.; Piehler, J.; Reggiori, F.; Ungermann, C. Function of the SNARE Ykt6 on autophagosomes requires the Dsl1 complex and the Atg1 kinase complex. EMBO Rep. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Yu, J.; Pan, L.; Wei, Z.; Weng, J.; Wang, W.; Ong, Y.S.; Tran, T.H.T.; Hong, W.; Zhang, M. Lipid-Induced Conformational Switch Controls Fusion Activity of Longin Domain SNARE Ykt6. Mol. Cell 2010, 37, 383–395. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.; Dergai, M.; Agarwal, S.; Chung, D.; Van Rossum, D.B.; Shevade, A.; Kuchin, S.; Zaichick, S.; Savas, J.N.; Fasshauer, D.; et al. A conformational switch driven by phosphorylation regulates Ykt6 activity in macroautophagy. BioRxiv 2020. Available Online: https://www.biorxiv.org/content/10.1101/2020.03.15.992727v1 (accessed on 16 November 2020). [CrossRef]
- Lau, C.G.; Takayasu, Y.; Rodenas-Ruano, A.; Paternain, A.V.; Lerma, J.; Bennett, M.V.L.; Zukin, R.S. SNAP-25 Is a Target of Protein Kinase C Phosphorylation Critical to NMDA Receptor Trafficking. J. Neurosci. 2010, 30, 242–254. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Salazar, G.; Patrushev, N.; Martin, A.S.; Hilenski, L.; Xiong, S.; Alexander, R.W. Early Endosomal Antigen 1 (EEA1) Is an Obligate Scaffold for Angiotensin II-induced, PKC-α-dependent Akt Activation in Endosomes. J. Biol. Chem. 2011, 286, 2886–2895. [Google Scholar] [CrossRef]
- Huang, S.; Tang, D.; Wang, Y. Monoubiquitination of Syntaxin 5 Regulates Golgi Membrane Dynamics during the Cell Cycle. Dev. Cell 2016, 38, 73–85. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A Proteome-wide, Quantitative Survey of In Vivo Ubiquitylation Sites Reveals Widespread Regulatory Roles. Mol. Cell. Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.-K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | GENE ID | Gene Accession | GI Number | Sequence |
|---|---|---|---|---|
| siGENOME Non-targeting Control_5 # | UGGUUUACAUGUCGACUAA | |||
| Ykt6 | 10652 | NM_006555 | 34304384 | GCUCAAAGCCGCAUACGAU |
| GUGAGAAGCUAGAUGACUU | ||||
| GAAGGUACUAGAUGAAUUC | ||||
| Alix | 10015 | NM_013374 | 371875333 | GAAGGAUGCUUUCGAUAAA |
| GAACAGAACCUGGAUAAUG | ||||
| GAGAGGGUCUGGAGAAUGA | ||||
| GCAGUGAGGUUGUAAAUGU | ||||
| EngrailedGAL4, UAS-GFP> | Mild Knockdown (RT) | Strong Knockdown (25 °C) | ||
|---|---|---|---|---|
| Larvae | Adult | Larvae | Adult | |
| ykt6 RNAi | Melanotic tumors | wing defects | lethal * | lethal |
| ykt6 RNAi; UAS-Ykt6-WT | Normal | viable | normal | viable |
| ykt6 RNAi; UAS Ykt6-4A | Normal | viable | normal | viable |
| ykt6 RNAi; UAS-Ykt6-4E | Melanotic tumors | wing defects | lethal | lethal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karuna M, P.; Witte, L.; Linnemannstoens, K.; Choezom, D.; Danieli-Mackay, A.; Honemann-Capito, M.; Gross, J.C. Phosphorylation of Ykt6 SNARE Domain Regulates Its Membrane Recruitment and Activity. Biomolecules 2020, 10, 1560. https://doi.org/10.3390/biom10111560
Karuna M P, Witte L, Linnemannstoens K, Choezom D, Danieli-Mackay A, Honemann-Capito M, Gross JC. Phosphorylation of Ykt6 SNARE Domain Regulates Its Membrane Recruitment and Activity. Biomolecules. 2020; 10(11):1560. https://doi.org/10.3390/biom10111560
Chicago/Turabian StyleKaruna M, Pradhipa, Leonie Witte, Karen Linnemannstoens, Dolma Choezom, Adi Danieli-Mackay, Mona Honemann-Capito, and Julia Christina Gross. 2020. "Phosphorylation of Ykt6 SNARE Domain Regulates Its Membrane Recruitment and Activity" Biomolecules 10, no. 11: 1560. https://doi.org/10.3390/biom10111560
APA StyleKaruna M, P., Witte, L., Linnemannstoens, K., Choezom, D., Danieli-Mackay, A., Honemann-Capito, M., & Gross, J. C. (2020). Phosphorylation of Ykt6 SNARE Domain Regulates Its Membrane Recruitment and Activity. Biomolecules, 10(11), 1560. https://doi.org/10.3390/biom10111560