Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches

 , , and
, , and
Abstract
1. Introduction
2. Theory
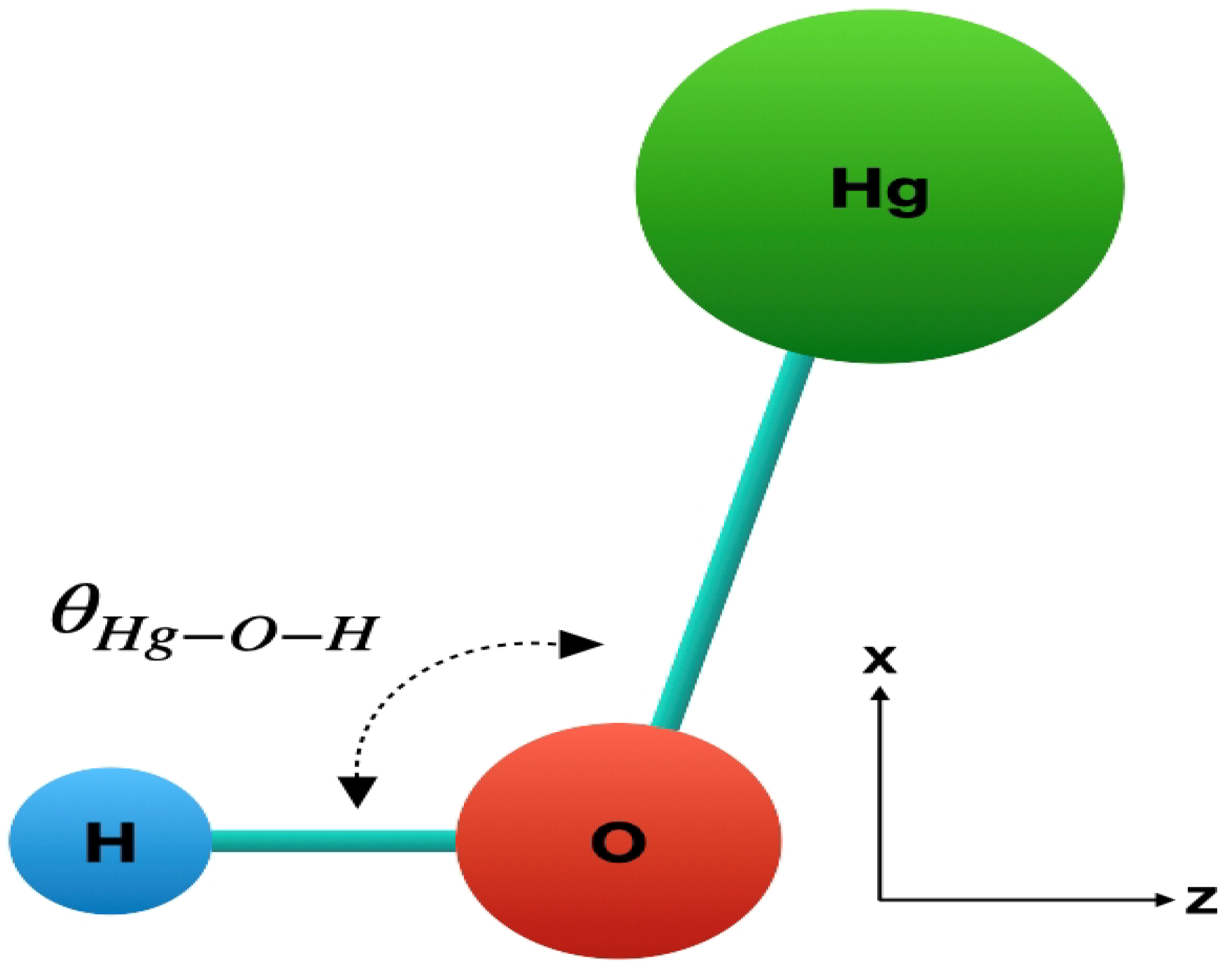
3. Ground State Geometry Optimization
4. Method of Calculation
5. Results and Discussion
6. Other Prospective Polyatomic Molecules for EDM Measurements
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Landau, L. On the conservation laws for weak interactions. Nucl. Phys. 1957, 3, 127131. [Google Scholar] [CrossRef]
- Ballentine, L.E. Quantum Mechanics: A Modern Development; World Scientific: Singapore, 1998; Volume 384386, Chapter 13; pp. 372–373. [Google Scholar]
- Luders, G. Proof of the TCP Theorem. Ann. Phys. 2000, 281, 1004. [Google Scholar] [CrossRef]
- Hoogeveen, F. DESY Reports 1990; DESY: Hamburg, Germany, 1990; pp. 6–90. [Google Scholar]
- Pospelov, M.; Ritz, A. CKM benchmarks for electron electric dipole moment experiments. Phys. Rev. D 2014, 89, 056006. [Google Scholar] [CrossRef]
- Kazarian, A.M.; Kuzmin, S.V.; Shaposhnikov, M.E. Cosmological lower bound on the EDM of the electron. Phys. Lett. B 1992, 276, 131. [Google Scholar] [CrossRef]
- Fuyuto, K.; Hisano, J.; Senaha, E. Toward verification of electroweak baryogenesis by electric dipole moments. Phys. Lett. B 2016, 755, 491. [Google Scholar] [CrossRef]
- Schiff, L.I. Measurability of nuclear electric dipole moments. Phys. Rev. 1963, 132, 2194. [Google Scholar] [CrossRef]
- Sandars, P.G.H. The electric-dipole moments of an atom II. The contribution from an electric-dipole moment on the electron with particular reference to the hydrogen atom. J. Phys. B 1968, 1, 511. [Google Scholar] [CrossRef]
- Abe, M.; Gopakumar, G.; Hada, M.; Das, B.P.; Tatewaki, H.; Mukherjee, D. Application of relativistic coupled-cluster theory to the effective electric field in YbF. Phys. Rev. A 2014, 90, 022501. [Google Scholar] [CrossRef]
- Prasannaa, V.S.; Vutha, A.C.; Abe, M.; Das, B.P. Mercury monohalides: Suitability for electron electric dipole moment searches. Phys. Rev. Lett. 2015, 114, 183001. [Google Scholar] [CrossRef]
- Skripnikov, L.V. Combined 4-component and relativistic pseudopotential study of ThO for the electron electric dipole moment search. J. Chem. Phys. 2016, 145, 214301. [Google Scholar] [CrossRef]
- Skripnikov, L.V. Communication: Theoretical study of HfF+ cation to search for the T,P-odd interactions. J. Chem. Phys. 2017, 147, 021101. [Google Scholar] [CrossRef] [PubMed]
- Andreev, V.; Ang, D.G.; DeMille, D.; Doyle, J.M.; Gabrielse, G.; Haefner, J.; Hutzler, N.R.; Lasner, Z.; Meisenhelder, C.; O’Leary, B.R.; et al. Improved limit on the electric dipole moment of the electron. Nature 2018, 562, 355. [Google Scholar]
- Cairncross, W.; Gresh, D.N.; Grau, M.; Cossel, K.C.; Roussy, T.S.; Ni, Y.; Zhou, Y.; Ye, J.; Cornell, E.A. Precision measurement of the electron’s electric dipole moment using trapped molecular ions. Phys. Rev. Lett. 2017, 119, 153001. [Google Scholar] [CrossRef]
- Kara, D.M.; Smallman, I.J.; Hudson, J.J.; Sauer, B.E.; Tarbutt, M.R.; Hinds, E.A. Measurement of the electron’s electric dipole moment using YbF molecules: Methods and data analysis. New J. Phys. 2012, 14, 103051. [Google Scholar] [CrossRef]
- The NL-EDM Collaboration; Aggarwal, P.; Bethlem, H.L.; Borschevsky, A.; Denis, M.; Esajas, K.; Haase, P.A.B.; Hao, Y.; Hoekstra, S.; Jungmann, K.; et al. Measuring the electric dipole moment of the electron in BaF. Eur. Phys. J. D 2018, 72, 197. [Google Scholar] [CrossRef]
- Vutha, A.C.; Horbatsch, M.; Hessels, E.A. Oriented polar molecules in a solid inert-gas matrix: A proposed method for measuring the electric dipole moment of the electron. Atoms 2018, 6, 3. [Google Scholar] [CrossRef]
- Meyer, E.R.; Bohn, J.L.; Deskevich, M.P. Candidate molecular ions for an electron electric dipole moment experiment. Phys. Rev. A 2006, 73, 062108. [Google Scholar] [CrossRef]
- Meyer, E.R.; Bohn, J.L. Electron electric-dipole-moment searches based on alkali-metal- or alkaline-earth-metal-bearing molecules. Phys. Rev. A 2009, 80, 042508. [Google Scholar] [CrossRef]
- Lee, J.; Meyer, E.R.; Paudel, R.; Bohn, J.L.; Leanhardt, A.E. An electron electric dipole moment search in the X3Δ1 ground state of tungsten carbide molecules. J. Mod. Opt. 2009, 56, 2005. [Google Scholar] [CrossRef]
- Kudashov, A.D.; Petrov, A.N.; Skripnikov, L.V.; Mosyagin, N.S.; Isaev, T.A.; Berger, R.; Titov, A.V. Coupled-cluster study of radium monofluoride, RaF, as a candidate to search for P- and T,P- violation effects. Phys. Rev. A 2014, 90, 052513. [Google Scholar] [CrossRef]
- Skripnikov, L.V.; Petrov, A.N.; Mosyagin, N.S.; Titov, A.V.; Flambaum, V.V. TaN molecule as a candidate for the search for a T,P-violating nuclear magnetic quadrupole moment. Phys. Rev. A 2015, 92, 012521. [Google Scholar] [CrossRef]
- Sunaga, A.; Prasannaa, V.S.; Abe, M.; Hada, M.; Das, B.P. Enhancement factors of parity- and time-reversal-violating effects for monofluorides. Phys. Rev. A 2018, 92, 040501(R). [Google Scholar] [CrossRef]
- Fazil, N.M.; Prasannaa, V.S.; Latha, K.V.P.; Abe, M.; Das, B.P. RaH as a potential candidate for electron electric-dipole-moment searches. Phys. Rev. A 2019, 99, 052502. [Google Scholar] [CrossRef]
- Kozyryev, I.; Hutzler, N.R. Precision measurement of time-reversal symmetry violation with laser-cooled polyatomic molecules. Phys. Rev. Lett. 2017, 119, 133002. [Google Scholar] [CrossRef] [PubMed]
- Augenbraun, B.L.; Lasner, Z.D.; Frenett, A.; Sawaoka, H.; Miller, C.; Steimle, T.C.; Doyle, J.M. Laser-cooled polyatomic molecules for improved electron electric dipole moment searches. N. J. Phys. 2020, 22, 022003. [Google Scholar] [CrossRef]
- Gaul, K.; Berger, R. Ab initio study of parity and time-reversal violation in laser-coolable triatomic molecules. arXiv 2018, arXiv:1811.05749. [Google Scholar] [CrossRef]
- Denis, M.; Haase, P.A.B.; Timmermans, R.G.E.; Eliav, E.; Hutzler, N.R.; Borschevsky, A. Enhancement factor for the electric dipole moment of the electron in the BaOH and YbOH molecules. Phys. Rev. A 2019, 99, 042512. [Google Scholar] [CrossRef]
- Prasannaa, V.S.; Shitara, N.; Sakurai, A.; Abe, M.; Das, B.P. Enhanced sensitivity of the electron electric dipole moment from YbOH: The role of theory. Phys. Rev. A 2019, 99, 062502. [Google Scholar] [CrossRef]
- Calvert, J.G.; Lindberg, S.E. Mechanisms of mercury removal by O3 and OH in the atmosphere. Atm. Environ. 2005, 39, 3355. [Google Scholar] [CrossRef]
- Goodsite, M.E.; Plane, J.M.C.; Skov, H. A theoretical study of the oxidation of HgO to HgBr2 in the troposphere. Environ. Sci. Technol. 2004, 38, 1772. [Google Scholar] [CrossRef]
- Ezarfi, N.; Benjelloun, A.T.; Sabor, S.; Benzakour, M.; Mcharfi, M. Theoretical investigations of structural, thermal properties and stability of the group 12 metal M(XH) isomers in atmosphere: M=(Zn, Cd, Hg) and XH=(OH, SH). Theory Chem. Acc. 2019, 138, 109. [Google Scholar] [CrossRef]
- Lindroth, E.; Lynn, B.W.; Sandars, P.G.H. Order α2 theory of the atomic electric dipole moment due to an electric dipole moment on the electron. J. Phys. B At. Mol. Opt. Phys. 1989, 22, 559. [Google Scholar] [CrossRef]
- Das, B.P. Aspects of Many-Body Effects in Molecules and Extended Systems; Mukherjee, D., Ed.; Springer: Berlin/Heidelberg, Germany, 1989; p. 411. [Google Scholar]
- Kozlov, M.G. New limit on the scalar P,T-odd electron-nucleus interaction. Phys. Lett. A 1988, 130, 426. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations: Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations: Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations: Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814. [Google Scholar] [CrossRef]
- Frisch, M.J. Gaussian 16; Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mozhayskiy, V.A.; Krylov, A.I. ezSpectrum. Available online: http://iopenshell.usc.edu/downloads (accessed on 14 October 2020).
- Yanai, T.; Nakano, H.; Nakajima, T.; Tsuneda, T.; Hirata, S.; Kawashima, Y.; Nakao, Y.; Kamiya, M.; Sekino, H.; Hirao, K. UTCHEM—A Program for ab initio Quantum Chemistry; Goos, G., Hartmanis, J., van Leeuwen, J., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2003; Volume 2660, p. 84. [Google Scholar]
- Yanai, T.; Nakajima, T.; Ishikawa, Y.; Hirao, K. A new computational scheme for the Dirac-Hartree-Fock method employing an efficient integral algorithm. J. Chem. Phys. 2001, 114, 6526–6538. [Google Scholar] [CrossRef]
- Visscher, L.; Lee, T.J.; Dyall, K.G. Formulation and implementation of a relativistic unrestricted coupled-cluster method including noniterative connected triples. J. Chem. Phys. 1996, 105, 8769. [Google Scholar] [CrossRef]
- Cizek, J. Correlation Effects in Atoms and Molecules; Advances in Chemical Physics; Lefebvre, W.C., Moser, C., Eds.; Interscience Publishers: New York, NY, USA, 1969. [Google Scholar]
- Zack, L.N.; Sun, M.; Bucchino, M.P.; Clouthier, D.J.; Ziurys, L.M. Gas-phase synthesis and structure of monomeric ZnOH: A model species for metalloenzymes and catalytic surfaces. J. Phys. Chem. A 2012, 116, 1542. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Andaloussi, M.B.D.; Nagashima, U.; Jensen, P. Electronic structure and rovibrational properties of ZnOH in the A′ electronic state: A computational molecular spectroscopy study. J. Chem. Phys. 2014, 141, 094308. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Lopez, A.; Acuna, U.; Trabelsi, T.; Carmona-Garcia, J.; Davalos, J.Z.; Rivero, D.; Cuevas, C.A.; Kinnison, D.E.; Sitkiewicz, S.P.; Roca-Sanjuan, D.; et al. Gas-phase photolysis of Hg(I) radical species: A new atmospheric mercury reduction process. J. Am. Chem. Soc. 2019, 141, 8698. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E.; Filatov, M. Bonding in mercury molecules described by the normalized elimination of the small component and coupled cluster theory. ChemPhysChem 2008, 9, 2510. [Google Scholar] [CrossRef] [PubMed]
- Saue, T. Relativistic hamiltonians for chemistry: A primer. ChemPhysChem 2011, 12, 3077. [Google Scholar] [CrossRef]
- Jensen, H.J.A.; Bast, R.; Saue, T.; Visscher, l. A Relativistic Ab Initio Electronic Structure Program. Available online: https://www.researchgate.net/publication/315699001DIRAC16DIRACarelativisticabinitioelectronicstructureprogramReleaseDIRAC162016 (accessed on 14 October 2020).
- Mitra, R.; Prasannaa, V.S.; Sahoo, B.K.; Tong, X.; Abe, M.; Das, B.P. Mercury hydroxide as a promising triatomic molecule to probe P,T-odd interactions. arXiv 2019, arXiv:1908.07360, unpublished. [Google Scholar]
- Dyall, K.G. Relativistic double-zeta, triple-zeta, and quadruple-zeta basis sets for the 5d elements Hf–Hg. Theor. Chem. Acc. 2004, 112, 403. [Google Scholar] [CrossRef]
- Dyall, K.G.; Gomes, A.S.P. Revised relativistic basis sets for the 5d elements Hf-Hg. Theory Chem. Acc. 2010, 125, 97. [Google Scholar] [CrossRef]
- Dyall, K.G. Relativistic double-zeta, triple-zeta, and quadruple-zeta basis sets for the light elements H-Ar. Theory Chem. Acc. 2016, 135, 128. [Google Scholar] [CrossRef]
- Wang, S.C. On the asymmetrical top in quantum mechanics. Phys. Rev. 1929, 34, 243. [Google Scholar] [CrossRef]
- Kivelson, D. A (K + 2)nd order formula for asymmetry doublets in rotational spectra. J. Chem. Phys. 1953, 21, 536. [Google Scholar] [CrossRef]
- Polo, S.R. Energy levels of slightly asymmetric top molecules. Can. J. Phys. 1957, 35, 8. [Google Scholar] [CrossRef]
- Khriplovich, I.B.; Lamoreaux, S.K. CP Violation Without Strangeness; Springer: Berlin/Heidelberg, Germany, 1997. [Google Scholar]
- Sawyer, B.C.; Lev, B.L.; Hudson, E.R.; Stuhl, B.K.; Lara, M.; Bohn, J.L.; Ye, J. Magnetoelectrostatic trapping of ground state OH molecules. Phys. Rev. Lett. 2007, 98, 253002. [Google Scholar] [CrossRef] [PubMed]
- Hutzler, N.R.; Lu, H.; Doyle, J.M. The buffer gas beam: An intense, cold, and slow source for atoms and molecules. Chem. Rev. 2012, 112, 4803. [Google Scholar] [CrossRef]
- Wu, X.; Han, Z.; Chow, J.; Ang, D.G.; Meisenhelder, C.; Panda, C.D.; West, E.P.; Gabrielse, G.; Doyle, J.M.; DeMille, D. The metastable Q3Δ2 state of ThO: A new resource for the ACME electron EDM search. New J. Phys. 2020, 22, 023013. [Google Scholar] [CrossRef]
- Grasdijk, O.; Timgren, O.; Kastelic, J.; Wright, T.; Lamoreaux, S.; DeMille, D.; Wenz, K.; Aitken, M.; Zelevinsky, T.; Winick, T.; et al. CeNTREX: A new search for time-reversal symmetry violation in the 205Tl nucleus. arXiv 2020, arXiv:2010.01451v1. [Google Scholar]
- Jadbabaie, A.; Pilgram, N.H.; Klos, J.; Kotochigova, S.; Hutzler, N.R. Enhanced molecular yield from a cryogenic buffer gas beam source via excited state chemistry. New J. Phys. 2020, 22, 022002. [Google Scholar] [CrossRef]
- Verma, M.; Jayich, A.M.; Vutha, A.C. Electron electric dipole moment searches using clock transitions in ultracold molecules. Phys. Rev. Lett. 2020, 125, 153201. [Google Scholar] [CrossRef]
- Filatov, M.; Cremer, D. Relativistically corrected hyperfine structure constants calculated with the regular approximation applied to correlation corrected ab initio theory. J. Chem. Phys. 2004, 121, 5618. [Google Scholar] [CrossRef]
- Filatov, M.; Zou, W.; Cremer, D. Calculation of response properties with the normalized elimination of the small component method. Int. J. Quant. Chem. 2014, 114, 993. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| State | Reference | |||
|---|---|---|---|---|
| Ground | 2.091 | 0.966 | 104.1 | Ref. [50] |
| 2.25 | 0.99 | 106.8 | Ref. [32] | |
| 2.181 | - | - | Ref. [51] | |
| 2.2079 | 0.9691 | 103.6 | Ref. [33] | |
| 2.2294 | 0.9633 | 104.83 | This work | |
| First-excited | 3.1458 | 0.9563 | 180 | This work |
| Second-excited | 2.0766 | 0.9615 | 102.5 | This work |
| Third-excited | 3.5482 | 1.0095 | 81.93 | This work |
| Geometry | ||||
|---|---|---|---|---|
| DHF | LECC | DHF | LECC | |
| From this work | ||||
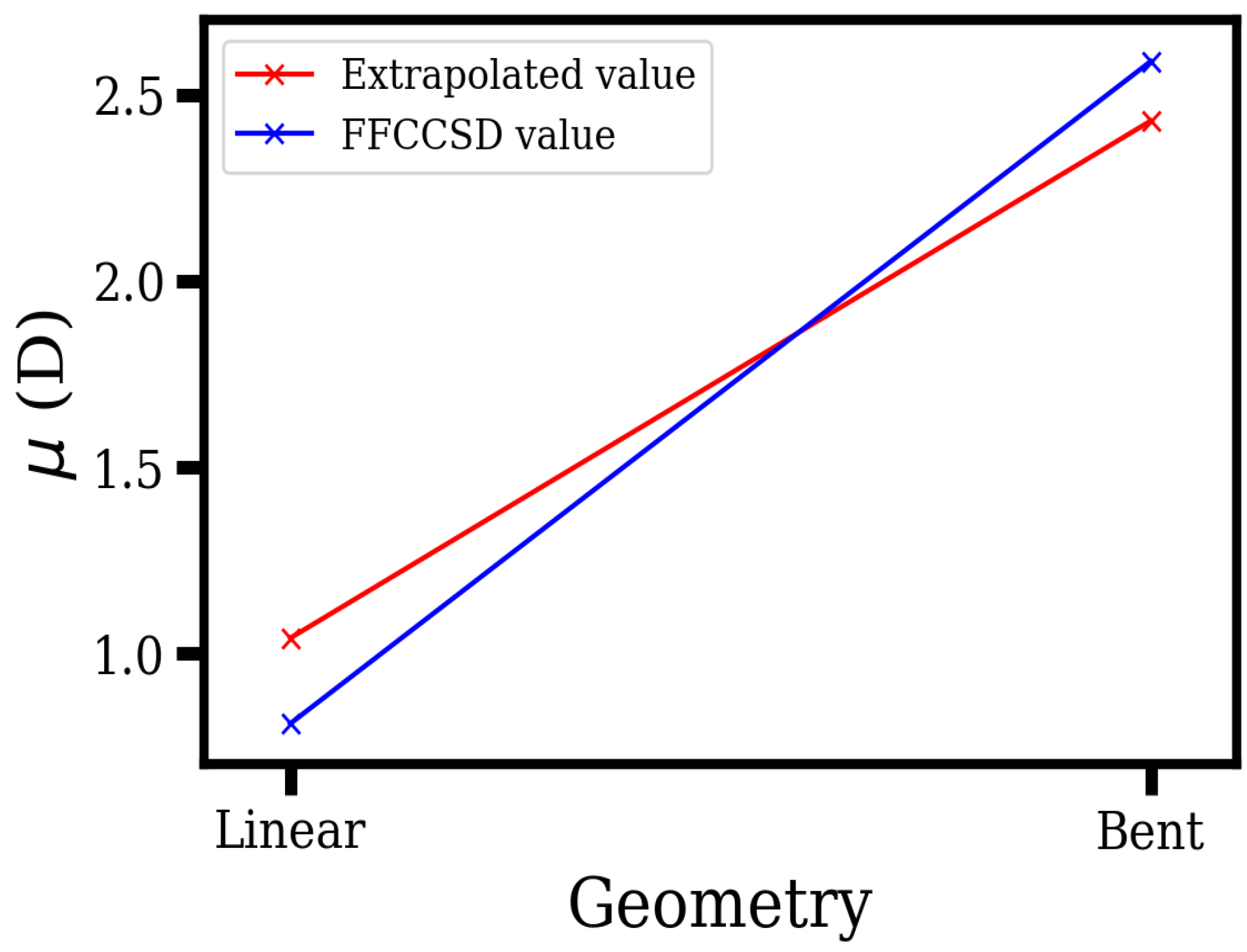
| Linear | 107.24 | 109.02 | 1.57 | 1.04 |
| Bent | 28.01 | 28.47 | 3.67 | 2.43 |
| From other works | ||||
| 1.89 [32] | ||||
| 1.92 [51] | ||||
| 1.96 [33] | ||||
| Term | (GV/cm) | (D) |
|---|---|---|
| O (DHF) | 107.24 | 1.57 |
| h.c. | 9.50 | −0.42 |
| −2.76 | −0.15 | |
| h.c. | −0.38 | 0.12 |
| −4.58 | −0.11 |
| Atom | AOs | Linear | Bent |
|---|---|---|---|
| Hg | 378.40 | 100.11 | |
| −270.26 | −71.81 | ||
| −31.40 | −8.07 | ||
| 30.19 | 7.77 | ||
| 0.79 | 0.19 | ||
| −0.78 | −0.18 | ||
| O | 2.78 | 1.44 | |
| −2.77 | −1.44 |
| Molecule | Reference(s) | ||
|---|---|---|---|
| ThO | 79.9 [12] | Ref. [14] | |
| HfF | 22.5 [13] | Ref. [15] | |
| YbF | 23.1 [10] | Ref. [16] | |
| HgOH | 28.47 | This work | |
| HgCH | 75.07 | This work | |
| HgCF | 60.95 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitra, R.; Prasannaa, V.S.; Sahoo, B.K.; Hutzler, N.R.; Abe, M.; Das, B.P. Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches. Atoms 2021, 9, 7. https://doi.org/10.3390/atoms9010007
Mitra R, Prasannaa VS, Sahoo BK, Hutzler NR, Abe M, Das BP. Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches. Atoms. 2021; 9(1):7. https://doi.org/10.3390/atoms9010007
Chicago/Turabian StyleMitra, Ramanuj, V. Srinivasa Prasannaa, Bijaya K. Sahoo, Nicholas R. Hutzler, Minori Abe, and Bhanu Pratap Das. 2021. "Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches" Atoms 9, no. 1: 7. https://doi.org/10.3390/atoms9010007
APA StyleMitra, R., Prasannaa, V. S., Sahoo, B. K., Hutzler, N. R., Abe, M., & Das, B. P. (2021). Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches. Atoms, 9(1), 7. https://doi.org/10.3390/atoms9010007