Synthetically Lethal Interactions of Heme Oxygenase-1 and Fumarate Hydratase Genes

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs and Reagents
2.3. Production of Lentiviral Vectors Encoding shRNA Sequences against HMOX1 and Transduction of Target Cells
2.4. MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) Viability Assay
2.5. BrdU (Bromodeoxyuridine) Incorporation Assay
2.6. Colony Formation Assay
2.7. Quantitative Real-Time PCR (qRT-PCR)
2.8. Determination of Protein Concentration
2.9. Fumarate Hydratase Activity Measurement
2.10. Western Blot
2.11. Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) Measurement
2.12. Statistical Analysis
3. Results
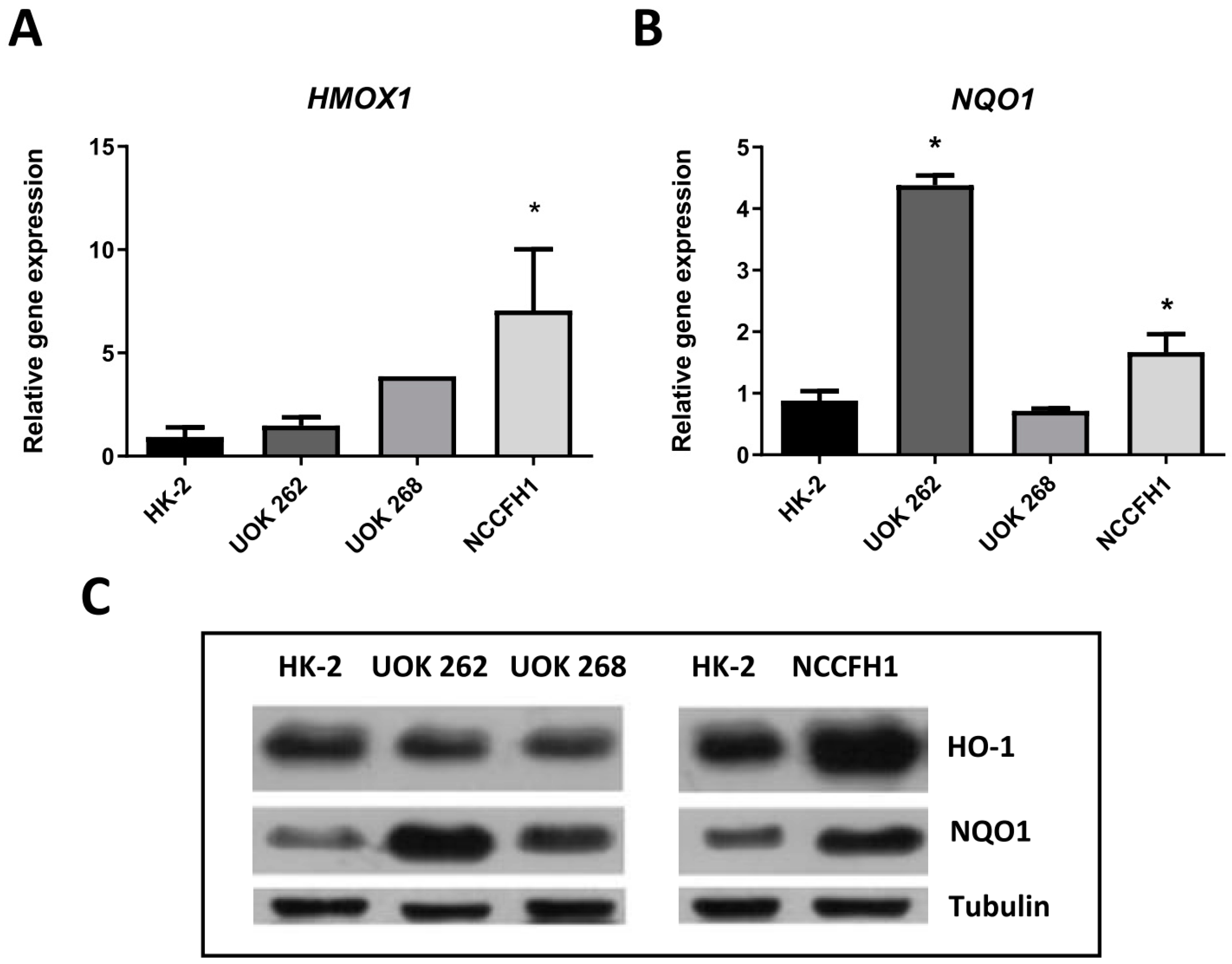
3.1. Characterization of FH-Deficient Cell Lines
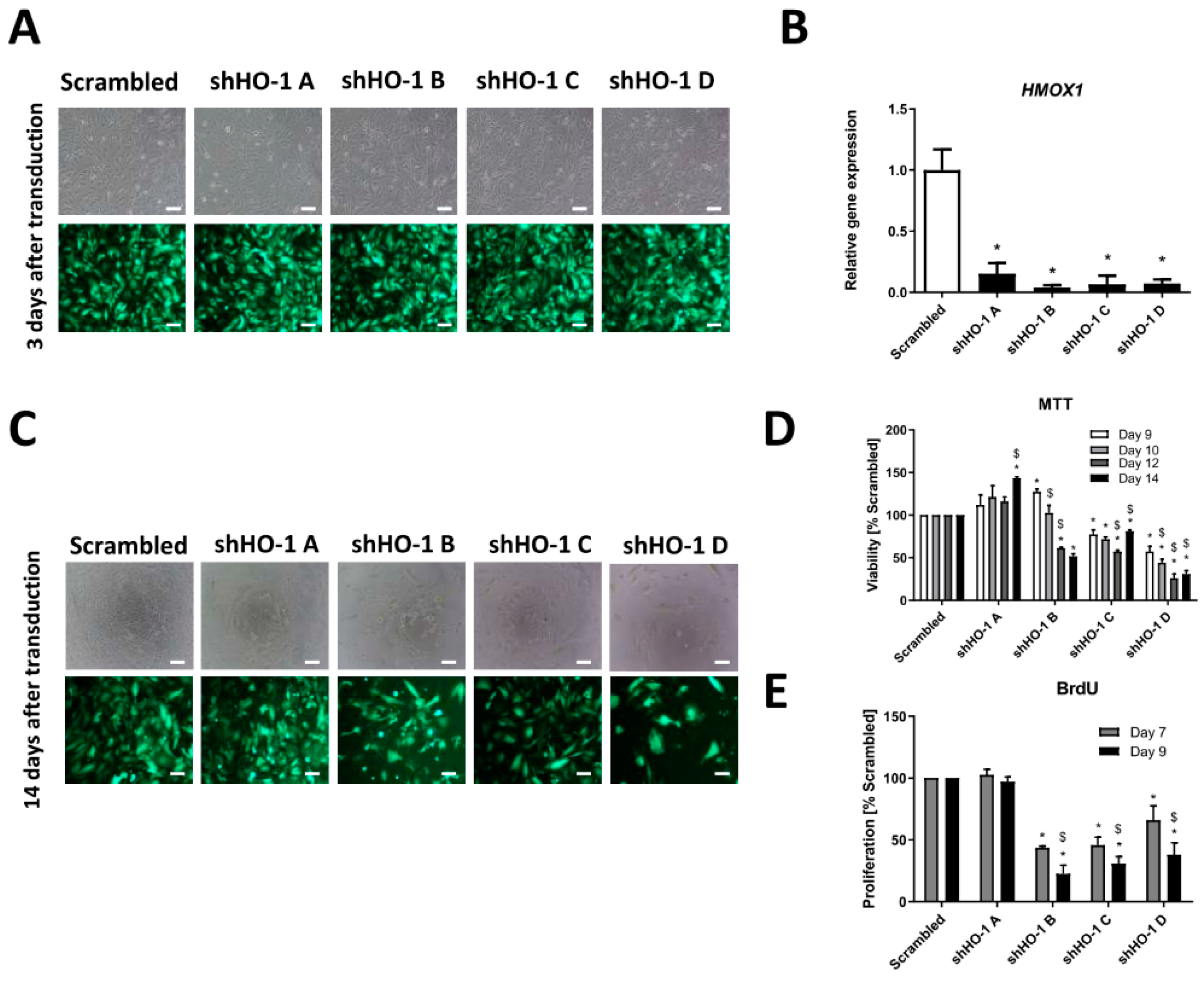
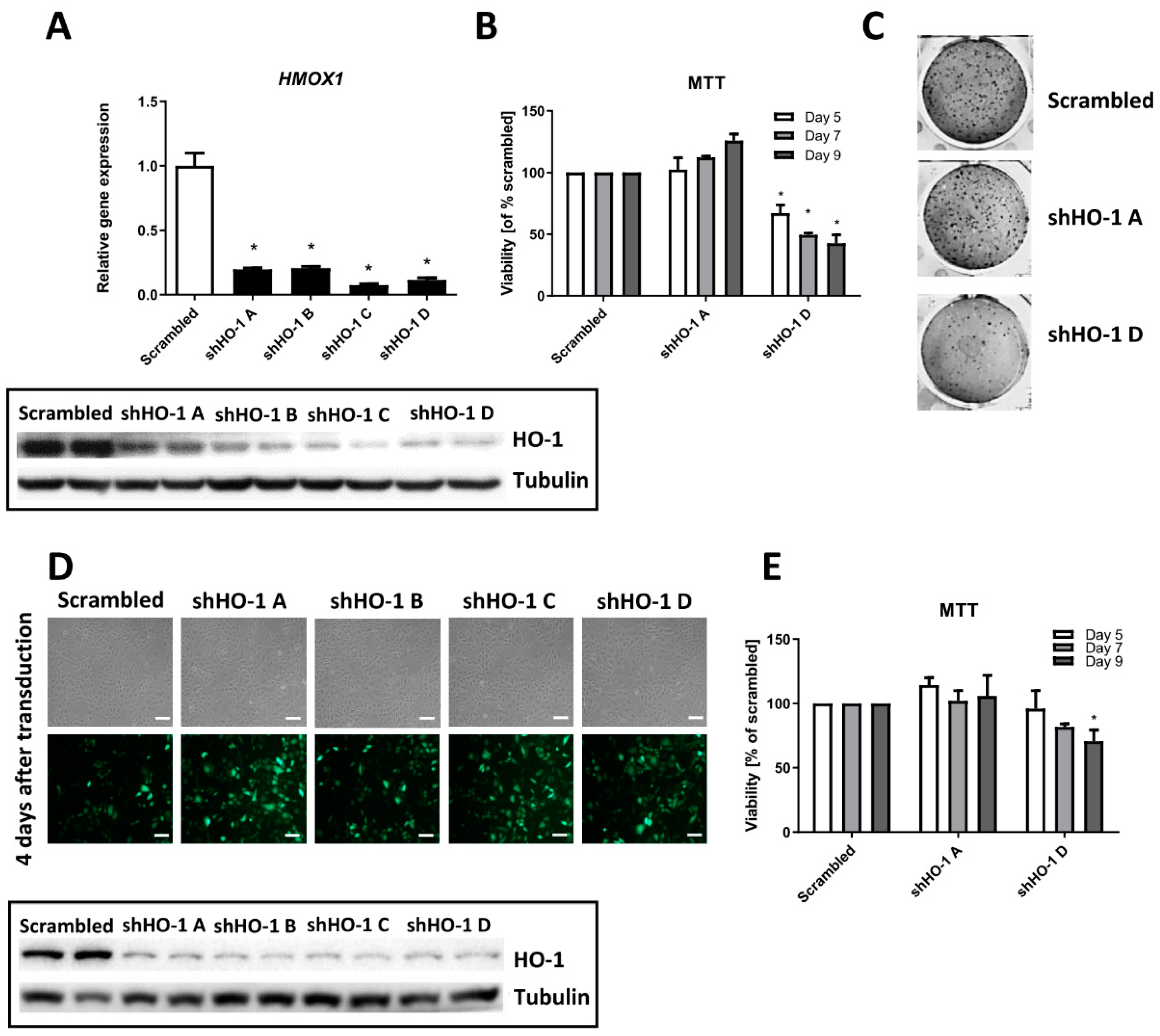
3.2. Genetic Inhibition of HMOX1 Decreases Viability and Proliferation of FH-Deficient Cell Lines
3.3. The Effect of Chemical Inhibition of HO Activity on Viability and Clonogenic Potential of FH-Deficient Cell Lines
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tomlinson, I.P.M.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.M.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [PubMed]
- Grubb, R.L.; Franks, M.E.; Toro, J.; Middelton, L.; Choyke, L.; Fowler, S.; Torres-Cabala, C.; Glenn, G.M.; Choyke, P.; Merino, M.J.; et al. Hereditary Leiomyomatosis and Renal Cell Cancer: A Syndrome Associated With an Aggressive Form of Inherited Renal Cancer. J. Urol. 2007, 177, 2074–2080. [Google Scholar] [CrossRef] [PubMed]
- Menko, F.H.; Maher, E.R.; Schmidt, L.S.; Middelton, L.A.; Aittomäki, K.; Tomlinson, I.; Richard, S.; Linehan, W.M. Hereditary leiomyomatosis and renal cell cancer (HLRCC): Renal cancer risk, surveillance and treatment. Fam. Cancer 2014, 13, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Valera, V.A.; Padilla-Nash, H.M.; Sourbier, C.; Vocke, C.D.; Vira, M.A.; Abu-Asab, M.S.; Bratslavsky, G.; Tsokos, M.; Merino, M.J.; et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: In vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet. Cytogenet. 2010, 196, 45–55. [Google Scholar] [CrossRef]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Sudarshan, S.; Sourbier, C.; Kong, H.S.; Block, K.; Valera Romero, V.A.; Yang, Y.; Galindo, C.; Mollapour, M.; Scroggins, B.; Goode, N.; et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol. Cell. Biol. 2009, 29, 4080–4090. [Google Scholar] [CrossRef]
- Sourbier, C.; Valera-Romero, V.; Giubellino, A.; Yang, Y.; Sudarshan, S.; Neckers, L.; Linehan, W.M. Increasing reactive oxygen species as a therapeutic approach to treat hereditary leiomyomatosis and renal cell carcinoma. Cell Cycle Georget. Tex. 2010, 9, 4183–4189. [Google Scholar] [CrossRef]
- Ratcliffe, P.J. Fumarate Hydratase Deficiency and Cancer: Activation of Hypoxia Signaling? Cancer Cell 2007, 11, 303–305. [Google Scholar] [CrossRef][Green Version]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Kulkarni, R.A.; Bak, D.W.; Wei, D.; Bergholtz, S.E.; Briney, C.A.; Shrimp, J.H.; Alpsoy, A.; Thorpe, A.L.; Bavari, A.E.; Crooks, D.R.; et al. A chemoproteomic portrait of the oncometabolite fumarate. Nat. Chem. Biol. 2019, 15, 391–400. [Google Scholar] [CrossRef]
- Yang, M.; Ternette, N.; Su, H.; Dabiri, R.; Kessler, B.; Adam, J.; Teh, B.; Pollard, P. The Succinated Proteome of FH-Mutant Tumours. Metabolites 2014, 4, 640–654. [Google Scholar] [CrossRef]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.H.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An Antioxidant Response Phenotype Shared between Hereditary and Sporadic Type 2 Papillary Renal Cell Carcinoma. Cancer Cell 2011, 20, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal Cyst Formation in Fh1-Deficient Mice Is Independent of the Hif/Phd Pathway: Roles for Fumarate in KEAP1 Succination and Nrf2 Signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Podkalicka, P.; Mucha, O.; Józkowicz, A.; Dulak, J.; Łoboda, A. Heme oxygenase inhibition in cancers: Possible tools and targets. Contemp. Oncol. Poznan Pol. 2018, 22, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO system in tumor growth, angiogenesis and metabolism—Targeting HO-1 as an anti-tumor therapy. Vascul. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef]
- Nijman, S.M.B. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef]
- Qiu, S.; Adema, C.M.; Lane, T. A computational study of off-target effects of RNA interference. Nucleic Acids Res. 2005, 33, 1834–1847. [Google Scholar] [CrossRef]
- Mucha, O.; Podkalicka, P.; Czarnek, M.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Stepniewski, J.; Jozkowicz, A.; Dulak, J.; Loboda, A. Pharmacological versus genetic inhibition of heme oxygenase-1—The comparison of metalloporphyrins, shRNA and CRISPR/Cas9 system. Acta Biochim. Pol. 2018, 65, 277–286. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxid. Med. Cell. Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef]
- He, J.Z.; Ho, J.J.D.; Gingerich, S.; Courtman, D.W.; Marsden, P.A.; Ward, M.E. Enhanced translation of heme oxygenase-2 preserves human endothelial cell viability during hypoxia. J. Biol. Chem. 2010, 285, 9452–9461. [Google Scholar] [CrossRef] [PubMed]
- Intagliata, S.; Salerno, L.; Ciaffaglione, V.; Leonardi, C.; Fallica, A.N.; Carota, G.; Amata, E.; Marrazzo, A.; Pittalà, V.; Romeo, G. Heme Oxygenase-2 (HO-2) as a therapeutic target: Activators and inhibitors. Eur. J. Med. Chem. 2019, 183, 111703. [Google Scholar] [CrossRef] [PubMed]
- Mucha, O.; Podkalicka, P.; Mikulski, M.; Barwacz, S.; Andrysiak, K.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Ryszawy, D.; Białas, A.; et al. Development and characterization of a new inhibitor of heme oxygenase activity for cancer treatment. Arch. Biochem. Biophys. 2019, 671, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Floresta, G.; Ciaffaglione, V.; Gentile, D.; Margani, F.; Turnaturi, R.; Rescifina, A.; Pittalà, V. Progress in the development of selective heme oxygenase-1 inhibitors and their potential therapeutic application. Eur. J. Med. Chem. 2019, 167, 439–453. [Google Scholar] [CrossRef]
- Wong, R.J.; Vreman, H.J.; Schulz, S.; Kalish, F.S.; Pierce, N.W.; Stevenson, D.K. In vitro inhibition of heme oxygenase isoenzymes by metalloporphyrins. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2011, 31, S35–S41. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Yang, Y.; Valera, V.; Sourbier, C.; Vocke, C.D.; Wei, M.; Pike, L.; Huang, Y.; Merino, M.A.; Bratslavsky, G.; Wu, M.; et al. A novel fumarate hydratase-deficient HLRCC kidney cancer cell line, UOK268: A model of the Warburg effect in cancer. Cancer Genet. 2012, 205, 377–390. [Google Scholar] [CrossRef]
- Perrier-Trudova, V.; Huimin, B.W.; Kongpetch, S.; Huang, D.; Ong, P.; LE Formal, A.; Poon, S.L.; Siew, E.Y.; Myint, S.S.; Gad, S.; et al. Fumarate Hydratase-deficient Cell Line NCCFH1 as a New In Vitro Model of Hereditary Papillary Renal Cell Carcinoma Type 2. Anticancer Res. 2015, 35, 6639–6653. [Google Scholar]
- Boettcher, M.; Lawson, A.; Ladenburger, V.; Fredebohm, J.; Wolf, J.; Hoheisel, J.D.; Frezza, C.; Shlomi, T. High throughput synthetic lethality screen reveals a tumorigenic role of adenylate cyclase in fumarate hydratase-deficient cancer cells. BMC Genomics 2014, 15, 158. [Google Scholar] [CrossRef]
- Rahman, M.N.; Vlahakis, J.Z.; Vukomanovic, D.; Lee, W.; Szarek, W.A.; Nakatsu, K.; Jia, Z. A novel, “double-clamp” binding mode for human heme oxygenase-1 inhibition. PLoS ONE 2012, 7, e29514. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef]
- Yang, Y.; Lane, A.N.; Ricketts, C.J.; Sourbier, C.; Wei, M.-H.; Shuch, B.; Pike, L.; Wu, M.; Rouault, T.A.; Boros, L.G.; et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS ONE 2013, 8, e72179. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, M.; Marona, P.; Kozakowska, M.; Jez, M.; Seczynska, M.; Loboda, A.; Bukowska-Strakova, K.; Szade, A.; Walawender, M.; Kusior, M.; et al. Heme Oxygenase-1 Controls an HDAC4-miR-206 Pathway of Oxidative Stress in Rhabdomyosarcoma. Cancer Res. 2016, 76, 5707–5718. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Greenside, P.G.; Natoli, T.; Lahr, D.L.; Wadden, D.; Tirosh, I.; Narayan, R.; Root, D.E.; Golub, T.R.; Subramanian, A.; et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017, 15, e2003213. [Google Scholar] [CrossRef]
- Rao, D.D.; Senzer, N.; Cleary, M.A.; Nemunaitis, J. Comparative assessment of siRNA and shRNA off target effects: What is slowing clinical development. Cancer Gene Ther. 2009, 16, 807–809. [Google Scholar] [CrossRef]
- Cullen, B.R. Enhancing and confirming the specificity of RNAi experiments. Nat. Methods 2006, 3, 677–681. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene of Interest | Primer Sequence |
|---|---|
| EEF2 forward | 5′-GAGATCCAGTGTCCAGAGCAG-3′ |
| EEF2 reverse | 5′-CTCGTTGACGGGCAGATAGG-3′ |
| FH forward | 5′-GTATTATGGCGCCCAGACC-3′ |
| FH reverse | 5′-ATCCTGGTTTACTTCAGCGG-3′ |
| HMOX1 forward | 5′-TTCTTCACCTTCCCCAACATT-3′ |
| HMOX1 reverse | 5′-CAGCTCCTGCAACTCCTCAAA-3′ |
| NQO1 forward | 5′-AGGACCCTTCCGGAGTAAGA-3′ |
| NQO1 reverse | 5′-CCAGGATTTGAATTCGGGCG-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podkalicka, P.; Mucha, O.; Kruczek, S.; Biela, A.; Andrysiak, K.; Stępniewski, J.; Mikulski, M.; Gałęzowski, M.; Sitarz, K.; Brzózka, K.; et al. Synthetically Lethal Interactions of Heme Oxygenase-1 and Fumarate Hydratase Genes. Biomolecules 2020, 10, 143. https://doi.org/10.3390/biom10010143
Podkalicka P, Mucha O, Kruczek S, Biela A, Andrysiak K, Stępniewski J, Mikulski M, Gałęzowski M, Sitarz K, Brzózka K, et al. Synthetically Lethal Interactions of Heme Oxygenase-1 and Fumarate Hydratase Genes. Biomolecules. 2020; 10(1):143. https://doi.org/10.3390/biom10010143
Chicago/Turabian StylePodkalicka, Paulina, Olga Mucha, Szczepan Kruczek, Anna Biela, Kalina Andrysiak, Jacek Stępniewski, Maciej Mikulski, Michał Gałęzowski, Kamil Sitarz, Krzysztof Brzózka, and et al. 2020. "Synthetically Lethal Interactions of Heme Oxygenase-1 and Fumarate Hydratase Genes" Biomolecules 10, no. 1: 143. https://doi.org/10.3390/biom10010143
APA StylePodkalicka, P., Mucha, O., Kruczek, S., Biela, A., Andrysiak, K., Stępniewski, J., Mikulski, M., Gałęzowski, M., Sitarz, K., Brzózka, K., Józkowicz, A., Dulak, J., & Łoboda, A. (2020). Synthetically Lethal Interactions of Heme Oxygenase-1 and Fumarate Hydratase Genes. Biomolecules, 10(1), 143. https://doi.org/10.3390/biom10010143