An Overview of the Ferroptosis Hallmarks in Friedreich’s Ataxia


 ,
, 
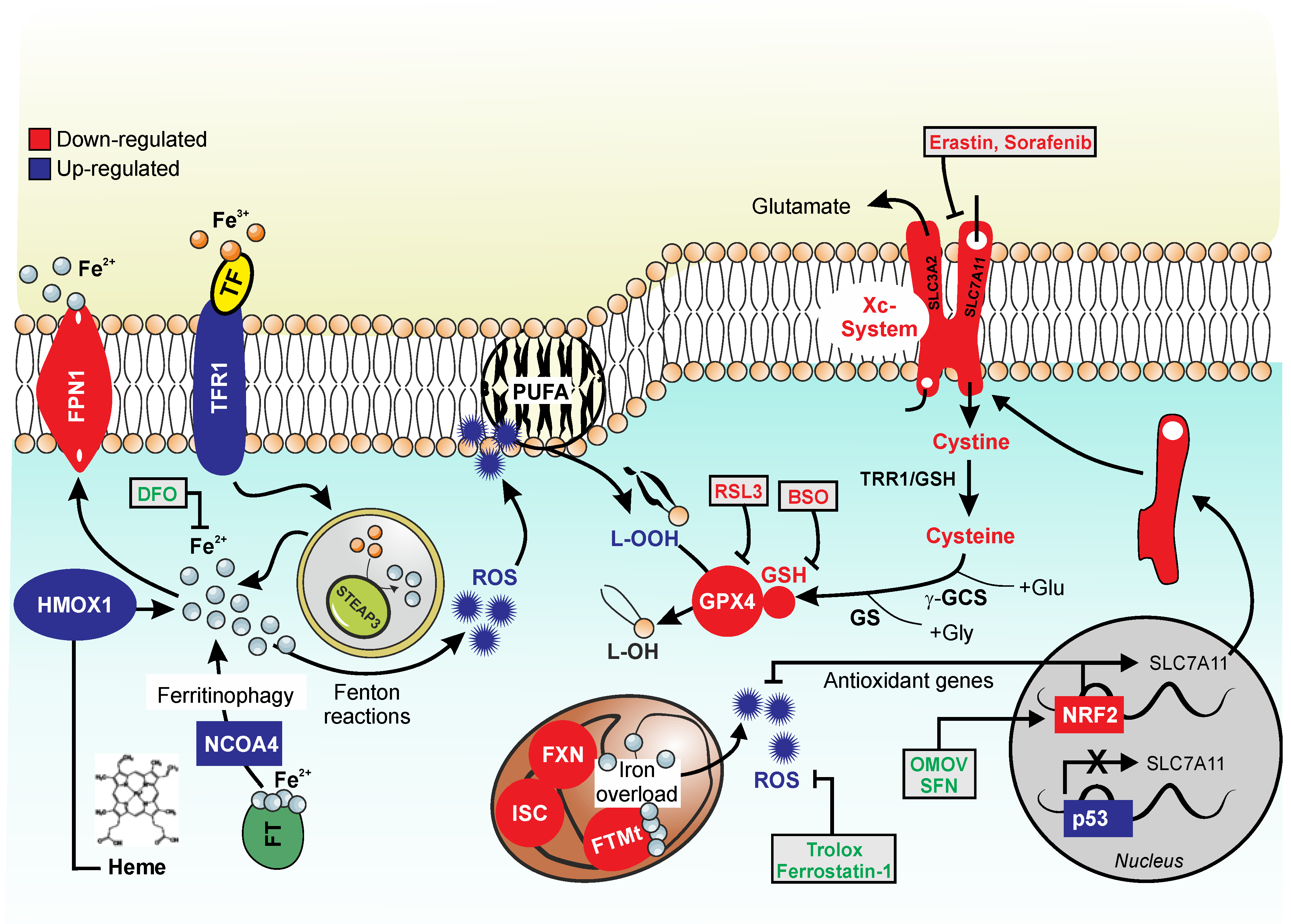{kind=link}
Abstract
1. Introduction
2. Ferroptosis
2.1. Lipid Metabolism in Ferroptosis
2.2. Iron Metabolism in Ferroptosis
3. Ferroptosis Markers in FRDA
3.1. Lipid Peroxidation in FRDA
3.2. Glutathione and Glutathione Peroxidases (GPXs) in FRDA
3.3. Iron Dysmetabolism in FRDA
3.4. NRF2 in FRDA
3.5. P53 in FRDA
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxynonenal; |
| AA | arachidonic acid; |
| ACSL4 | Acyl-CoA synthetase long-chain family member 4; |
| AD | Alzheimer’s disease; |
| ALS | Amyotrophic lateral sclerosis; |
| BSO | L-buthionine-(S,R)-sulfoximine; |
| CNS | central nervous system; |
| DFO | Deferoxamine mesylate; |
| DMF | dimethyl fumarate; |
| FPN | ferroportin; |
| FRDA | Friedreich’s ataxia; |
| FT | ferritin; |
| FTL | ferritin light chain; |
| FSP1 | ferroptosis suppressor protein 1; |
| FTMt | mitochondrial ferritin; |
| FXN | frataxin; |
| GPX | glutathione peroxidase; |
| GSH | glutathione; |
| HCC | hepatocellular carcinoma cells; |
| Hmox1 | Heme Oxygenase 1; |
| HSPB1 | heat shock beta-1 protein; |
| IRE | iron response element; |
| IREB2 | RNA-binding protein 2; |
| IRP | iron-responsive element-binding protein; |
| ISC | iron-sulfur clusters; |
| LIP | labile iron pool; |
| LOOH | lipid peroxides; |
| MDA | malondialdehyde; |
| NAC | N-acetylcysteine; |
| NCOA4 | nuclear receptor coactivator 4; |
| NQO1 | quinone oxidoreductase; |
| NRF2 | Nuclear factor erythroid 2-related factor 2; |
| OMOV | Omaveloxolone; |
| PE | phosphatidylethanolamine; |
| PUFAs | Polyunsatured fatty acids; |
| ROS | reactive oxygen species; |
| SeC | Selenocysteine; |
| SFN | sulforaphane; |
| Steap3 | six-transmembrane epithelial antigen of the prostate 3; |
| TF | transferrin; |
| TFR1 | transferrin receptor complex; |
| UTRs | untranslated regions |
References
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Levi, S.; Santambrogio, P.; Parsons, P.J.; Kruger, P.C.; Yang, K.X.; Feustel, P.J.; et al. The Pathogenesis of Cardiomyopathy in Friedreich Ataxia. PLoS ONE 2015, 10, e0116396. [Google Scholar] [CrossRef] [PubMed]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sidossis, L.; Kajimura, S. Brown and beige fat in humans: Thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 2015, 125, 478–486. [Google Scholar] [CrossRef]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef]
- Abeti, R.; Uzun, E.; Renganathan, I.; Honda, T.; Pook, M.A.; Giunti, P. Targeting lipid peroxidation and mitochondrial imbalance in Friedreich’s ataxia. Pharmacol. Res. 2015, 99, 344–350. [Google Scholar] [CrossRef]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.L.; Homayoun, S.; Hart, P.E.; Schapira, A.H.V.; Cooper, J.M. Role of Oxidative Damage in Friedreich’s Ataxia. Neurochem. Res. 2004, 29, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Sato, H.; Kuriyama-Matsumura, K.; Hashimoto, T.; Sasaki, H.; Wang, H.; Ishii, T.; Mann, G.E.; Bannai, S. Effect of oxygen on induction of the cystine transporter by bacterial lipopolysaccharide in mouse peritoneal macrophages. J. Biol. Chem. 2001, 276, 10407–10412. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kölle, P.; Banjac Canak, A.; Förster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 2010, 285, 22244–22253. [Google Scholar] [CrossRef]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Roberts, L.J.; Fessel, J.P. The biochemistry of the isoprostane, neuroprostane, and isofuran pathways of lipid peroxidation. Chem. Phys. Lipids 2004, 128, 173–186. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Pinot, M.; Vanni, S.; Pagnotta, S.; Lacas-Gervais, S.; Payet, L.-A.; Ferreira, T.; Gautier, R.; Goud, B.; Antonny, B.; Barelli, H. Lipid cell biology. Polyunsaturated phospholipids facilitate membrane deformation and fission by endocytic proteins. Science 2014, 345, 693–697. [Google Scholar] [CrossRef]
- Stanley, W.C.; Khairallah, R.J.; Dabkowski, E.R. Update on lipids and mitochondrial function: Impact of dietary n-3 polyunsaturated fatty acids. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 122. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef]
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thévenod, F. Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free. Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.-H.; Bae, D.-H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1468–1483. [Google Scholar] [CrossRef]
- LaVaute, T.; Smith, S.; Cooperman, S.; Iwai, K.; Land, W.; Meyron-Holtz, E.; Drake, S.K.; Miller, G.; Abu-Asab, M.; Tsokos, M.; et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001, 27, 209–214. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef]
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J. Alzheimers Dis. 2013, 37, 127–136. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimers Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Kyheng, M.; Garçon, G.; Rolland, A.S.; Blasco, H.; Gelé, P.; Lenglet, T.T.; Veyrat-Durebex, C.; Corcia, P.; et al. A ferroptosis–based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 2918. [Google Scholar] [CrossRef]
- Spasić, S.; Nikolić-Kokić, A.; Miletić, S.; Oreščanin-Dušić, Z.; Spasić, M.B.; Blagojević, D.; Stević, Z. Edaravone May Prevent Ferroptosis in ALS. Curr. Drug Targets 2020, 21, 776–780. [Google Scholar] [CrossRef]
- Coppola, G.; Marmolino, D.; Lu, D.; Wang, Q.; Cnop, M.; Rai, M.; Acquaviva, F.; Cocozza, S.; Pandolfo, M.; Geschwind, D.H. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich’s ataxia. Hum. Mol. Genet. 2009, 18, 2452–2461. [Google Scholar] [CrossRef]
- Obis, È.; Irazusta, V.; Sanchís, D.; Ros, J.; Tamarit, J. Frataxin deficiency in neonatal rat ventricular myocytes targets mitochondria and lipid metabolism. Free. Radic. Biol. Med. 2014, 73, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rozwadowska, N.; Clark, A.; Fil, D.; Napierala, J.S.; Napierala, M. Excision of the expanded GAA repeats corrects cardiomyopathy phenotypes of iPSC-derived Friedreich’s ataxia cardiomyocytes. Stem Cell Res. 2019, 40, 101529. [Google Scholar] [CrossRef] [PubMed]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar] [CrossRef] [PubMed]
- Cotticelli, M.G.; Crabbe, A.M.; Wilson, R.B.; Shchepinov, M.S. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 2013, 1, 398–404. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Tsui, H.S.; Milne, G.L.; Shmanai, V.V.; Bekish, A.V.; Fomich, M.A.; Pham, M.N.; Nong, Y.; Murphy, A.N.; Clarke, C.F.; et al. Isotope-reinforced polyunsaturated fatty acids protect mitochondria from oxidative stress. Free Radic. Biol. Med. 2015, 82, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium: Biochemical role as a component of glutathione peroxidase. Science 1973, 179, 588–590. [Google Scholar] [CrossRef]
- Florian, S.; Krehl, S.; Loewinger, M.; Kipp, A.; Banning, A.; Esworthy, S.; Chu, F.-F.; Brigelius-Flohé, R. Loss of GPx2 increases apoptosis, mitosis, and GPx1 expression in the intestine of mice. Free Radic. Biol. Med. 2010, 49, 1694–1702. [Google Scholar] [CrossRef]
- Baez-Duarte, B.G.; Mendoza-Carrera, F.; García-Zapién, A.; Flores-Martínez, S.E.; Sánchez-Corona, J.; Zamora-Ginez, I.; Torres-Rasgado, E.; León-Chávez, B.A.; Pérez-Fuentes, R. Multidisciplinary Research Group on Diabetes of the Instituto Mexicano del Seguro Social Glutathione peroxidase 3 serum levels and GPX3 gene polymorphisms in subjects with metabolic syndrome. Arch. Med. Res. 2014, 45, 375–382. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Ufer, C.; Kühn, H.; Borchert, A. Molecular biology of glutathione peroxidase 4: From genomic structure to developmental expression and neural function. Biol. Chem. 2007, 388, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Tramer, F.; Micali, F.; Sandri, G.; Bertoni, A.; Lenzi, A.; Gandini, L.; Panfili, E. Enzymatic and immunochemical evaluation of phospholipid hydroperoxide glutathione peroxidase (PHGPx) in testes and epididymal spermatozoa of rats of different ages. Int. J. Androl. 2002, 25, 72–83. [Google Scholar] [CrossRef]
- Aitken, R.J. Gpx5 protects the family jewels. J. Clin. Investig. 2009, 119, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim Biophys Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Rusolo, F.; Capone, F.; Pasquale, R.; Angiolillo, A.; Colonna, G.; Castello, G.; Costantini, M.; Costantini, S. Comparison of the seleno-transcriptome expression between human non-cancerous mammary epithelial cells and two human breast cancer cell lines. Oncol. Lett. 2017, 13, 2411–2417. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, Y.; Guo, S.; Wang, G. Glutathione peroxidases as oncotargets. Oncotarget 2017, 8, 80093–80102. [Google Scholar] [CrossRef]
- Auchère, F.; Santos, R.; Planamente, S.; Lesuisse, E.; Camadro, J.-M. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich’s ataxia. Hum. Mol. Genet. 2008, 17, 2790–2802. [Google Scholar] [CrossRef]
- Jauslin, M.L.; Wirth, T.; Meier, T.; Schoumacher, F. A cellular model for Friedreich Ataxia reveals small-molecule glutathione peroxidase mimetics as novel treatment strategy. Hum. Mol. Genet. 2002, 11, 3055–3063. [Google Scholar] [CrossRef]
- Richardson, T.E.; Yang, S.-H.; Wen, Y.; Simpkins, J.W. Estrogen Protection in Friedreich’s Ataxia Skin Fibroblasts. Endocrinology 2011, 152, 2742–2749. [Google Scholar] [CrossRef][Green Version]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef]
- Tozzi, G.; Nuccetelli, M.; Bello, M.L.; Bernardini, S.; Bellincampi, L.; Ballerini, S.; Gaeta, L.M.; Casali, C.; Pastore, A.; Federici, G.; et al. Antioxidant enzymes in blood of patients with Friedreich’s ataxia. Arch. Dis. Child. 2002, 86, 376–379. [Google Scholar] [CrossRef]
- Helveston, W.; Hurd, R.; Uthman, B.; Wilder, B.J. Abnormalities of glutathione peroxidase and glutathione reductase in four patients with Friedreich’s disease. Mov. Disord. 1996, 11, 106–107. [Google Scholar] [CrossRef] [PubMed]
- Höfer, A.; Kirches, E.; Mawrin, C. Antioxidative and tumourigenic phenotype of frataxin-expressing glioma cells. Akt. Neurol. 2009, 36, P653. [Google Scholar] [CrossRef]
- Kirches, E.; Andrae, N.; Hoefer, A.; Kehler, B.; Zarse, K.; Leverkus, M.; Keilhoff, G.; Schonfeld, P.; Schneider, T.; Wilisch-Neumann, A.; et al. Dual role of the mitochondrial protein frataxin in astrocytic tumors. Lab. Investig. 2011, 91, 1766–1776. [Google Scholar] [CrossRef] [PubMed]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration—Cause or Consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Xu, S. Iron and Atherosclerosis: The Link Revisited. Trends Mol. Med. 2019, 25, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Gao, G.; Chang, Y.-Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharmacol. 2014, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Isaya, G.; O’Neill, H.A.; Santambrogio, P.; Cozzi, A.; Arosio, P.; Levi, S. The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Hum. Mol. Genet. 2004, 13, 2279–2288. [Google Scholar] [CrossRef]
- Campanella, A.; Rovelli, E.; Santambrogio, P.; Cozzi, A.; Taroni, F.; Levi, S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 2009, 18, 1–11. [Google Scholar] [CrossRef]
- Desmyter, L.; Dewaele, S.; Reekmans, R.; Nystrom, T.; Contreras, R.; Chen, C. Expression of the human ferritin light chain in a frataxin mutant yeast affects ageing and cell death. Exp. Gerontol. 2004, 39, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.B.; Lynch, D.R.; Farmer, J.M.; Brooks, D.G.; Fischbeck, K.H. Increased serum transferrin receptor concentrations in Friedreich ataxia. Ann. Neurol. 2000, 47, 659–661. [Google Scholar] [CrossRef]
- Cavadini, P.; O’Neill, H.A.; Benada, O.; Isaya, G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002, 11, 217–227. [Google Scholar] [CrossRef]
- Li, K.; Besse, E.K.; Ha, D.; Kovtunovych, G.; Rouault, T.A. Iron-dependent regulation of frataxin expression: Implications for treatment of Friedreich ataxia. Hum. Mol. Genet. 2008, 17, 2265. [Google Scholar] [CrossRef]
- Huang, M.L.-H.; Becker, E.M.; Whitnall, M.; Rahmanto, Y.S.; Ponka, P.; Richardson, D.R. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. USA 2009, 106, 16381–16386. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Chen, X.; Li, J.; Comish, P.; Kang, R.; Tang, D. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchère, F.; Lönn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef]
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L.-H. Molecular Alterations in a Mouse Cardiac Model of Friedreich Ataxia: An Impaired Nrf2 Response Mediated via Upregulation of Keap1 and Activation of the Gsk3β Axis. Am. J. Pathol. 2017, 187, 2858–2875. [Google Scholar] [CrossRef]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell. Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. Nrf2 Pathway in Age-Related Neurological Disorders: Insights into MicroRNAs. Cell. Physiol. Biochem. 2018, 47, 1951–1976. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Gahir, S.S.; Lee, C.-Y.I.; Proksch, J.W.; Sakamoto, M.; Ward, K.W. Pharmacokinetics and pharmacodynamics of the novel Nrf2 activator omaveloxolone in primates. Drug Des. Dev. Ther. 2019, 13, 1259–1270. [Google Scholar] [CrossRef]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef] [PubMed]
- Ast, T.; Meisel, J.D.; Patra, S.; Wang, H.; Grange, R.M.H.; Kim, S.H.; Calvo, S.E.; Orefice, L.L.; Nagashima, F.; Ichinose, F.; et al. Hypoxia Rescues Frataxin Loss by Restoring Iron Sulfur Cluster Biogenesis. Cell 2019, 177, 1507–1521.e16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hickman, J.H.; Wang, S.-J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015, 14, 2881–2885. [Google Scholar] [CrossRef]
- Palomo, G.M.; Cerrato, T.; Gargini, R.; Diaz-Nido, J. Silencing of frataxin gene expression triggers p53-dependent apoptosis in human neuron-like cells. Hum. Mol. Genet. 2011, 20, 2807–2822. [Google Scholar] [CrossRef]
- Loría, F.; Díaz-Nido, J. Frataxin knockdown in human astrocytes triggers cell death and the release of factors that cause neuronal toxicity. Neurobiol. Dis. 2015, 76, 1–12. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turchi, R.; Faraonio, R.; Lettieri-Barbato, D.; Aquilano, K. An Overview of the Ferroptosis Hallmarks in Friedreich’s Ataxia. Biomolecules 2020, 10, 1489. https://doi.org/10.3390/biom10111489
Turchi R, Faraonio R, Lettieri-Barbato D, Aquilano K. An Overview of the Ferroptosis Hallmarks in Friedreich’s Ataxia. Biomolecules. 2020; 10(11):1489. https://doi.org/10.3390/biom10111489
Chicago/Turabian StyleTurchi, Riccardo, Raffaella Faraonio, Daniele Lettieri-Barbato, and Katia Aquilano. 2020. "An Overview of the Ferroptosis Hallmarks in Friedreich’s Ataxia" Biomolecules 10, no. 11: 1489. https://doi.org/10.3390/biom10111489
APA StyleTurchi, R., Faraonio, R., Lettieri-Barbato, D., & Aquilano, K. (2020). An Overview of the Ferroptosis Hallmarks in Friedreich’s Ataxia. Biomolecules, 10(11), 1489. https://doi.org/10.3390/biom10111489