ATM Inhibitor Suppresses Gemcitabine-Resistant BTC Growth in a Polymerase θ Deficiency-Dependent Manner


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Research Design and Methods
2.1. Cells and Reagents
2.2. Virus Production and Infection
2.3. Analysis of DDR Gene Expression
2.4. RNA Extraction and Quantitative RT-PCR (RT-qPCR)
2.5. Cytotoxicity, Cell Proliferation, and Cell Viability Assays
2.6. Immunoblotting and Immunofluorescent Staining
2.7. Comet Assay
2.8. Micronucleus Assay
2.9. Colony Formation Assay
2.10. Statistics
3. Results
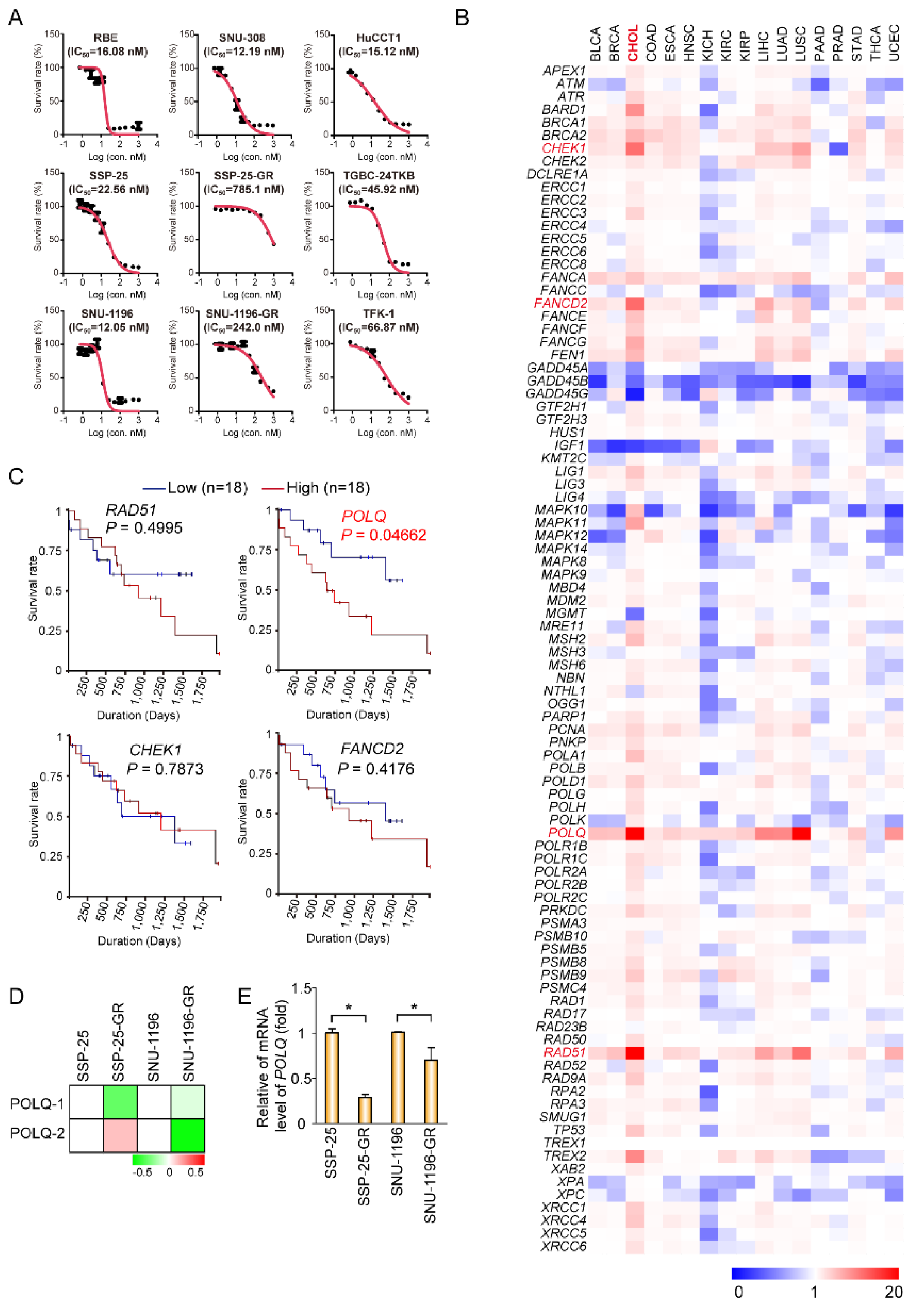
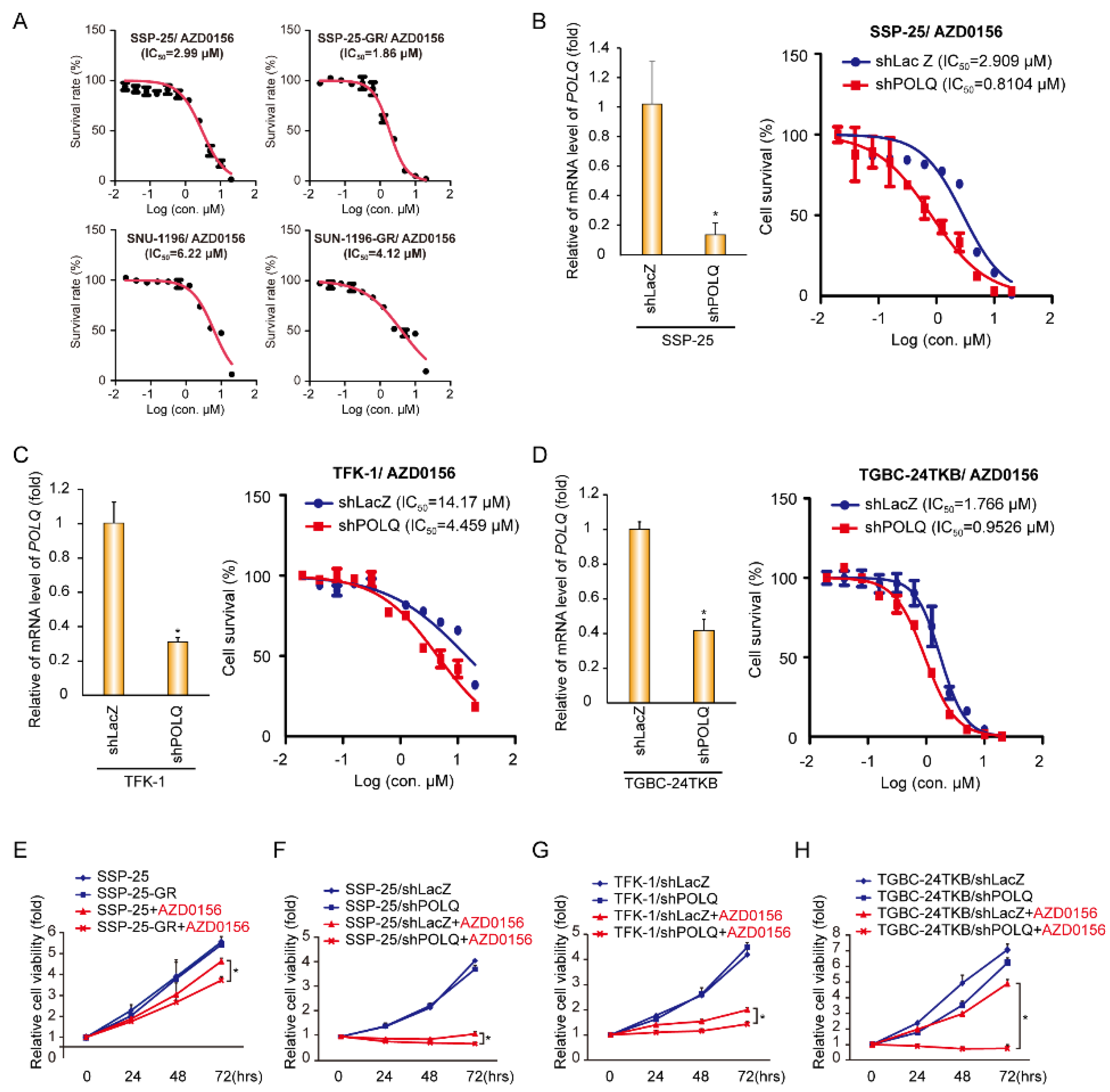
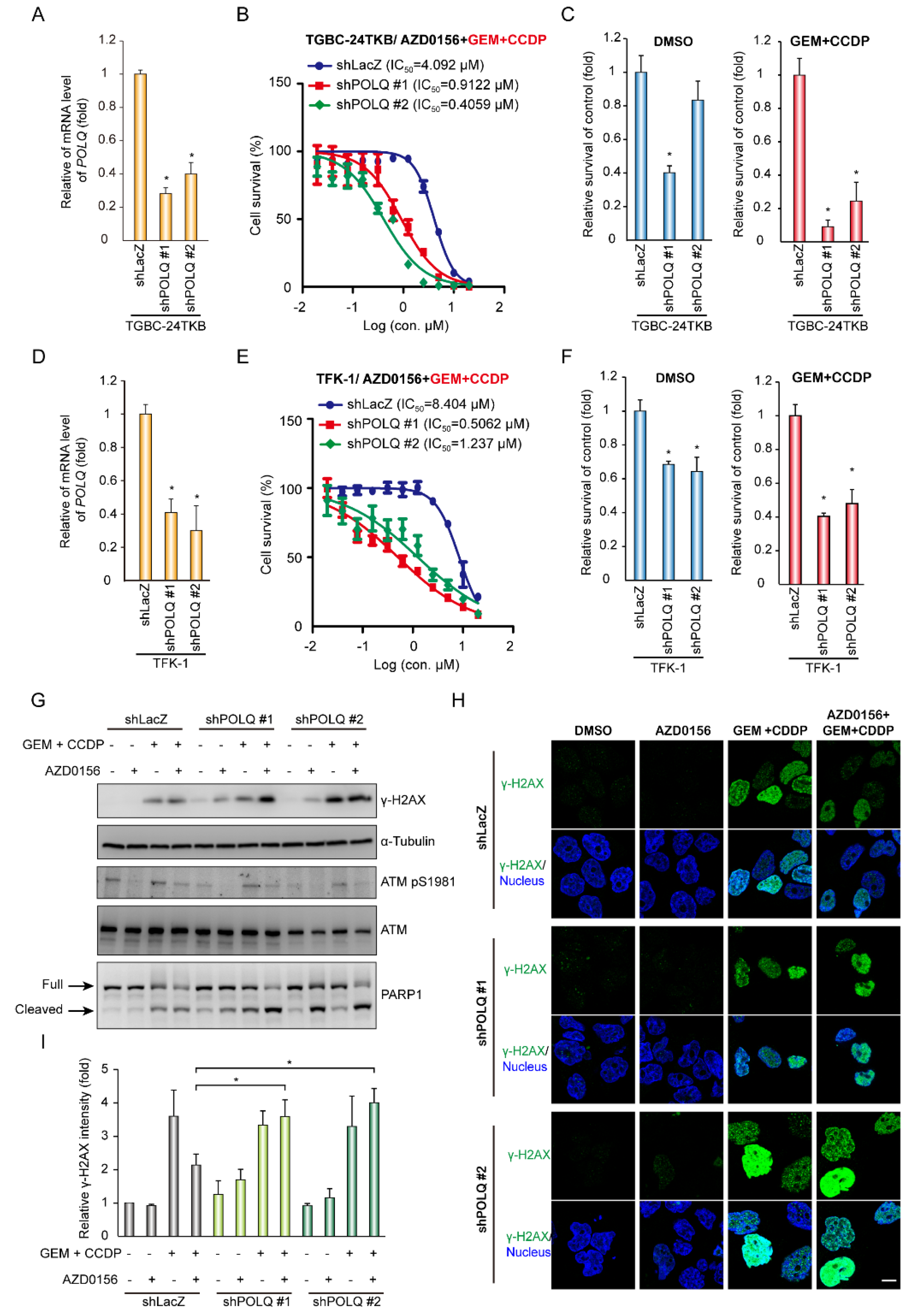
3.1. The ATM Inhibitor (AZD0156) Decreases Tumor Survival in Gemcitabine-Resistant or DNA Polymerase θ-Depleted BTC Cells
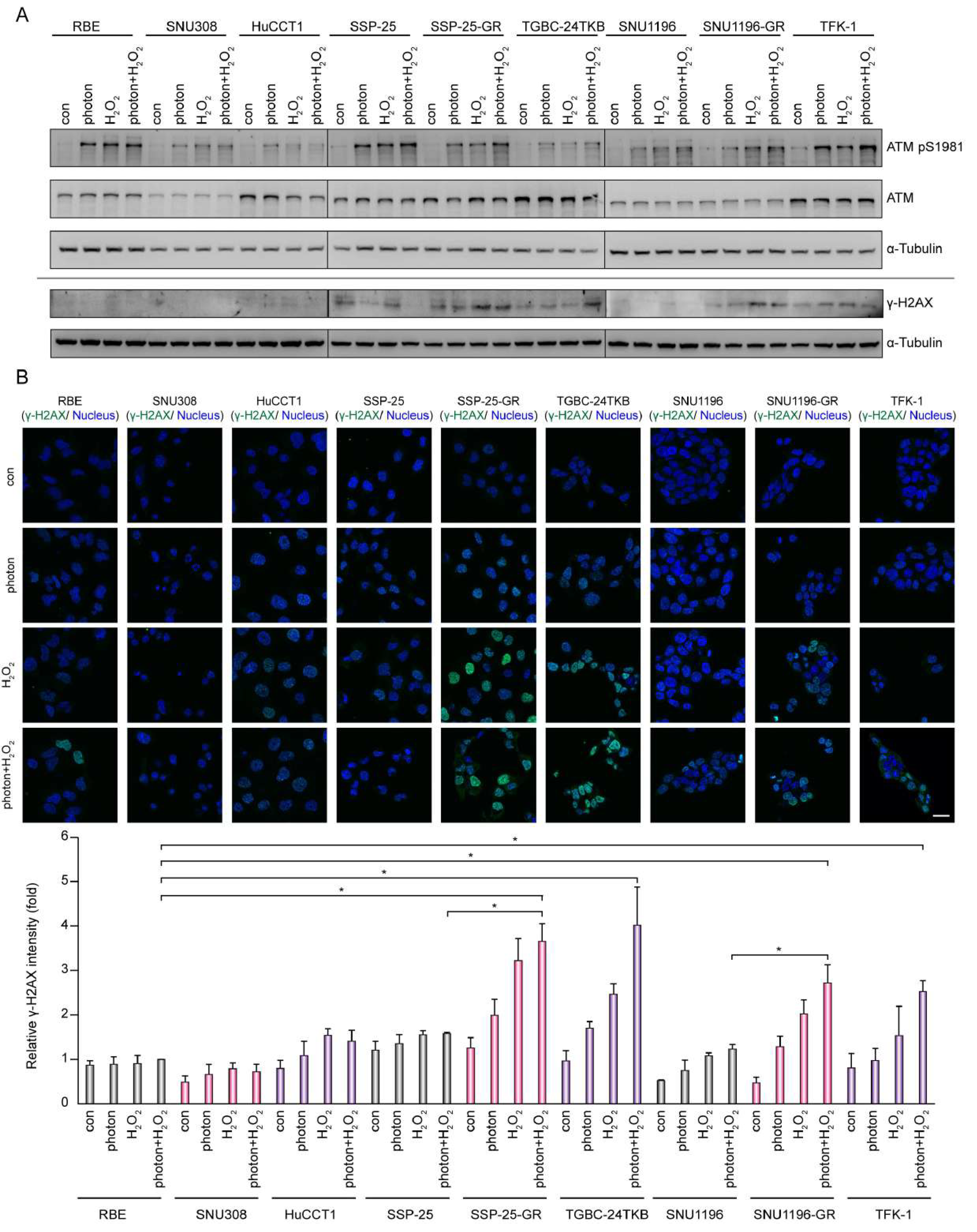
3.2. DNA Damage Enhances the Effect of AZD0156-Mediated Cell Death in DNA Polymerase θ-Depleted BTC Cells
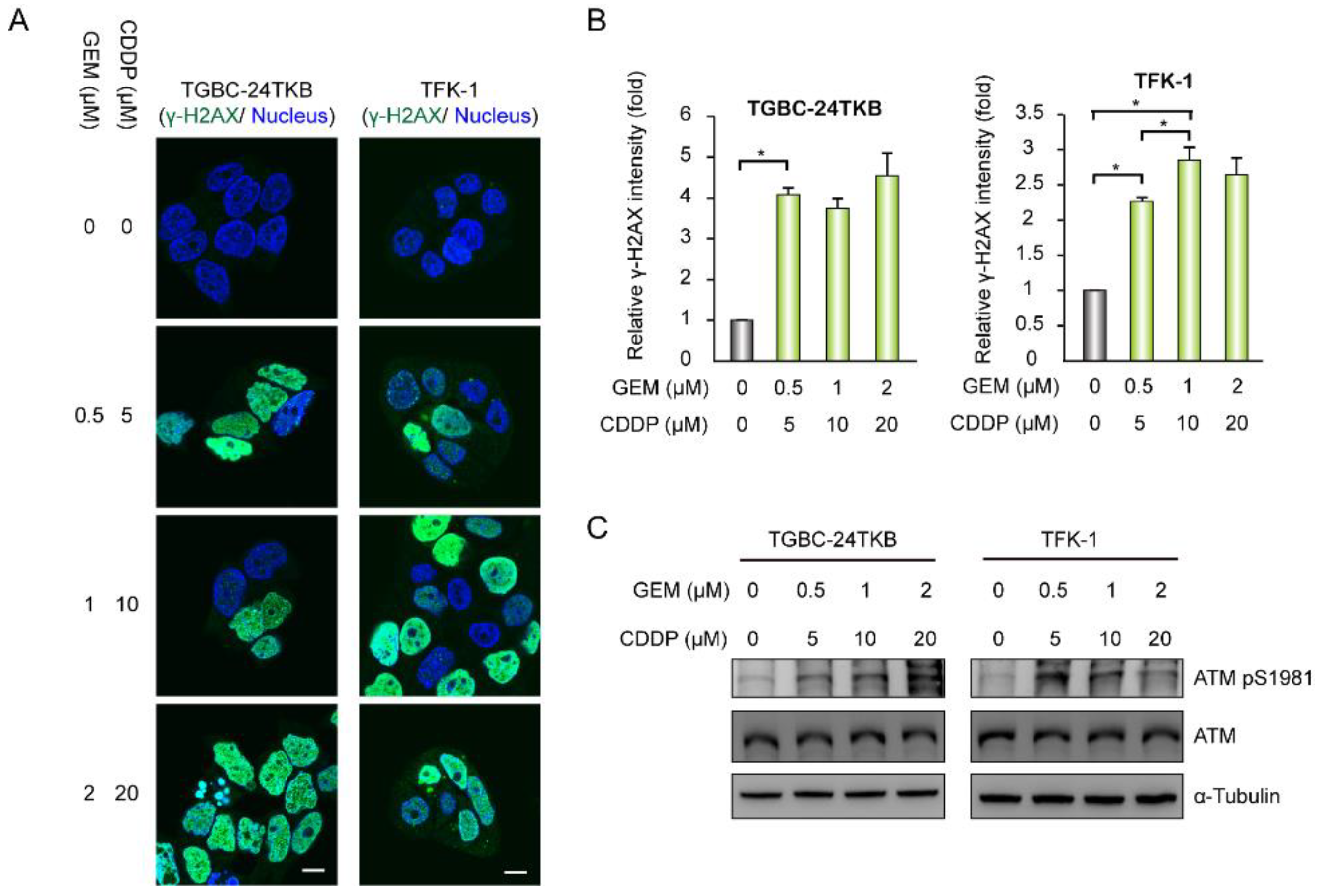
3.3. A Combination of ATM Inhibition and DNA Polymerase θ Depletion Leads to a Synergistic Effect on Tumor Growth Upon Chemotherapy Drug-Induced DNA Damage in BTC Cells
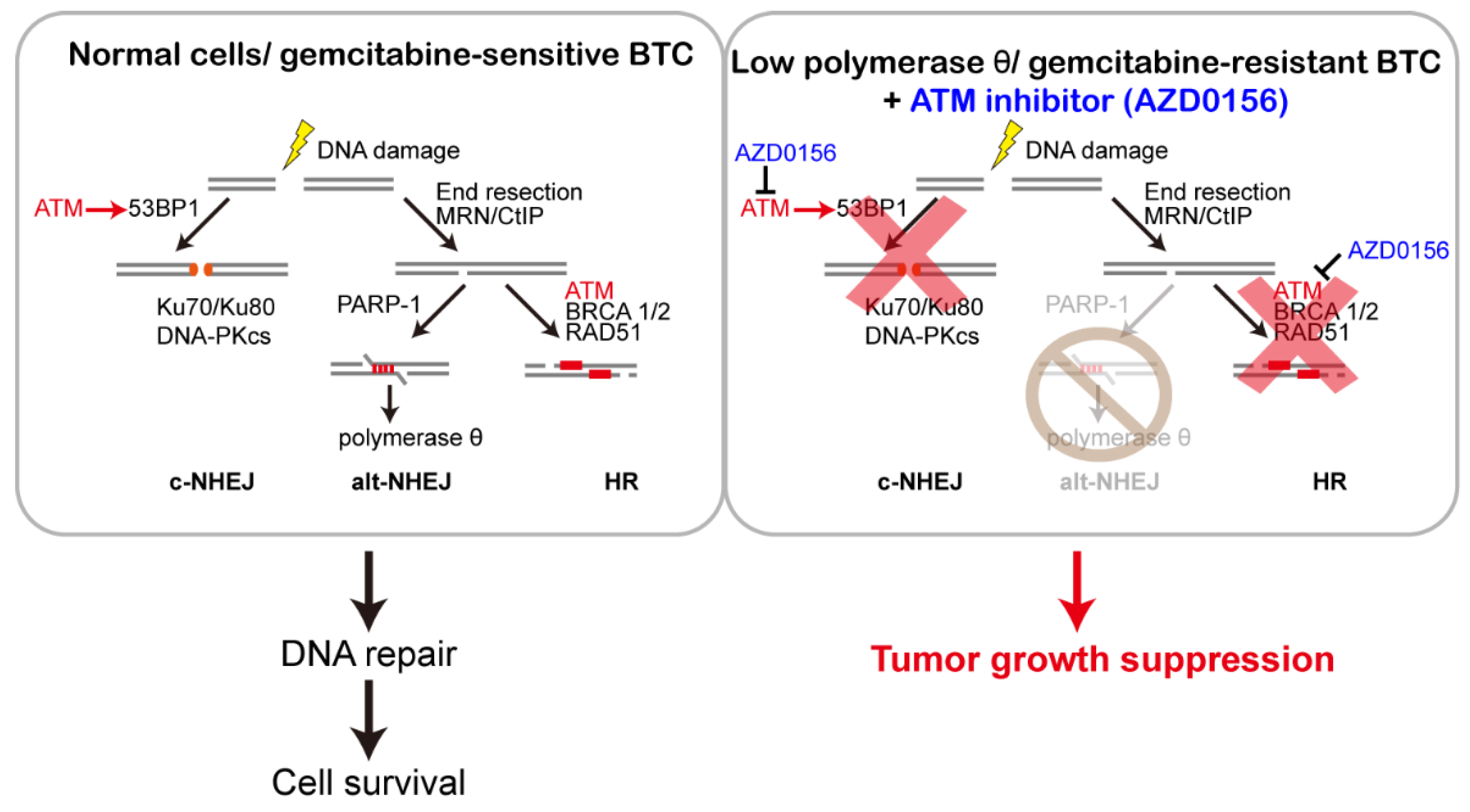
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary Tract Cancer: State of the Art and potential role of DNA Damage Repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.W.; Askan, G.; Daniel, T.D.; Lowery, M.; Klimstra, D.S.; Abou-Alfa, G.K.; Shia, J. Biliary carcinomas: Pathology and the role of DNA mismatch repair deficiency. Chin. Clin. Oncol. 2016, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Liu, Q.; Lopez, K.; Murnane, J.; Humphrey, T.; Barcellos-Hoff, M.H. Misrepair in Context: TGFbeta Regulation of DNA Repair. Front. Oncol. 2019, 9, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, R.D.; Doublie, S. DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair (Amst.) 2016, 44, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Garrett, M.D.; Ashworth, A. Targeting the double-strand DNA break repair pathway as a therapeutic strategy. Clin. Cancer Res. 2006, 12, 4463–4468. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Coackley, C.; Bristow, R.G. ATM-dependent phosphorylation of 53BP1 in response to genomic stress in oxic and hypoxic cells. Radiother. Oncol. 2011, 99, 307–312. [Google Scholar] [CrossRef]
- Gatei, M.; Scott, S.P.; Filippovitch, I.; Soronika, N.; Lavin, M.F.; Weber, B.; Khanna, K.K. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000, 60, 3299–3304. [Google Scholar]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Shima, N.; Munroe, R.J.; Schimenti, J.C. The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol. Cell Biol. 2004, 24, 10381–10389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Z. Understanding the Genetic Mechanisms of Cancer Drug Resistance Using Genomic Approaches. Trends Genet. 2016, 32, 127–137. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ge, Y.; Wang, T.; Edwards, H.; Ren, Q.; Jiang, Y.; Quan, C.; Wang, G. Inhibition of ATR potentiates the cytotoxic effect of gemcitabine on pancreatic cancer cells through enhancement of DNA damage and abrogation of ribonucleotide reductase induction by gemcitabine. Oncol. Rep. 2017, 37, 3377–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsels, L.A.; Morgan, M.A.; Tanska, D.M.; Parsels, J.D.; Palmer, B.D.; Booth, R.J.; Denny, W.A.; Canman, C.E.; Kraker, A.J.; Lawrence, T.S.; et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol. Cancer Ther. 2009, 8, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Ewald, B.; Sampath, D.; Plunkett, W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol. Cancer Ther. 2007, 6, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Jayachandran, G.; Ueda, K.; Wang, B.; Roth, J.A.; Ji, L. NPRL2 sensitizes human non-small cell lung cancer (NSCLC) cells to cisplatin treatment by regulating key components in the DNA repair pathway. PLoS ONE 2010, 5, e11994. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Touat, M.; Sourisseau, T.; Dorvault, N.; Chabanon, R.M.; Garrido, M.; Morel, D.; Krastev, D.B.; Bigot, L.; Adam, J.; Frankum, J.R.; et al. DNA repair deficiency sensitizes lung cancer cells to NAD+ biosynthesis blockade. J. Clin. Investig. 2018, 128, 1671–1687. [Google Scholar] [CrossRef]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wild, A.T.; Turcan, S.; Wu, W.H.; Sigel, C.; Klimstra, D.S.; Ma, X.; Gong, Y.; Holland, E.C.; Huse, J.T.; et al. Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci. Adv. 2020, 6, eaaz3221. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Goff, J.P.; Shields, D.S.; Seki, M.; Choi, S.; Epperly, M.W.; Dixon, T.; Wang, H.; Bakkenist, C.J.; Dertinger, S.D.; Torous, D.K.; et al. Lack of DNA polymerase theta (POLQ) radiosensitizes bone marrow stromal cells in vitro and increases reticulocyte micronuclei after total-body irradiation. Radiat. Res. 2009, 172, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Song, Y.; Li, S.; Kurian, S.; Xiang, R.; Chiba, T.; Wu, X. DNA polymerase theta (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. J. Biol. Chem. 2019, 294, 3909–3919. [Google Scholar] [CrossRef]
- Wang, X.; Simon, R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med. Genom. 2013, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748.e6–760.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, A.R.; Jin, M.H.; Park, J.E.; Bang, J.H.; Oh, D.Y.; Bang, Y.J. Therapeutic Targeting of the DNA Damage Response Using an ATR Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2019, 51, 1167–1179. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.-R.; Wu, C.-E.; Yeh, C.-N. ATM Inhibitor Suppresses Gemcitabine-Resistant BTC Growth in a Polymerase θ Deficiency-Dependent Manner. Biomolecules 2020, 10, 1529. https://doi.org/10.3390/biom10111529
Pan Y-R, Wu C-E, Yeh C-N. ATM Inhibitor Suppresses Gemcitabine-Resistant BTC Growth in a Polymerase θ Deficiency-Dependent Manner. Biomolecules. 2020; 10(11):1529. https://doi.org/10.3390/biom10111529
Chicago/Turabian StylePan, Yi-Ru, Chiao-En Wu, and Chun-Nan Yeh. 2020. "ATM Inhibitor Suppresses Gemcitabine-Resistant BTC Growth in a Polymerase θ Deficiency-Dependent Manner" Biomolecules 10, no. 11: 1529. https://doi.org/10.3390/biom10111529
APA StylePan, Y.-R., Wu, C.-E., & Yeh, C.-N. (2020). ATM Inhibitor Suppresses Gemcitabine-Resistant BTC Growth in a Polymerase θ Deficiency-Dependent Manner. Biomolecules, 10(11), 1529. https://doi.org/10.3390/biom10111529