Characterization of the Prion Protein Binding Properties of Antisense Oligonucleotides

Abstract
1. Introduction
2. Materials and Methods
2.1. Recombinant Protein Preparation
2.2. Test Compounds
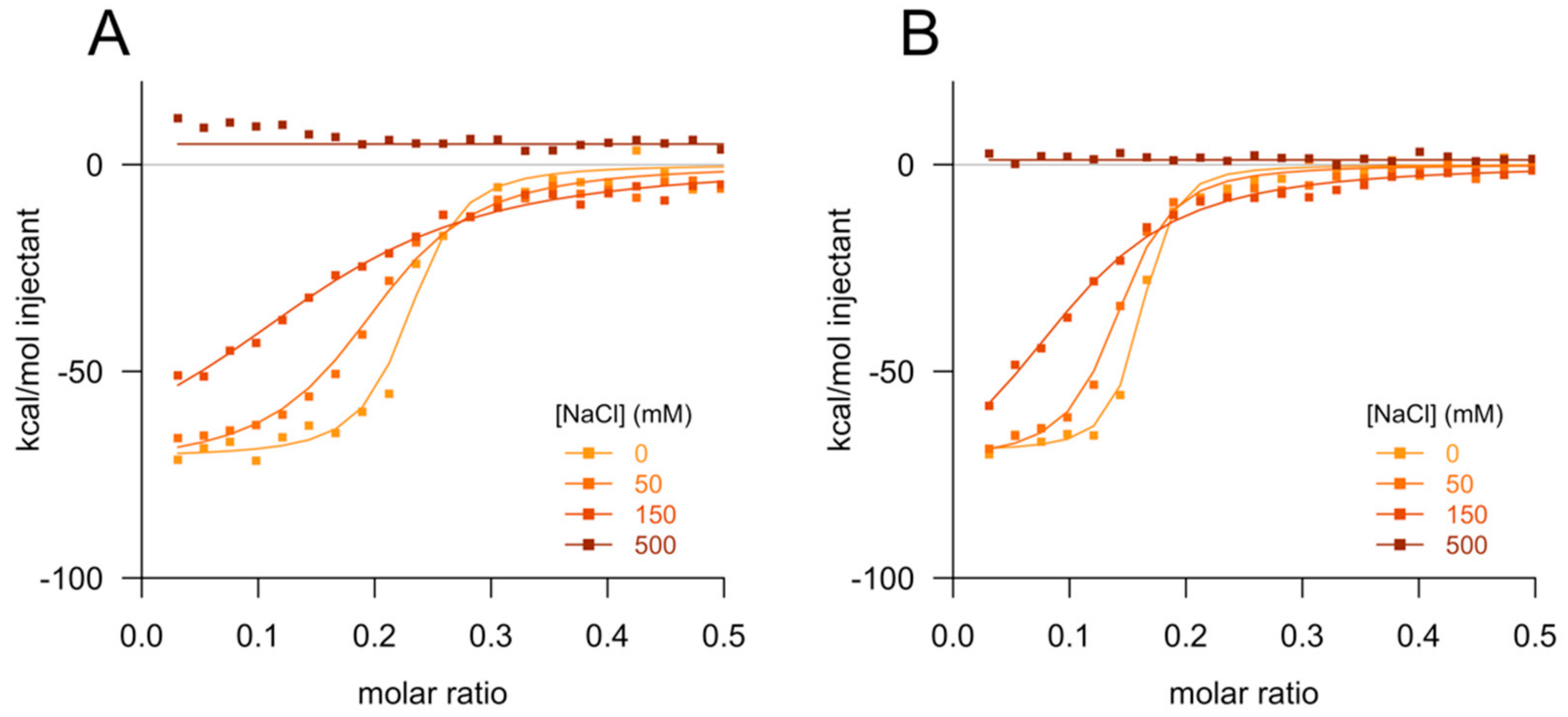
2.3. Isothermal Titration Calorimetry
2.4. Nuclear Magnetic Resonance
2.5. Dynamic Light Scattering
2.6. Tissue Culture
2.7. qPCR
2.8. Immunoblotting and Proteinase K Digest
2.9. Enzyme-linked immunosorbent assay (ELISA)
2.10. Data Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Vallabh, S.M. Antisense Oligonucleotides for the Prevention of Genetic Prion Disease. Ph.D. Dissertation, Harvard University, Cambridge, MA, USA, 2019. [Google Scholar]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.F.; Rose, J.B.; Nichols, J.G.; Wu, H.; Migawa, M.T.; Wyrzykiewicz, T.K.; Siwkowski, A.M.; Crooke, S.T. Human RNase H1 discriminates between subtle variations in the structure of the heteroduplex substrate. Mol. Pharmacol. 2007, 71, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.F.; Murray, H.M.; Damle, S.S.; Hart, C.E.; Hung, G.; De Hoyos, C.L.; Liang, X.-H.; Crooke, S.T. Viable RNaseH1 knockout mice show RNaseH1 is essential for R loop processing, mitochondrial and liver function. Nucleic Acids Res. 2016, 44, 5299–5312. [Google Scholar] [CrossRef] [PubMed]
- Nazor Friberg, K.; Hung, G.; Wancewicz, E.; Giles, K.; Black, C.; Freier, S.; Bennett, F.; Dearmond, S.J.; Freyman, Y.; Lessard, P.; et al. Intracerebral Infusion of Antisense Oligonucleotides Into Prion-infected Mice. Mol. Ther. Nucleic Acids 2012, 1, e9. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, G.J. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 1993, 67, 643–650. [Google Scholar] [PubMed]
- Gabizon, R.; Meiner, Z.; Halimi, M.; Ben-Sasson, S.A. Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J. Cell. Physiol. 1993, 157, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Macedo, B.; Cordeiro, Y. Unraveling Prion Protein Interactions with Aptamers and Other PrP-Binding Nucleic Acids. Int. J. Mol. Sci. 2017, 18, 1023. [Google Scholar] [CrossRef]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- Kocisko, D.A.; Vaillant, A.; Lee, K.S.; Arnold, K.M.; Bertholet, N.; Race, R.E.; Olsen, E.A.; Juteau, J.-M.; Caughey, B. Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob. Agents Chemother. 2006, 50, 1034–1044. [Google Scholar] [CrossRef]
- Karpuj, M.V.; Giles, K.; Gelibter-Niv, S.; Scott, M.R.; Lingappa, V.R.; Szoka, F.C.; Peretz, D.; Denetclaw, W.; Prusiner, S.B. Phosphorothioate oligonucleotides reduce PrP levels and prion infectivity in cultured cells. Mol. Med. 2007, 13, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.J.; Zhao, H.T.; Race, B.; Raymond, L.D.; Williams, K.; Swayze, E.E.; Graffam, S.; Le, J.; Caron, T.; Stathopoulos, J.; et al. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Vallabh, S.M.; Nobuhara, C.K.; Llorens, F.; Zerr, I.; Parchi, P.; Capellari, S.; Kuhn, E.; Klickstein, J.; Safar, J.G.; Nery, F.C.; et al. Prion protein quantification in human cerebrospinal fluid as a tool for prion disease drug development. PNAS 2019, 116, 7793–7798. [Google Scholar] [CrossRef] [PubMed]
- Minikel, E.V.; Kuhn, E.; Cocco, A.R.; Vallabh, S.M.; Hartigan, C.R.; Reidenbach, A.G.; Safar, J.G.; Raymond, G.J.; McCarthy, M.D.; O’Keefe, R.; et al. Domain-specific quantification of prion protein in cerebrospinal fluid by targeted mass spectrometry. Mol. Cell Proteom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vallabh, S.M.; Minikel, E.V.; Williams, V.J.; Carlyle, B.C.; McManus, A.J.; Wennick, C.D.; Bolling, A.; Trombetta, B.A.; Urick, D.; Nobuhara, C.K.; et al. Cerebrospinal fluid and plasma biomarkers in individuals at risk for genetic prion disease. medRxiv 2019. [Google Scholar] [CrossRef]
- Orrù, C.D.; Groveman, B.R.; Hughson, A.G.; Manca, M.; Raymond, L.D.; Raymond, G.J.; Campbell, K.J.; Anson, K.J.; Kraus, A.; Caughey, B. RT-QuIC Assays for Prion Disease Detection and Diagnostics. Methods Mol. Biol. 2017, 1658, 185–203. [Google Scholar]
- Butler, D.A.; Scott, M.R.; Bockman, J.M.; Borchelt, D.R.; Taraboulos, A.; Hsiao, K.K.; Kingsbury, D.T.; Prusiner, S.B. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J. Virol. 1988, 62, 1558–1564. [Google Scholar]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a central nervous system delivered 2’-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef]
- Geary, R.S.; Watanabe, T.A.; Truong, L.; Freier, S.; Lesnik, E.A.; Sioufi, N.B.; Sasmor, H.; Manoharan, M.; Levin, A.A. Pharmacokinetic properties of 2’-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J. Pharmacol. Exp. Ther. 2001, 296, 890–897. [Google Scholar]
- Pervushin, K.; Riek, R.; Wider, G.; Wüthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371. [Google Scholar] [CrossRef] [PubMed]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Swayze, E.E.; Siwkowski, A.M.; Wancewicz, E.V.; Migawa, M.T.; Wyrzykiewicz, T.K.; Hung, G.; Monia, B.P.; Bennett, C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007, 35, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Skotte, N.H.; Southwell, A.L.; Østergaard, M.E.; Carroll, J.B.; Warby, S.C.; Doty, C.N.; Petoukhov, E.; Vaid, K.; Kordasiewicz, H.; Watt, A.T.; et al. Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: providing a therapeutic option for all Huntington disease patients. PLoS ONE 2014, 9, e107434. [Google Scholar] [CrossRef] [PubMed]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef]
- Matos, C.O.; Passos, Y.M.; do Amaral, M.J.; Macedo, B.; Tempone, M.; Bezerra, O.C.L.; Moraes, M.O.; Almeida, M.S.; Weber, G.; Missailidis, S.; et al. Liquid-liquid phase separation and aggregation of the prion protein globular domain modulated by a high-affinity DNA aptamer. bioRxiv 2019, 659037. [Google Scholar] [CrossRef]
- Kostylev, M.A.; Tuttle, M.D.; Lee, S.; Klein, L.E.; Takahashi, H.; Cox, T.O.; Gunther, E.C.; Zilm, K.W.; Strittmatter, S.M. Liquid and Hydrogel Phases of PrPC Linked to Conformation Shifts and Triggered by Alzheimer’s Amyloid-β Oligomers. Mol. Cell 2018, 72, 426–443.e12. [Google Scholar] [CrossRef]
- Cordeiro, Y.; Machado, F.; Juliano, L.; Juliano, M.A.; Brentani, R.R.; Foguel, D.; Silva, J.L. DNA converts cellular prion protein into the beta-sheet conformation and inhibits prion peptide aggregation. J. Biol. Chem. 2001, 276, 49400–49409. [Google Scholar] [CrossRef]
- Lima, L.M.T.R.; Cordeiro, Y.; Tinoco, L.W.; Marques, A.F.; Oliveira, C.L.P.; Sampath, S.; Kodali, R.; Choi, G.; Foguel, D.; Torriani, I.; et al. Structural insights into the interaction between prion protein and nucleic acid. Biochemistry 2006, 45, 9180–9187. [Google Scholar] [CrossRef]
- Gomes, M.P.B.; Millen, T.A.; Ferreira, P.S.; e Silva, N.L.C.; Vieira, T.C.R.G.; Almeida, M.S.; Silva, J.L.; Cordeiro, Y. Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J. Biol. Chem. 2008, 283, 19616–19625. [Google Scholar] [CrossRef] [PubMed]
- Macedo, B.; Millen, T.A.; Braga, C.A.C.A.; Gomes, M.P.B.; Ferreira, P.S.; Kraineva, J.; Winter, R.; Silva, J.L.; Cordeiro, Y. Nonspecific prion protein-nucleic acid interactions lead to different aggregates and cytotoxic species. Biochemistry 2012, 51, 5402–5413. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.-H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Salamat-Miller, N.; Fang, J.; Seidel, C.W.; Assenov, Y.; Albrecht, M.; Middaugh, C.R. A network-based analysis of polyanion-binding proteins utilizing human protein arrays. J. Biol. Chem. 2007, 282, 10153–10163. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, L.D.; Raymond, G.J.; Maxson, L.; Silveira, J.; Baron, G.S. Inhibition of protease-resistant prion protein accumulation in vitro by curcumin. J. Virol. 2003, 77, 5499–5502. [Google Scholar] [CrossRef]
- Kocisko, D.A.; Baron, G.S.; Rubenstein, R.; Chen, J.; Kuizon, S.; Caughey, B. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J. Virol. 2003, 77, 10288–10294. [Google Scholar] [CrossRef]
- Riemer, C.; Burwinkel, M.; Schwarz, A.; Gültner, S.; Mok, S.W.F.; Heise, I.; Holtkamp, N.; Baier, M. Evaluation of drugs for treatment of prion infections of the central nervous system. J. Gen. Virol. 2008, 89, 594–597. [Google Scholar] [CrossRef]
- Kocisko, D.A.; Morrey, J.D.; Race, R.E.; Chen, J.; Caughey, B. Evaluation of new cell culture inhibitors of protease-resistant prion protein against scrapie infection in mice. J. Gen. Virol. 2004, 85, 2479–2483. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Annotated Sequence | nt | Chemistry | MW (kDa) |
|---|---|---|---|---|
| control ASO 0 | mCmCTTmCmCmCTGAAGGTTmCmCTmCmC | 20 | PS MOE | 7.15 |
| active ASO 1 | mCToAoTTTAATGTmCAoGoTmCT | 17 | PS/PO MOE/cEt | 5.99 |
| active ASO 2 | TToGomCAATTmCTATmComCoAAA | 17 | PS/PO MOE/cEt | 5.98 |
| control ASO 3 | mCGomCoTTATAmCTAATmCoAoTAT | 17 | PS/PO MOE/cEt | 5.98 |
| control ASO 4 | mCmCoToAoTAGGAmCTATmCmCAoGoGoAA | 20 | PS/PO MOE | 7.13 |
| active ASO 6 | mCToTomCoTATTTAATGTmCAoGoTmCT | 20 | PS/PO MOE | 7.07 |
| active ASO 7 | TAoGomComCTTTGTACCTTAoAomCmCA | 20 | PS/PO MOE | 7.08 |
| active ASO 8 | GmComCoAAGGTTmCGmCmCoAoTGA | 17 | PS/PO MOE | 6.12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reidenbach, A.G.; Minikel, E.V.; Zhao, H.T.; Guzman, S.G.; Leed, A.J.; Mesleh, M.F.; Kordasiewicz, H.B.; Schreiber, S.L.; Vallabh, S.M. Characterization of the Prion Protein Binding Properties of Antisense Oligonucleotides. Biomolecules 2020, 10, 1. https://doi.org/10.3390/biom10010001
Reidenbach AG, Minikel EV, Zhao HT, Guzman SG, Leed AJ, Mesleh MF, Kordasiewicz HB, Schreiber SL, Vallabh SM. Characterization of the Prion Protein Binding Properties of Antisense Oligonucleotides. Biomolecules. 2020; 10(1):1. https://doi.org/10.3390/biom10010001
Chicago/Turabian StyleReidenbach, Andrew G., Eric Vallabh Minikel, Hien T. Zhao, Stacy G. Guzman, Alison J. Leed, Michael F. Mesleh, Holly B. Kordasiewicz, Stuart L. Schreiber, and Sonia M. Vallabh. 2020. "Characterization of the Prion Protein Binding Properties of Antisense Oligonucleotides" Biomolecules 10, no. 1: 1. https://doi.org/10.3390/biom10010001
APA StyleReidenbach, A. G., Minikel, E. V., Zhao, H. T., Guzman, S. G., Leed, A. J., Mesleh, M. F., Kordasiewicz, H. B., Schreiber, S. L., & Vallabh, S. M. (2020). Characterization of the Prion Protein Binding Properties of Antisense Oligonucleotides. Biomolecules, 10(1), 1. https://doi.org/10.3390/biom10010001