Abstract
In this study, we analyzed the light absorption by diatomic molecules or colliding atoms in a spectral region dominated by an avoided crossing of adiabatic state levels or crossing of the corresponding diabatic state levels. Our attention was focused on the low-resolution spectrum at a higher gas temperature under local thermodynamic equilibrium conditions. The absorption measurements of mixed vapors of potassium (≈80%) and cesium (≈20%) were made in the temperature range of 542–715 K and the infrared spectral range 900–1250 nm. In this area, the main spectral contributions were the broad bands of K2, Cs2, and KCs molecules. There was a crossing of and state potential curves and the coupling of this state was due to the matrix element of the spin–orbit interaction. Using data for relevant electronic potential curves and transition dipole moments existing in the literature, the spectra of the molecular bands of K2, Cs2, and KCs molecules were calculated. Full quantum mechanical and semi-quantum coupled channel calculations were done and compared with their non-coherent adiabatic or diabatic approximations. Through the comparison of our theoretical and experimental spectra, we identified all observed spectral features and determined the atoms’ number density and gas temperature.
1. Introduction
Several approaches to the theoretical simulation of low-resolution, high-temperature optical spectra of diatomic molecules under local thermodynamic equilibrium conditions have been analyzed in detail by Beuc et al. [1]. Using relevant data for molecular potential curves and transition dipole moments, theoretical simulations can be powerful tools for identifying spectral features, gas temperatures, and atom number densities. Whereas in Beuc et al. [1], the optical transition between two well-isolated adiabatic states is analyzed, in this paper, the photon absorption from the lower adiabatic state into two excited non-radiative coupled electronic states is studied.
Many years ago, Devdariani et al. [2,3,4] and O’Callaghan et al. [5] studied this problem within semiclassical theory using a Landau-Zener approximation [6,7]. Although their analysis focused on the absorption and emission of light by colliding atoms, their conclusions can be qualitatively applied when interpreting quantum mechanical calculations of bound–bound molecular transitions.
The focus of this study was the spectral phenomenon that exists for all alkali metal homo- and hetero-nuclear molecular spectra. On the red side of the first resonant atomic transition doublet, the spectrum is dominated by molecular bands (shorter bands). The brackets in the sub-script contain the gerade or ungerade symmetry mark, which is valid only for homo-nuclear molecules. The potential curve of the singlet state crosses with the potential curve of one component of the triplet electronic state . These electronic states are coupled by the matrix element of the spin-orbit (SO) interaction, which has as a consequence the perturbation of the spectrum.
For several decades, low-resolution spectra of the band at high temperatures have been widely experimentally and theoretically investigated, for example: Li2 [8,9,10,11], Na2 [1,8,11,12], K2 [11,12,13,14,15], Rb2 [16], and Cs2 [1,5,17,18]. The influence of coupling is directly observed only in the case of the heavy alkali dimers Rb2 and Cs2 due to their large SO interaction.
Many more studies of mixing in alkali dimer spectra have been done using high-resolution spectroscopic techniques, such as laser-induced fluorescence Fourier transform spectroscopy and optical–optical double resonance polarization spectroscopy. By using deperturbation analysis, accurate spectroscopic data have reproduced the potential curves of electronic states and coupling matrix element in the case of homonuclear dimers K2 [19,20,21], Rb2 [22,23], and Cs2 [24,25], and heteronuclear dimers LiCs [26,27], NaCs [28], RbCs [29,30], and KCs [31,32,33]. In these theoretical analyses of spectroscopic data, the rotational couplings of state components whose angular moments differ by 1 are included. The theoretical analysis in this study was focused on the low-resolution spectrum; therefore, this type of coupling can be neglected.
To our knowledge, there is no study of the high-temperature low-resolution spectra of heteronuclear dimers in the literature. We measured the absorption spectrum of a potassium and cesium vapor mixture at temperatures of 542–715 K. In the infrared spectra from 900–1250 nm, the dominant contributions were the bands of K2, Cs2, and KCs dimers. Using existing relevant potential curves and transition dipole moments obtained by quantum chemical calculations and the analysis of spectroscopic data, we calculated theoretical spectra in the measured wavelength range. We compared coupled channel quantum mechanical spectra calculations with coupled channel semi-quantum calculations and showed that a time-efficient semi-quantum approach yielded results comparable to a full quantum approach. Using semi-classical theory [2,3,4,5], we defined the conditions under which coupled channel calculations can be approximated by non-coherent adiabatic or diabatic approximations.
By comparing the experimental absorption coefficients with the theoretical simulations, we determined the temperature and potassium and cesium number densities in the gas mixture. The numerically time-efficient semi-quantum coupled channel calculus gave satisfactory good low-resolution spectra for K2, Cs2, and KCs dimers and can be used as an efficient alkali gas diagnostic tool.
2. Theoretical Background and Methods
The total Hamiltonian of a diatomic molecule is , wherein is the nuclei kinetic energy operator and µ is the molecular reduced mass. is the electronic part of the Hamiltonian containing the electron kinetic energy operator, all electrostatic interactions in a molecule, and the spin–orbit interaction, all of which depend on the interatomic distance R and positions vectors of all electrons r. The eigenvector of the Hamiltonian is the function:
which is represented as a sum of the product of the wavefunctions that describe electrons and the functions that describe the motion of the atoms. By including the wavefunctions into the Schrödinger equation, one obtains a series of coupled equations:
where , . In a spherical coordinate system, Xij can be written as , where depends on electronic wavefunction changes related to the interatomic distance and depends on changes related to the rotation of the molecule (for details, see Janev et al. [34]). The functions belong to a complete set of orthogonal normalized functions that can be chosen in various ways.
The adiabatic representation is a uniquely defined base of wavefunctions that diagonalize the electronic Hamiltonian . In the adiabatic representation, Equation (2) has the form:
Potential curves for adiabatic states of the same symmetry cannot intersect (Wigner-Neuman theorem) and they avoid crossing. In the region of avoided crossing, adiabatic states of the same symmetry are coupled by the matrix element (radial coupling). Note that the adiabatic or diabatic states whose electronic angular momentum differ by 1 are coupled by the matrix element (angular coupling). In the region where one can neglect all matrix elements (Born-Oppenheimer adiabatic approximation BOA), Equation (2) has the simple form:
and the molecular wave function has the simple form . Please note, in the case of molecules with large reduced masses, which is true for the dimers analyzed in this paper, the matrix elements are negligible.
The diabatic representation according to Lichten [35] is the base of electronic wavefunctions for which the matrix elements are negligible () and the Equation (2) has the simple form , where . The diabatic base for which is called the radial diabatic base. Smith [36] has shown that the adiabatic and the radial diabatic base are connected through a unitary transformation. Unlike the uniquely defined adiabatic base, there is a class of radial diabatic bases that can be obtained using a unitary transformation (R independent) of one of them. In this study, we chose a diabatic base that diagonalized the electronic Hamiltonian at small interatomic distances.
The probability of an optical transition between the lower energy molecular state and higher energy state is proportional to the square of the molecular dipole moment matrix element:
where is the electronic dipole moment for the transition between the adiabatic or diabatic states i and j.
2.1. One Excited Electronic State
If one assumes that the conditions for the Born-Oppenheimer approximation are satisfied, the wave function of the molecular state is given by one product of the nuclear and electronic wave function . Here, the electronic state is labeled with (Λ usually refers to the axial component of the electronic angular momentum). The motion of the atoms is characterized by vibrational and rotational quantum numbers. After the separation of the variables, the rovibrational wavefunction can be expressed as a product of the rotational and vibrational wavefunctions . The Schrödinger equation for the vibrational wavefunction is:
where is the electronic potential of the electronic state . The same equation gives the energies and the energy-normalized wavefunctions of the free states.
The spectra of the averaged absorption from the lower to the upper electronic state comprise contributions from the transitions between all rovibrational states of a lower and the upper electronic state.
The absorption cross-section for the transition from a rovibrational state to the rovibrational state is [8,9]:
where is the electronic transition dipole moment, is the line-shape function, is the transition energy, and is the Hönl–London factor. At thermodynamic equilibrium, the reduced absorption coefficient is [8,9,12]:
is a statistical factor that depends on the symmetry of the electronic states and the temperature:
where is the spin and is the angular momentum of atom A, B. is a statistical factor that depends on the atomic nuclear spin and parity of the molecular angular momentum with the averaged value for the homonuclear molecules and for heteronuclear molecules.
In each electronic state , there is a finite number of bound and quasi-bound states with unity-normalized wavefunctions and an infinite continuum of free rovibrational states with energy-normalized wavefunctions . Therefore, the sum over the rovibrational transitions in Equation (8) formally includes the integration over the bound-free, free-bound, and bound-bound transitions.
To calculate the energies and wavefunctions of rovibrational states, the FGH method (Fourier grid Hamiltonian) is used, where functions are represented on a finite number of grid points Ri (i = 1, …, N) [37]. On the grid of uniformly spaced points, where for the electronic state S with an electronic angular moment and electronic potential , the N × N Hamiltonian matrix is defined as:
Energies and radial wavefunctions can be determined using the diagonalization of the Hamiltonian matrix . Eigenvalues of the matrix represent the energies of rovibrational states and eigenvectors of the Hamiltonian matrix containing the corresponding unity-normalized wavefunction on the grid points. This method, in addition to the bound-state energies, gives a discrete set of free-state continuum energies, but in the range spanned by the grid, the corresponding unity-normalized wavefunctions do represent the states of a continuum.
The quantum calculation (QC) of spectra using Equation (8) are time-consuming and therefore not suitable for fast diagnoses (see the discussion in Beuc et al. [1]). If one needs to analyze low-resolution spectra using the Q-branch approximation (only transitions) by replacing the summation over with the summation over the intervals containing n neighbor values, the efficient form of the reduced absorption coefficient is [1]:
The number of intervals is the nearest integer of , is the larger value of and , and . In this study, we use the absorption coefficient in Equation (11) in the wavelength domain as a QC result:
In the above equation, the line profile is approximated with a Heaviside pi (or boxcar) function , where the optical transition wavelength is and is equal to or larger than the line profile half-width and smaller than the instrumental profile half-width.
Semi-quantum approximation (SQA) [1,15] give an even faster algorithm for calculating the low-resolution reduced absorption coefficient, which in the wavelength domain, has a form:
where is equal to the larger value of and . This expression was formally obtained using a completely semi-classical procedure but was in a quantum-like form. An evaluation of the applicability, accuracy, and numerical efficiency of Equations (8), (11), (12), and (13) was extensively discussed in Beuc et al. [1].
One can describe the radial movement of atoms using a classical trajectory with the radial velocity , where is the energy of the molecule and is the collision impact parameter. The wavefunction of the molecule in the electronic state “dressed” [38] with photon frequency is and the wavefunction of the molecule in the excited state is . The absorption cross-section of the optical transition between the lower () and the upper () molecular state is:
At thermodynamic equilibrium, the reduced absorption coefficient is given by the averaged absorption cross-section over parameters of the statistical ensemble and :
where is the electronic transition difference potential.
The non-coherent quasi-static approximation is done using the first-order stationary phase approximation of the time-dependent integral in Equation (15), partial integration over , and neglecting the rapidly oscillating terms [1,39]:
where , , and the summation is over all real Condon points satisfying . This approximation gives a good description of the spectra but diverges at the difference potential extremes, which are most often the consequence of the avoided crossing of the adiabatic electronic states’ potential curves.
In the case where the difference potential has only one extreme point , a coherent uniform Airy approximation of the spectral profile is defined [1,39]. In the classically allowed region, the reduced absorption coefficient has the form:
where and are the Condon points satisfying , and the mapping parameter is . The functions and are integrals of the square of the Airy function and its first derivative, respectively. The uniform Airy approximation can be extended to the case where the transition difference potential has several extremes [1].
2.2. Two Coupled Excited Electronic States
In this paragraph, an optical transition from the ground electronic state satisfying BOA to two coupled electronic states and is analyzed. It is assumed that the electronic wavefunction of the excited states belonging to the radial diabatic base diagonalizes the electronic Hamiltonian at small interatomic distances. The electronic wavefunction and potential for the ground state are and for the excited states and , respectively. The excited electronic diabatic states are coupled via the matrix element and all other coupling matrix elements and are neglected. The difference potentials for the transitions between the ground and excited states are and the corresponding transition dipole moments are . The potential curves and difference potential curves intersect at an interatomic distance , , and .
The corresponding adiabatic states’ wavefunctions and potentials are given by diagonalizing the matrix :
where . There is a simple relationship between the diabatic state potential difference and the adiabatic state potential difference .
Difference potentials and the corresponding transition dipole moments for the transition between the ground state and excited adiabatic states are:
In the neighborhood of the diabatic state potential curves’ crossing point, the potential curves and difference potential curves have an avoided crossing, and the transition dipole moments significantly depend on the interatomic distance. The electronic adiabatic states and are coupled via a radial matrix element, and their couplings with all other electronic states were assumed to be negligible.
The molecular wavefunction of the excited state can be written as a sum of coupled states functions in a diabatic (d) or adiabatic (a) representation:
The energies and rovibrational radial wavefunctions in both representations are the solutions of the coupled channel Schrödinger equation:
It is assumed here that both electronic states have the same electronic angular moment . The coupling matrix element in the diabatic representation is , and in the adiabatic representation, it is . To solve the coupled channel Equation (21) numerically, it is suitable to use the diabatic representation and the FGH method [40]. Energies and radial wavefunctions can be determined via diagonalization of the 2N × 2N Hamiltonian matrix:
where is an N × N matrix defined by Equation (10) and is an N × N matrix.
The absorption cross-section for the transition from a rovibrational state of the ground electronic state to the rovibrational states of the coupled excited electronic states is:
where the transition energy is . The reduced absorption coefficient for the transition from the lower electronic state to the excited coupled electronic states in the adiabatic or diabatic representation can be written as:
This is a full quantum mechanical coupled channel approach (QCC) in the adiabatic or diabatic representation. The first contribution represents an optical transition to the electronic state , the second contribution represents the transition to the electronic state , and third contribution represents their interference. The calculations of , , and can be done using Equations (8) or (12) by replacing with , , and , respectively.
The same Equation (24) holds for the semi-quantum coupled channel approximation (SQCC), where in Equation (13), the transition energy is and is replaced by , , and for each contribution, respectively.
If one neglects the coupling element , the coupled Equation (21) is simplified into two ordinary Schrödinger equations:
Because , the interference contribution similar to the one in Equation (23) is negligible and the absorption coefficient can be written as the non-coherent sum of transitions to the excited states and :
where each contribution in Equation (26) can be calculated using Equations (8) or (12) by substituting and replacing with . If the adiabatic (diabatic) representation is used in Equation (26), one gets a non-coherent quantum-mechanical adiabatic (diabatic) approximation QAA (QDA) of QCC.
Equation (26) also describes an approximation of SQCC, where each contribution is calculated using Equation (13) by substituting and replacing with . Depending on the choice of the electronic wavefunctions’ basis, one obtains a non-coherent semi quantum adiabatic SQAA or diabatic SQDA approximation
2.3. The Landau–Zener Model
To analyze the essential properties of the absorption spectrum for transitions in the neighborhood of the crossing of diabatic states’ potential curves, simplifications inherent to the Landau–Zener model have been made. In the neighborhood of the crossing point , the potentials of the excited diabatic states can be approximated: (note that ), and for the potential of the ground electronic state, , where ,, and . The transition difference potentials are where and . Additionally, the following approximations can be used: for the coupling matrix element, , and for the transition dipole moments,.
Using the same approximation, the difference potentials for the transition between the ground and the excited adiabatic states and the corresponding transition dipole moments are:
The difference potentials curves and are two branches of a hyperbola. If , the potential curves are monotonic with only one Condon point at each frequency. When , the difference potential curves and have a minimum and a maximum, respectively, and both adiabatic states have two Condon points at each frequency.
To calculate the spectral profile in the neighborhood of the diabatic potential curves’ crossing point, a satisfactory approximation is , where is the radial velocity at the crossing point. Using Equation (16), a non-coherent sum of the absorption coefficients for transitions to excited diabatic states is:
This is the non-coherent semi-classical diabatic approximation (SCDA) of the spectra, which is valid in both cases and .
In the case , a non-coherent sum of the semi-classical absorption coefficient for the transition to the excited adiabatic states’ semi-classical adiabatic approximation (SCAA) has the form .
SCAA in the case can be obtained using the uniform Airy approximation (Equation (16)) [41]:
The absorption coefficient is a function of the dimensionless reduced frequency (energy) suggested in Девдариани and Cебякин [2]. The crossing of diabatic states difference potentials is at and the maximum and minimum of the adiabatic state difference potentials are at . The profile is a symmetric function of , where , parameter , and is the Massey parameter of nonadiabatic mixing. The interference profile is an anti-symmetric function of , with and as a consequence. The following are asymptotically valid: and . In the neighborhood of the potential minimum , parameter z has a simple form such that in the classically forbidden region , functions and can be approximated using and by analytical continuation.
The semi-classical time-dependent molecular wavefunction in the case of two non-radiative coupled diabatic electronic states is:
Devdariani and coworkers [2,3,4] and O’Callaghan et al. [5] have studied the influence of non-adiabatic electronic states mixing on the shape of the spectrum within semi-classical atomic collision theory. The atomic collision can be divided into two half-collisions: the first refers to the motion from to the turning point , and the second from the turning point to . Within the framework of the Landau–Zener model, each half-collision takes place over a time interval , where is at the crossing point . The coefficients are the solutions of the coupled equations:
There are two scenarios involving a collision process: the initial conditions of the first half-collision are or .
The cross-section for the transition between the lower molecular state dressed with photon to the higher state for each half-collision is:
Both groups of authors have calculated the spectral contribution for each half-collision and both scenarios of the initial conditions. After non-coherent summation of all contributions and averaging over the statistical ensemble at thermodynamic equilibrium, they obtained the relation for the absorption coefficient:
In O’Callaghan et al. [5], all calculations of spectral profiles and were made numerically, but in Devdariani and coworkers [2,3,4], the authors found analytical solutions based on the parabolic cylinder function .
In the case , both groups of authors obtained the same result for spectral profiles and . As a consequence, the reduced absorption coefficient obtained using the semi-classical coupled channel approach SCCC is equal to that obtained using SCAA and SCDA, namely . This conclusion can be generalized to the spectrum in the neighborhood of the diabatic curves’ crossing point if this point is isolated from other crossing points and the condition is satisfied.
For , both groups of authors obtained a similar behavior of the profile functions and . The functions and have the same symmetry property concerning the sign of the frequency and the asymptotic behavior and , as well as the corresponding functions in the adiabatic representation. In Devdariani and coworkers [2,3,4], the functions and were calculated analytically as follows:
The function refers to a temperature-averaged probability of a molecule moving in a potential of the initial diabatic electron state. Similarly, the probability of the molecule moving in the initial adiabatic state potential is .
Figure 1a,c shows the spectral profiles and , respectively, as functions of the frequency and parameter . Due to the symmetry properties, the spectral profiles are presented for positive frequencies only. The maximum value of the profiles is near , corresponding to the position of the minimum of the adiabatic state difference potential (satellite rainbow). It can be seen that at low values of parameter , both profiles tend toward the values of SCDA and .
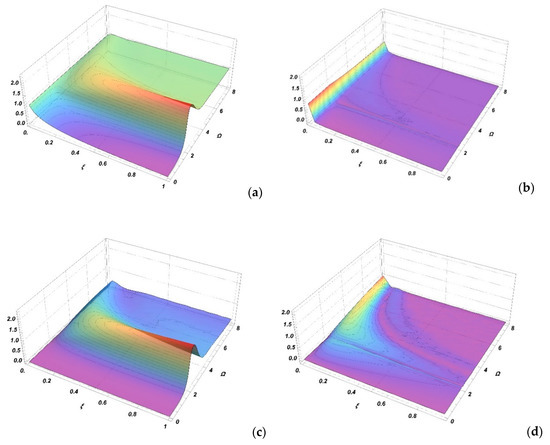
Figure 1.
Absorption spectra contributions and as a function of the Massey parameter and reduced frequency are shown in (a,c). The differences between these contributions and their adiabatic approximations and are shown in (b,d).
The differences of the spectral profiles in SCAA and SCCC and are shown in Figure 1b,d, respectively. It is seen that the differences between these two approaches are small or negligible for . The differences are slightly larger in the case of profiles but this is less important since it is very common that a scalar product is small or equal to zero and the same holds for the second contribution in Equation (33). From Figure 1 it can be concluded that SCCC can be approximated for using SCDA and for using SCAA.
We can generalize the conclusion to any case of diabatic electronic states’ potential crossing, as long as this crossing is isolated from other crossings and the condition is satisfied, and the Massey parameter is:
The criteria of applicability for SCAA and SCDA obtained in semi-classical theory can be applied to both quantum mechanical and semi-quantum approaches.
3. Results
3.1. Experiment
A T-type all-sapphire cell (ASC) of 4 cm in length and 1 cm in inner diameter was used for the transmission measurements in the K-Cs mixture vapor. The mixture in the sealed cell was 80% potassium and 20% cesium. The number density of atoms and molecules in the cell depends on the temperature at the tip of the side-arm finger (Tf) and the cell body (Tc). Tc was higher than Tf (at least 30 K) to prevent the condensation of the vapors on the inner side of the sapphire cell. The temperatures were controlled using two Chromel-Alumel Thermocouples.
In Figure 2a, we present the transmission intensities for several cells’ Tc and finger Tf temperatures in the infrared spectral region between 900 nm and 1250 nm.
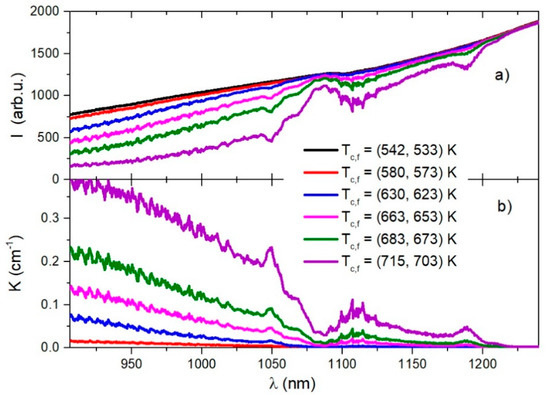
Figure 2.
Relative intensity (in arbitrary units) of the light transmitted through a sapphire absorption cell at several temperatures is shown in (a). The alkali mixture absorption coefficients for the corresponding temperatures are shown in (b).
Assuming a low light intensity and a uniform medium inside the absorption cell of length L, using Beer-Lambert’s Law , one can determine the linear absorption coefficient , where is the intensity at the entrance of the cell. Following the measurements shown in Figure 2a, we approximated . The absorption coefficients for several temperatures are shown in Figure 2b.
Assuming the local thermodynamic equilibrium at an effective temperature T, the linear absorption coefficient of the mixture of potassium and cesium vapor for a low-pressure binary approximation was:
where , , and are the reduced absorption coefficients of K2, Cs2, and KCs molecules, respectively, and and are the number densities of K and Cs atoms, respectively. To analyze the spectra shown in Figure 2a,b, it was necessary to determine the reduced absorption coefficients of the K2, Cs2, and KCs molecules in the near-infrared part of the spectrum.
3.2. Near-Infrared Spectra of K2, KCs, and Cs2 Molecules
The dominant contribution to the near-infrared spectrum of alkali dimers is the band. This molecular band is the result of optical transitions between all rovibrational states of the excited ( state) and the ground electronic states (X state). The potential curve of the state crosses with the potential curve of the triplet electronic states . One component of the triplet state (b state) is coupled with the state via the matrix element of the SO interaction . The electronic transition dipole moment for the transition is and the transition is dipole-forbidden:. The consequence of the coupling is the perturbation of the spectrum. In the case of heteronuclear KCs dimer, there is an allowed transition but its contribution in the spectral region of interest is negligible and will not be analyzed in this study.
Most quantum-chemical ab-initio calculations are usually non-relativistic calculations in which the electronic Hamiltonian does not include SO interactions. As a result of these calculations, the electronic wavefunctions in Hund‘s case (a) are the representation and the corresponding electronic potentials, which are , and and , respectively. The SO interaction removes the triplet state component’s degeneration [42] such that the diabatic state potential is . The coupling matrix element is . Functions and have the same asymptotic behavior, namely , where is the first doublet fine structure splitting (potassium 57.7103 cm−1, cesium 554.0388 cm−1). In References [20,21,22,23,24,25,26,27,28,29,30,31,32,33], the functions and are fit to the Morse potential form and differ slightly at small interatomic distances. For the sake of simplicity, in this study, we used the approximation .
Using the diabatic states’ wavefunctions and potentials, one can construct the corresponding adiabatic ( and ) state potentials and wavefunctions by diagonalizing matrix :
where . Because the transition is dipole-forbidden, the adiabatic transition dipole moment and transitions have the simple form:
To obtain the electronic potential curves and transition dipole moments required in our calculations, we used existing theoretical and experimental data from the literature. In the case of the K2 molecule, to construct potential curves, we combined ab-initio results [43], experimental data [19,20,21], and long-range region analytical results [44]. The spin–orbit function is taken from Manaa et al. [21] and the transition dipole moment was from Yan and Meyer [43]. For the KCs molecule, all data were taken from the Supplementary Materials of Borsalino et al. [33]. In the case of the Cs2 molecule, for the diabatic state potentials, we used ab-initio results [45,46] (for states with 6S + 6P asymptote energies was shifted by +14 cm−1) and experimental data [24,25]. In the long-range region, the potential curves were smoothly matched with analytical curves [44]. The function was taken from Bai et al. [25] and the transition dipole moment was from Allouche and Aubert-Frécon [47].
All relevant potential curves for K2, KCs, and Cs2 molecules are shown in Figure 3. It can be seen that the SO splitting function in the K2 molecule was approximately ten times smaller than in the case of the KCs and Cs2 molecules. The diabatic states’ potential curves and had two well-separated crossing points for each molecule: K2 (9.0 Bohr, 46.3 Bohr), KCs (9.6 Bohr, 20.9 Bohr), and Cs2 (10.9 Bohr, 24.1 Bohr). In the same region, the potential curves of the adiabatic states avoided crossing.
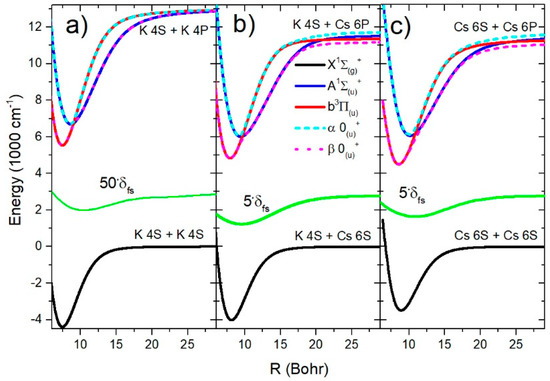
Figure 3.
The electronic states’ potentials curves relevant for the analysis of the infrared spectra of K2, KCs, and Cs2 molecules are shown in (a–c), respectively. The green curve shows the spin–orbit splitting function responsible for the coupling of the A and b states.
At the long-range crossing point, , where and are the difference potentials of the and transitions, respectively. According to the discussion in Section 2.3, in the neighborhood of the diabatic state potentials’ crossing point, the adiabatic state difference potentials were monotonic functions and the absorption spectra could be found using a non-coherent adiabatic approximation.
Figure 4a–c shows the short-range region difference potentials of the , , , and transitions for the K2, KCs, and Cs2 molecules, respectively. At the short-range crossing point, and the difference potentials of the and transitions had extrema in the neighborhood of the crossing point. As pointed out in Section 2.1 and Equation (16), the extremes of the difference potentials indicate the positions of the satellite rainbow in the spectrum. The difference potential of had one minimum each at 985 nm, 1072 nm, and 1126 nm for the K2, KCs, and Cs2 molecules, respectively. The transition difference potential had maxima and minima at (996 nm, 1055 nm), (1107 nm, 1192 nm), and (1197 nm, 1205 nm) for K2, KCs, and Cs2, respectively. The minima of the transition difference potential approximately coincided with the minima of transition difference potential. Figure 4d–f shows the relevant transitions dipole moments. In the neighborhood of the diabatic potentials’ crossing point, the dipole moments of the and transitions changed significantly with the interatomic distance, especially in the case of the K2 dimer.
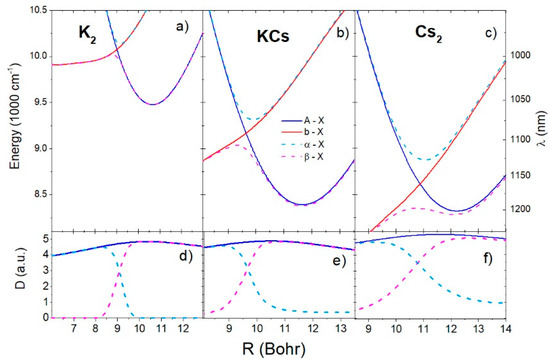
Figure 4.
Difference potential curves of the A-X, b-X, α-X, and β-X transitions for K2, KCs, and Cs2 molecules are shown in (a–c), respectively. The corresponding transition dipole moments are shown in (d–f). The labels on the left side of the upper panel denote energy, and the right-side denotes the wavelength of the transition.
At the short-range crossing point , the Massey parameter at a typical experimental temperature 700 K is given in Table 1 for all dimers. The coupling for the K2 dimer was much smaller than for Cs2 even for the KCs dimer. In Table 1, the probability of atomic motion in diabatic potential and the probability of atomic motion in adiabatic potential is given. It is evident that at the temperature of 700 K, in the K2 molecule, the atoms mainly moved in a diabatic potential, and for the KCs and especially Cs2 molecules, movement was in an adiabatic potential.

Table 1.
The first row gives the crossing points of the potential curves of electronic states A and b for K2, KCs, and Cs2 molecules. In the second row, the Massey parameters at the crossing points for the temperature T = 700 K are given. In the third and fourth rows, the averaged probabilities P of atoms moving in diabatic potentials and the probabilities for atoms moving in adiabatic electronic potentials are given, respectively.
The behavior of the rotational constants also indicated the influence of the mixing of the two electronic states. The energy-dependent constants for ,,, and , as well as the coupled and states for all dimers, are shown in Figure 5a–c. For the K2 molecule, the coupled states constants were grouped around the diabatic states constants, but in the case of KCs and especially Cs2 molecules, they were grouped around the adiabatic states constants. These facts are consistent with the Massey parameter shown in Table 1.
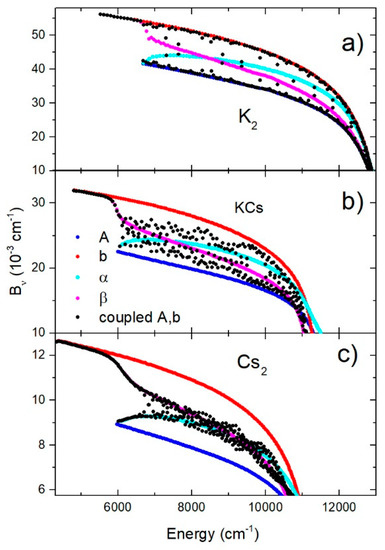
Figure 5.
Constants of A, b, , and , as well as the coupled A and b electronic states, in the case of K2, KCs, and Cs2 molecules are shown in (a–c), respectively.
Using the full quantum (semi-quantum) approach, we calculated the reduced absorption coefficients for transitions between the X state and the coupled A and b states QCC (SQCC), non-coherent adiabatic approximation QAA (SQAA), and non-coherent diabatic approximation QDA (SQDA) of this transition. The rovibrational energies and radial wavefunctions were calculated using the FGH method via diagonalization of Hamiltonian matrices (Equations (10) and (22)). The number of grid points was N = 800 in the quantum mechanical calculation and N = 2000 in the semi-quantum calculation.
Using Equations (12) and (13), the rovibrational transition contributions were collected in bins of 0.4 nm and the spectra were smoothed using a Gaussian with a half-width of 0.9 nm for the quantum approach and 3.0 nm for the semi-quantum approach. Summation over the rotational quantum number J in the quantum approach was replaced by summation over the groups of n = 3 neighbor J values.
The numerical evaluation in this study was done using the Wolfram Mathematica 12.1 computing system. To calculate all the rovibrational contributions needed for the SQAA and SQDA spectra, 27 s of computer time was required, and for SQCC, 48 s was required. Using these data to calculate the absorption coefficient (Equation (13)) at a given temperature, 1 s of computer time was required for SQAA and SQCC, and 0.5 s was required for the SQDA spectra. For the full quantum mechanical approach, 340 s of computer time was required to calculate all the rovibrational contributions of the QAA and QDA spectra, and 830 s was required to calculate the QCC spectra. For the absorption coefficient evaluation at a given temperature, 70 s was required for the QCC and QAA spectra, and 38 s was required for the QDA spectra.
The reduced absorption coefficients at 700 K obtained using different approaches are shown in Figure 6. Each column in Figure 6 shows the different theoretical approaches for each of the dimers (K2, KCs, Cs2). The first row of Figure 6 shows the QCC and SQCC spectra, the second row shows the QAA and SQAA spectra, while the third row shows the QDA and SQDA spectra. It is noticeable that the non-coherent diabatic approximation (QDA, SQDA) gave an almost identical result as the coupled channel calculation (QCC, SQCC) in the case of K2. In contrast, the non-coherent adiabatic approximation (QAA, SQAA) gave very similar results to the coupled channel calculation (QCC, SQCC) in the case of KCs and an almost identical result in the case of the Cs2 molecule. Furthermore, excellent agreement of the semi quantum approximation (SQCC, SQAA, SQDA) with the full quantum calculation (QCC, QAA, QDA) was evident, especially in the case of the Cs2 molecule.
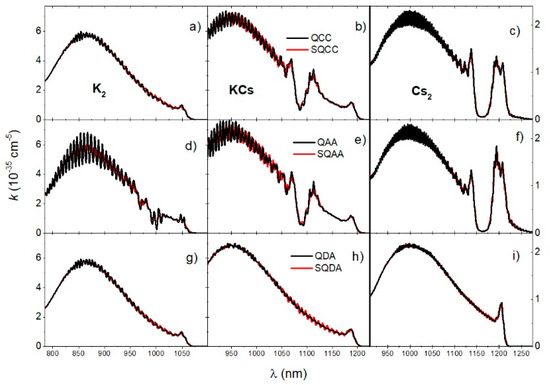
Figure 6.
Reduced absorption coefficients of K2, KCs, and Cs2 molecules at temperature are shown in the first (a,d,g), second (b,e,h), and third columns (c,f,i), respectively. The first row (a–c) shows the QCC and SQCC spectra, the second row (d–f) shows the QAA and SQAA spectra, and the last row (g–i) shows the QDA and SQDA spectra.
3.3. The Comparison of the Experimental and Theoretical Absorption Coefficient
The absorption coefficient is temperature dependent, especially in the case of bound–bound transitions. Figure 7a–c shows the theoretical absorption coefficients of transition calculated using the QCC approach at several experimental temperatures for K2, KCs, and Cs2 molecules. Using Equation (36), we aimed to obtain the best fit of the experimental spectrum and theoretical simulation by iteratively changing the following parameters: temperature T, potassium atom number density NK, and cesium atoms number density NCs. The iterative procedure started with the experimental temperature but the best agreement was obtained at the end of the iteration with a temperature that was about 20 K higher than the initial one. Theoretical simulations were done for two temperatures, as shown in Figure 7d,e. The sapphire cell was filled with potassium and cesium in an approximate ratio of . Figure 7d,e show the results obtained for and , respectively.
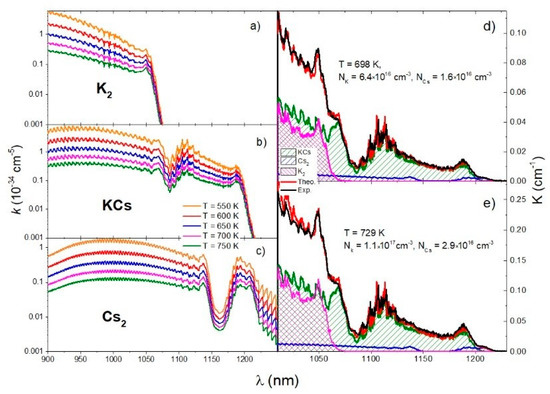
Figure 7.
The first column shows the reduced absorption coefficients of K2, KCs, and Cs2 molecules (a–c, respectively) in the temperature range T = 550 K–750 K. The comparison of the experimental absorption coefficient (black curve) with the theoretical simulation (red curve) is in the second column (d,e). The theoretical simulations of the contributions of KCs, Cs2, and K2 molecules are shown in green, blue, and magenta, respectively.
In Figure 7d,e, the spectral contributions of K2, KCs, and Cs2 molecules calculated using the QCC approach are shown in magenta, green, and blue, respectively. By comparing the experimental and theoretical spectra, all important features in the experimental spectrum were identified. The peak at 1048.5 nm was related to the minimum of the K2 transition difference potential at 1055 nm. The shoulder at 1068 nm was related to the minimum of the KCs transition difference potential at 1072 nm. The broad oscillating structure around 1106 nm was related to the maximum of the KCs transition difference potential at 1107 nm, and the peak at 1189 nm was related to the minimum at 1192 nm. The peak at 1208 nm was related to the minimum of the Cs2 transition difference potential at 1205 nm.
4. Discussion and Conclusions
We studied the influence of the spin–orbit coupling of two excited diabatic electronic states of the same symmetry whose potential curves intersected on the high-temperature spectrum of the optical transition. In the neighborhood of the diabatic state potential curves’ crossing point, the potential curves of the corresponding adiabatic states of the same symmetry avoided crossing and these states were mixed via the matrix element of radial non-adiabatic coupling. To obtain the correct spectrum of transitions from the isolated ground electronic state to the excited coupled diabatic or adiabatic states, the energies and wave functions of the excited states needed to be determined using a coupled channel calculus (QCC).
We distinguished two cases of the spectrum. In the neighborhood of an isolated diabatic state potential curves’ crossing point, the transition difference potential curves of the excited adiabatic states were monotonic in the first case and had a minimum and a maximum in the second case. In the first case, based on the semi-classical analysis in Section 2.3, we concluded that a non-coherent adiabatic approach was a satisfactory approximation of the coupled channel calculation. Based on the discussion in Section 2.3, for the second case, we concluded that if the Massey parameter (Equation (35)) satisfied , a non-coherent diabatic approach was a correct description of the spectrum, and if , a non-coherent adiabatic approach was justified. The potentials of the adiabatic electronic states are often available in the literature but there is no data on the corresponding diabatic state potentials and the spin–orbit mixing of these states. Assuming that the Landau–Zener approximation is valid in the neighborhood where the adiabatic state potentials avoided crossing, the Massey parameter can be written:
where is the adiabatic state potentials difference, and the crossing point is defined via the condition . If only the adiabatic state potential curves are available, by using Equation (39), one can assess whether the non-coherent adiabatic approximation gives a satisfactory theoretical simulation of the spectrum.
The semi-quantum approach is extensively studied in References [1,15,18], and in this paper, we analyzed its applicability in coupled channel calculations. As discussed in References [1,15], the semi-quantum approximation describes the vibrational structure of the molecular band well but neglects the rotational structure. In the high-temperature, low-resolution spectra of molecules with a larger reduced mass, such as K2, KCs, and Cs2, the rotational structure was not resolved and the semi-quantum coupled channel approach had a very good agreement with the quantum coupled channel calculation.
Due to the low computer time consumption, a semi-quantum approach was found to be an appropriate tool for gas diagnostics. In our experimental study of a potassium and cesium vapor mixture absorption spectrum, we used the semi-quantum method to determine the atom’s number density and temperature. The alkali mixture investigated in this article was suitable for analyzing the influence of the diabatic state SO coupling on the A–X transition spectrum. In the case of the K2 molecule, due to the small SO interaction, the Massey parameter was small with , and the non-coherent diabatic approximation described the spectrum well. In contrast, in the case of the Cs2 molecule, the SO interaction and the Massey parameter were large with , and the spectrum could be calculated well using a non-coherent adiabatic approximation.
Low-resolution spectroscopy is applicable in the plasma diagnostics but can also be useful in the study of some fundamental phenomena and processes in the alkali gas. The experimental and theoretical methods presented here should also be applicable in the analysis of the SO interaction influence on the A-X transition spectra in the case of other heteronuclear alkali molecules, such as NaCs and RbCs.
Author Contributions
R.B. discussed the problem, analyzed the experiment, made the calculations, prepared the figures, and wrote the paper; G.P. made and analyzed the experiment, discussed the problem, and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beuc, R.; Movre, M.; Pichler, G. High Temperature Optical Spectra of Diatomic Molecules at Local Thermodynamic Equilibrium. Atoms 2018, 6, 67. [Google Scholar] [CrossRef]
- Девдариани, А.З.; Cебякин Ю., Н. Температурная зависимoсть фoрмы Ландау-Зинерoвскoгo сателита спектральнoй линии. Oпт. и Cпектр. 1976, 48, 1018–1021. [Google Scholar]
- Девдариани, А.З.; Острoвский, В.Н.; Cебякин, Ю.Н. Осoбеннoсти в спектрах электрoнoв и фoтoнoв, свазанные c взаимпдействием квазистациoнарных термoв. ЖЕТФ 1979, 76, 529–542. [Google Scholar]
- Девдариани, А.З.; Cебякин, Ю.Н. Оптическая cпектрoскoпия Ландау-Зинерoвских перехoдoв. ЖЕТФ 1989, 96, 1997–2008. [Google Scholar]
- O’Callaghan, M.J.; Gallagher, A.; Holstein, T. Absorption and emission of radiation in the region of an avoided level crossing. Phys. Rev. A 1985, 32, 2754–2768. [Google Scholar] [CrossRef] [PubMed]
- Landau, L.D. On the theory of energy transmission in collisions I. Phys. Z. Sowjet. 1932, 1, 88. [Google Scholar]
- Zener, C. Non-adiabatic crossing of energy levels. Proc. R. Soc. Lond. A 1932, 137, 696–702. [Google Scholar] [CrossRef]
- Lam, L.K.; Gallagher, A.; Hessel, M.M. The intensity distribution in the Na2 and Li2 A–X bands. J. Chem. Phys. 1977, 66, 3550–3556. [Google Scholar] [CrossRef]
- Chung, H.-K.; Kirby, K.; Babb, J.F. Theoretical study of the absorption spectra of the lithium dimer. Phys. Rev. A 1999, 60, 2002–2008. [Google Scholar] [CrossRef]
- Erdman, P.S.; Larson, C.W.; Fajardo, M.; Sando, K.M.; Stwalley, W.C. Optical absorption of lithium metal vapor at high temperatures. J. Quant. Spectrosc. Radiat. Transf. 2004, 88, 447–481. [Google Scholar] [CrossRef]
- Beuc, R.; Peach, G.; Movre, M.; Horvatić, B. Lithium, sodium and potassium resonance lines pressure broadened by helium atoms. Astron. Astrophys. Trans. 2018, 30, 315–322. [Google Scholar]
- Chung, H.-K.; Kirby, K.; Babb, J.F. Theoretical study of the absorption spectra of the sodium dimer. Phys. Rev. A 2001, 63, 1–8. [Google Scholar] [CrossRef]
- Vadla, C.; Beuc, R.; Horvatic, V.; Movre, M.; Quentmeier, A.; Niemax, K. Comparison of theoretical and experimental red and near infrared absorption spectra in overheated potassium vapor. Eur. Phys. J. D 2006, 37. [Google Scholar] [CrossRef]
- Talbi, F.; Bouledroua, M.; Alioua, K. Theoretical determination of the potassium far-wing photoabsorption spectra. Eur. Phys. J. D 2008, 50, 141–151. [Google Scholar] [CrossRef]
- Beuc, R.; Movre, M.; Horvatić, B. Time-efficient numerical simulation of diatomic molecular spectra. Eur. Phys. J. D 2014, 68. [Google Scholar] [CrossRef]
- Beuc, R.; Movre, M.; Horvatic, V.; Vadla, C.; Dulieu, O.; Aymar, M. Absorption spectroscopy of the rubidium dimer in an overheated vapor: An accurate check of molecular structure and dynamics. Phys. Rev. A 2007, 75. [Google Scholar] [CrossRef]
- Benedict, R.P.; Drummond, D.L.; Schlie, L.A. Fluorescence spectra and kinetics of Cs2. J. Chem. Phys. 1979, 70, 3155. [Google Scholar] [CrossRef]
- Horvatić, B.; Beuc, R.; Movre, M. Numerical simulation of dense cesium vapor emission and absorption spectra. Eur. Phys. J. D 2015, 69, 113. [Google Scholar] [CrossRef]
- Ross, A.J.; Crozeti, P.; Effantint, C.; D’incani, J.; Barrow, R.F. Interactions between the and states of K2. J. Phys. B 1987, 20, 6225–6231. [Google Scholar] [CrossRef]
- Lisdat, C.; Dulieu, O.; Knockel, H.; Tiemann, E. Inversion analysis of K2 coupled electronic states with the Fourier grid method. Eur. Phys. J. D 2001, 17, 319–328. [Google Scholar] [CrossRef]
- Manaa, M.R.; Ross, A.J.; Martin, F.; Crozet, P.; Lyyra, A.M.; Li, L.; Amiot, C.; Bergeman, T. Spin–orbit interactions, new spectral data, and deperturbation of the coupled and states of K2. J. Chem. Phys. 2002, 117, 11208. [Google Scholar] [CrossRef]
- Salami, H.; Bergeman, T.; Beser, B.; Bai, J.; Ahmed, E.H.; Kotochigova, S.A.; Lyyra, M.; Huennekens, J.; Lisdat, C.; Stolyarov, A.V.; et al. Spectroscopic observations, spin-orbit functions, and coupled-channel deperturbation analysis of data on the and of Rb2. Phys. Rev. A 2009, 80, 022515. [Google Scholar] [CrossRef]
- Drozdova, A.N.; Stolyarov, A.V.; Tamanis, M.; Ferber, R.; Crozet, P.; Ross, A.J. Fourier transform spectroscopy and extended deperturbation treatment of the spin-orbit-coupled co and states of the Rb2 molecule. Phys. Rev. A 2013, 88, 022504. [Google Scholar] [CrossRef]
- Verges, J.; Amiot, C. The Cs2 fluorescence excited by the Ar+ 1.09-μm laser line. J. Mol. Spectrosc. 1987, 126, 393–404. [Google Scholar] [CrossRef]
- Bai, J.; Ahmed, E.H.; Beser, B.; Guan, Y.; Kotochigova, S.; Lyyra, A.M.; Ashman, S.; Wolfe, C.M.; Huennekens, J.; Xie, F.; et al. Global analysis of data on the spin-orbit-coupled and states of Cs2. Phys. Rev. A 2011, 83, 032514. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Jastrzebski, W.; Szczepkowski, J.; Pazyuk, E.A.; Stolyarov, A.V. Direct coupled-channels deperturbation analysis of the complex in LiCs with experimental accuracy. J. Chem. Phys. 2015, 142, 234308. [Google Scholar] [CrossRef] [PubMed]
- Grochola, A.; Szczepkowski, J.; Jastrzebski, W.; Kowalczyk, P. Study of the and (0) states in LiCs by a polarization labelling spectroscopy technique. J. Quant. Spectrosc. Radiat. Transf. 2014, 145, 147–152. [Google Scholar] [CrossRef]
- Zaharova, J.; Tamanis, M.; Ferber, R.; Drozdova, A.N.; Pazyuk, E.A.; Stolyarov, A.V. Solution of the fully-mixed-state problem: Direct deperturbation analysis of the complex in a NaCs dimer. Phys. Rev. A 2009, 79, 012508. [Google Scholar] [CrossRef]
- Bergeman, T.; Fellows, C.E.; Gutterres, R.F.; Amiot, C. Analysis of strongly coupled electronic states in diatomic molecules: Low-lying excited states of RbCs. Phys. Rev. A 2003, 67, 050501(R). [Google Scholar] [CrossRef]
- Docenko, O.; Tamanis, M.; Ferber, R.; Bergeman, T.; Kotochigova, S.; Stolyarov, A.V.; Nogueira, A.; Fellows, C.E. Spectroscopic data, spin-orbit functions, and revised analysis of strong perturbative interactions for the and states of RbCs. Phys. Rev. A 2010, 81, 042511. [Google Scholar] [CrossRef]
- Kruzins, A.; Klincare, I.; Nikolayeva, O.; Tamanis, M.; Ferber, R.; Pazyuk, E.A.; Stolyarov, A.V. Fourier-transform spectroscopy of (4) ,, (1)3 transitions in KCs and deperturbation treatment of and . J. Chem. Phys. 2013, 139, 244301. [Google Scholar] [CrossRef] [PubMed]
- Tamanis, M.; Klincare, I.; Kruzins, A.Ž.; Nikolayeva, O.; Ferber, R.; Pazyuk, E.A.; Stolyarov, A.V. Direct excitation of the “dark” state predicted by deperturbation analysis of the complex in KCs. Phys. Rev. A 2010, 82, 032506. [Google Scholar] [CrossRef]
- Borsalino, D.; Vexiau, R.; Aymar, M.; Luc-Koenig, E.; Dulieu, O.; Bouloufa-Maafa, N. Prospects for the formation of ultracold polar ground state KCs molecules via an optical process. J. Phys. B 2016, 49, 055301. [Google Scholar] [CrossRef]
- Janev, R.K. Nonadiabatic Transitions between Ionic and Covalent States. Adv. At. Mol. Phys. 1976. [Google Scholar] [CrossRef]
- Lichten, W. Resonant Charge Exchange in Atomic Collisions. Phys. Rev. 1963, 131, 229. [Google Scholar] [CrossRef]
- Smith, F.T. Diabatic and Adiabatic Representations for Atomic Collision Problems. Phys. Rev. 1969, 179, 111. [Google Scholar] [CrossRef]
- Colbert, D.T.; Miller, W.H. A novel discrete variable representation for quantum mechanical reactive scattering via the S -matrix Kohn method. J. Chem. Phys. 1992, 96, 1982–1991. [Google Scholar] [CrossRef]
- Lam, K.; George, T.F. Semiclassical approach to spontaneous emission of molecular collision systems: A dynamical theory of fluorescence line shapes. J. Chem. Phys. 1982, 76, 3396. [Google Scholar] [CrossRef]
- Beuc, R.; Horvatic, V. The investigation of the satellite rainbow in the spectra of diatomic molecules. J. Phys. B 1992, 25, 1497–1510. [Google Scholar] [CrossRef]
- Kokoouline, V.; Dulieu, O.; Kosloff, R.; Masnou-Seeuws, F. Theoretical treatment of channel mixing in excitedRb2 and Cs2 ultracold molecules: Determination of predissociation lifetimes with coordinate mapping. Phys. Rev. A 2000, 62. [Google Scholar] [CrossRef]
- Beuc, R.; Movre, M.; Mihajlov, A.A. Nonadiabatic affects in absorption line shape. In Proceedings of the 9th International Conference on Spectral Line Shapes, Torun, Poland, 25–29 July 1988; p. D11. [Google Scholar]
- Ovchinnikova, M.Y. Transitions between fine-structure components in resonance interactions for alkali metals. Theor. Exp. Chem. 1965, 1, 12–16. [Google Scholar] [CrossRef]
- Yan, L.; Meyer, W. Electronic state potential curves and electronic transition dipole moments of K2 molecules. Unpublished Results.
- Marinescu, M.; Dalgarno, A. Dispersion forces and long-range electronic transition dipole moments of alkali-metal dimer excited states. Phys. Rev. A 1995, 52, 311. [Google Scholar] [CrossRef] [PubMed]
- Spies, N. Theoretische Untersuchung von Elektronisch Angeregten Zuständen der Moleküle Li2 und Cs2. Ph.D. Thesis, Fachbereich Chemie, Kaiserslautern University, Kaiserslautern, Germany, 1990. [Google Scholar]
- Meyer, W.; Spies, N. Electronic state potential curves of Cs2 molecules. Unpublished Results.
- Allouche, A.R.; Aubert-Frécon, M. Transition dipole moments between the low-lying states of the Rb2 and Cs2 molecules. J. Chem. Phys. 2012, 136, 114302. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).