Abstract
Molecular properties of the thallium monocyanide (Tl·CN) system in its ground electronic state are studied using high-precision ab initio relativistic two-component pseudopotential replacing 60 inner-core electrons of Tl. A relativistic coupled-cluster method with single, double and perturbative triple amplitudes is employed to account for electronic correlations. Extrapolation of results to the complete basis set limit is used for all studied properties. The global potential energy minimum of Tl·CN corresponds to the linear cyanide (TlCN) isomer, while the non-rigid isocyanide-like (TlNC) structure lies by approximately 11 kJ/mol higher in energy. The procedure of restoration of the wavefunction in the “core” region of Tl atom was applied to calculate the interaction of the Tl nuclear Schiff moment with electrons. The parameter X of the interaction of the Tl nuclear Schiff moment with electrons in the linear TlCN molecule equals 7150 a.u. The prospects of using the TlCN molecule for the experimental detection of the nuclear Schiff moment are discussed.
Keywords:
molecular electronic structure; parity and time-reversal invariance violating interactions; Schiff moment; relativistic coupled cluster calculation PACS:
31.30.-i; 37.10.Mn; 12.15.Mm; 21.10.Ky
1. Introduction
Linear triatomic molecules with heavy atoms have recently attracted great attention in connection with the prospects for searching for the effects not described in the framework of the Standard Model of elementary particles. Some triatomic molecules with open-shell ground electronic states have been proposed for the search for the nuclear spin-dependent effects, violating space parity ( effects) and scalar electron-nucleon interaction violating both space parity and time reversibility (-odd effects) [1,2] and search for permanent electron electric dipole moment [3]. It turns out that linear triatomic molecules with ground electronic state (like the proposed RaOH and YbOH), being excited to the first bending vibrational state, possess so-called l-doubling of spin-rotational levels, similar to -doubling for spin-rotational levels for linear molecules with ground electronic state. The -doubling can be, for example, used with great efficiency for suppressing of the different systematic effects in ultra-precise molecular spectroscopy (see e.g., Reference [4]). Recently, the search for the Schiff moment of the Tl nucleus has started using the TlF molecule [5] and the spin-rotational spectrum of this molecule was studied for both ground and several excited states [6]. In Reference [1] we have pointed to the molecule TlCN as a prospective candidate for the search for and -odd effects (note that the nuclear Schiff moment arises due to -odd nuclear forces). Both experimental and theoretical information concerning the molecular structure and isomerism of TlCN is scarce. Three vibrational frequencies detected in the gas-phase experiment [7] were allegedly assigned to cyanide and isocyanide isomers. The DFT electronic structure calculations [7] using large-(68-electron-) core one-component pseudopotential of Tl and a very restricted basis set, confirmed the existence of linear TlCN, whereas the detected stationary point of the potential energy surface presumably associated with the isocyanide was a saddle-type point.
According to electronic structure calculations and experiments in the gas phase and in inert matrices, molecules consisting of a group 13 or group 1 metal atom M and the CN fragment, M·CN, which can be considered respectively as analogs and pseudo analogs of Tl·CN, can have linear or slightly bent isomers (the cyanide MCN and isocyanide MNC) and a T-shaped one usually denoted as M[CN]. In the first approximation, the relative stability of isomers is determined by the type of the M–CN bond. If this bond is essentially ionic, the T-shaped structure is normally the most stable one due to the optimal Coulomb attraction and the isomerization barriers are low [8]. This situation occurs in the Na·CN [9,10,11] and K·CN [10,11,12,13] molecules. In the latter case, the KNC isomer does not exist [11]. In contrast, when the bond has a significant covalent component, isocyanide structures are more stable, isomerization barriers are relatively large and the T-shaped isomers usually do not exist [14]. It seems natural to suppose the existence of linear TlCN isomers similar to those of other cyanides of 13th groups elements.
In the present paper we report a study of ground-state properties and isomerism of Tl·CN by accurate relativistic electronic structure methods and for the first time calculate the parameter X of the interaction of the Tl nuclear Schiff moment [15] with electrons in linear triatomic molecule, TlCN.
2. Computational Methods
The electron subsystem of Tl·CN is described using the accurate shape-consistent two-component relativistic pseudopotential replacing 60 inner-core (–) electrons of the Tl atom [16]. The remaining external electrons of Tl and all the electrons of the light atoms are treated explicitly. The many-electron problem is solved using the relativistic coupled-cluster method with single, double and perturbative triple cluster amplitudes, RCCSD(T) [17] with the triple zeta (TZ) and quadruple zeta (QZ) quality basis sets. The TZ set includes aug-cc-pVTZ bases for C, N [18] and one for Tl [19]; the QZ set consists of aug-cc-pVQZ for C, N [18] and for Tl [19]. One-electron molecular spinors are constructed by solving the relativistic Kramers-restricted analog of the Hartree–Fock equations. Final energy estimates are obtained by the two-point extrapolation to the complete basis set limit (CBS) [20]. Note that the extrapolation efficiently supresses the basis set superposition errors [21]. A separate set of calculations is performed using the two-component relativistic density functional theory (RDFT) [22] with the hybrid exchange-correlation functional PBE0 [23] and the basis sets for C, N [24] and uncontracted for Tl [16].
We started with scanning the RCCSD(T) potential energy surface in a wide range of the internal coordinates R and (see Figure 1) in order to approximately locate the stationary points. At this stage, the C–N distance was frozen at Å. Then the configurations and energies of all linear structure were refined using the Broyden-Fletcher-Goldfarb-Shanno’s algoritm [25].
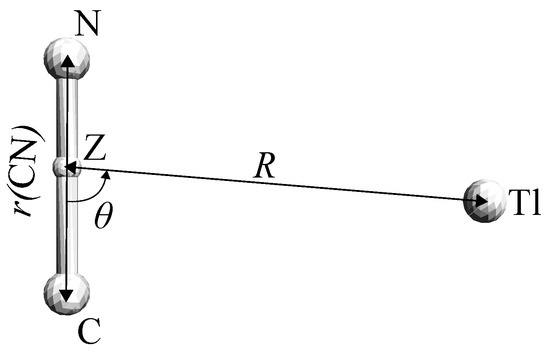
Figure 1.
Internal coordinates R, , for the Thallium Monocyanide (Tl·CN) system. Z is the center of mass of the CN fragment, and correspond to the linear TlCN and TlNC structures, respectively.
The vibrational frequencies for the TlCN isomer (Tl–C stretching , bending and C [,0.6]N stretching ) are evaluated within the harmonic approximation. Other molecular properties of this isomer are calculated for the equilibrium geometry obtained at the CBS limit. The dipole moment D is evaluated within the central finite-difference approximation, fitting the dependence of the total energy on the strength of the applied uniform electric field along the molecular axis in the range a.u.
The effective interaction of electrons with the Tl nuclear Schiff moment in the TlCN molecule is described by the following Hamiltonian [15,26]:
where = S is the Schiff moment of Tl, is Tl nuclear spin, is the unit vector along the internuclear axis z from Tl to C and X is the parameter given by:
where is the electronic density at the point , where corresponds to the center of the Tl nucleus. To calculate this parameter determined by the behavior of the valence wave function inside the nucleus, we employed the two-step method described in detail in References [27,28,29,30,31]. At the first step, the valence and outer-core part of the molecular wave function is treated within the generalized relativistic pseudopotential method [32,33,34]. The inner-core electrons are excluded from the explicit treatment. The resulting valence (pseudo) wave functions are smoothed in the spatial inner core region of a heavy atom that leads to considerable computational savings [35,36]. The second step is the nonvariational restoration of the correct 4-component behavior of the valence wave function in the spatial core region of the heavy atom [27,28,29,30,31].
The calculations are carried out using the dirac17 [37], mrcc [38,39], and two-component relativistic DFT [22] codes.
3. Results and Discussion
Molecular properties. The relative energies of stationary points for the Tl·CN potential energy surface are listed and compared with the corresponding values for the cyanides of group 13 and 1 elements in Table 1. At all levels of correlation treatment (RCCSD(T)/TZ,QZ,CBS, RDFT/PBE0) the global energy minimum corresponds to the linear TlCN isomer. The dependence of the ground-state energy on the bending angle in the region of isocyanide-like structures is extremely flat. The cross section of Tl·CN potential energy surface along the approximate isomerization path obtained at the RCCSD(T)/CBS level of theory is displayed in Figure 2.

Table 1.
Relative energies (kJ/mol) of isomers and transition states (TS) for M·CN systems. PW: present work.
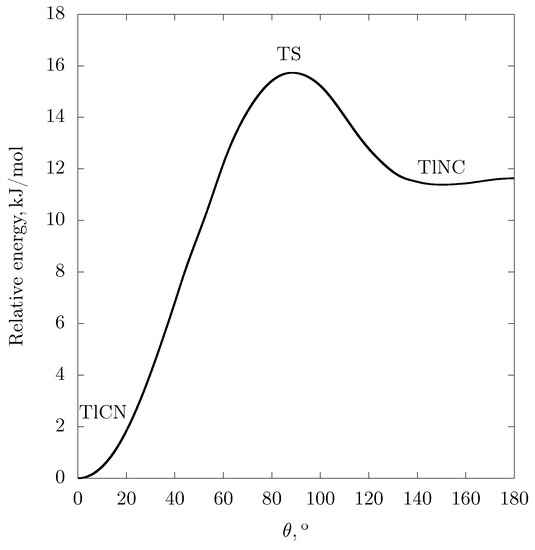
Figure 2.
The cross section of RCCSD(T)/CBS potential energy surface for Tl·CN along the approximate isomerization path. TS: transition state. The energy is given with respect to the global minimum. The distance Å is frozen at the equilibrium value for the TlCN isomer.
Due to the non-rigidity of the TlNC-like structure, the position of the local minimum is very sensitive to the level of correlation treatment; the harmonic approximation for the vibrations of this isomer is senseless.
The situation with no minima corresponding to the linear isocyanide structure and low isomerization barriers is typical for systems with predominantly ionic bonds M–CN. At the same time, for strongly ionic alkali metal cyanides the most stable isomers are T-shaped. In contrast, light analogues of the thallium compounds, such as the Al·CN and Ga·CN, are stable in the isocyanide configurations and have no T-shaped isomers. Along the series Al–Ga–In–Tl, the relative stability of the isocyanide isomers decreases monotonically, so that the TlCN isomer is significantly more stable than the isocyanide. The isomerisation barriers decrease along the series, becoming for the Tl·CN comparable to those in Na·CN, K·CN and Rb·CN systems. The results of simple DFT modeling of the indium system (Table 1) indicate that the linear InNC should not exist; however, this disagrees with the microwave spectroscopy data [42]. Anyway, according to the infrared spectroscopy study [7] the linear or slightly bent InNC is slightly more abundant than the cyanide.
The calculated equilibrium geometries, harmonic vibrational frequencies and dipole moment for the TlCN isomer, along with the corresponding available experimental data for other analogue and pseudo analogue M·CN systems, are collected in Table 2. The TlCN harmonic vibrational frequencies were calculated for the most abundant isotopomer (). In all cases, the dipole moment direction corresponds to the charge redistribution M(CN).
Table 2.
Molecular parameters of M·CN isomers for M belonging to groups 13 and 1. Internuclear distances in ångstroms, vibrational frequencies () in cm, dipole moments (D) in atomic units, for cyanides and r(MN) for isocyanides. PW: present work.
Unusually flat bending potentials and large amplitudes of zero-point vibrations in Al, Ga and In isocyanides were discussed in Reference [42]. The shape of the bending potential of the TlNC isomer (Figure 2) apparently indicates that the bending motion is fairly free in the range . Note that the computed TlCN bending frequency cm also corresponds to the significant zero-point vibration amplitude.
The calculated TlCN equilibrium geometry corresponds to the rotational constant value = 2.406 GHz. The l doubling interval can be roughly estimated as MHz. This value is significantly smaller than its counterparts for the systems recently proposed for the search for and -odd effects, RaOH (3.1 MHz) [1] and YbOH, where the value of l-doubling splitting was estimated to be ≈10 MHz [3]. The TlCN l-doubling interval is expected to be the smallest one for group 13 (iso)cyanide molecules; even for the rather heavy and flexible InNC molecule, the l-doubling interval should be about 4.6 MHz. Thus full polarisation of the TlCN molecule can be reached in electric fields smaller than 100 V/cm, which allows much better control of the systematic effects in high-precision experiments.
For all group 13 and group 1 atoms, the C [,0.6]N stretching frequencies in M·CN are in the ranges ca. 2125–2150 cm for cyanide isomers and ca. 2040–2050 cm for isocyanide or T-shaped isomers. The only unexpected experimental value for the AlCN molecule obtained by Fukushima [41] ( cm) disagrees with another experimental estimate ( cm [7]) and the results of electronic structure calculations. Our estimate cm for TlCN is in line with this pattern.
The RCCSD(T) dipole moment values for TlCN seem unexpectedly small even compared to that of AlCN and AlNC molecules where the covalent contribution to Al-CN bonding is believed significant.
Schiff moment. The values of the X parameter for the TlCN molecule obtained within different basis sets and methods are given in Table 3. One can see a good convergence of the results with increasing basis set size. In particular, the contribution of the perturbative triple cluster amplitudes is almost independent of basis set size, being smaller than 3%. This also demonstrates a good convergence with respect to the level of the electron correlation treatment.
Table 3.
Calculated values of the X parameter for TlCN. The recommended estimate is given in boldface.
The final X value (7150 a.u.) is close to that obtained earlier for the TlF molecule (7635 a.u., [27]).
If the properties of the excited electronic states in TlCN resemble those in TlF, one would also expect amenability of TlCN for cooling with lasers. This would open an exciting perspective for the ultra-precision spectroscopy of polyatomic molecules with closed electronic shells. The resulting value of the X parameter is also close to that for the RaO (7532 a.u. [46]) and PbO [30] molecules and about two times larger than in the RaF molecule [47]. Just to compare the Schiff moment enhancement factor with non-molecular systems, we notice that the X parameter, for example, liquid xenon is almost 40 times smaller [48].
4. Conclusions
The results of relativistic calculations on the ground electronic state of the Tl·CN system in the framework of the coupled cluster (RCCSD(T)) method with the extrapolation to the complete basis set limit demonstrate unambiguously that the lowest-energy isomer has a linear cyanide-like (TlCN) equilibrium structure. The isocyanide-like isomer has equilibrium energy 11 kJ/mol above the global minimum and is extremely non-rigid with respect to bending deformations. Similar results were obtained within the simple relativistic density functional computational scheme. In the Al–Tl row of cyanides and isocyanides the Tl·CN system is probably the first wherein the cyanide TlCN is the most stable isomer. Along this row, the rigidity of cyanides and isocyanides gradually decreases. Our estimate of the parameter characterising the interaction of the Tl nuclear Schiff moment with electrons in the TlCN isomer is close to that obtained earlier for TlF [27] and the analogous parameter for the Ra nucleus in RaO [46]. We conclude that the TlCN molecule has good prospects for searching for effects outside of the Standard Model.
Author Contributions
Conceptualization, A.Z. and T.A.I.; Data curation, A.V.K. and D.E.M.; Formal analysis, A.V.K. and D.E.M.; Methodology, A.Z. and L.V.S.; Supervision, A.Z. and T.A.I.
Funding
This research was funded by RSF grant number 18-12-00227.
Acknowledgments
We are indebted to C. van Wüllen for providing us with his relativistic DFT code [22]. Schiff moment calculation including basis set generation are supported by RSF Grant No. 18-12-00227. T.A.I. thanks participants of the NPCCM2018 workshop (MITP, Mainz, 26-30 November 2018) for inspiring discussions.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Isaev, T.A.; Zaitsevskii, A.V.; Eliav, E. Laser-coolable polyatomic molecules with heavy nuclei. J. Phys. B At. Mol. Opt. Phys. 2017, 50, 225101. [Google Scholar] [CrossRef]
- Norrgard, E.B.; Barker, D.S.; Eckel, S.P.; Fedchak, J.A.; Klimov, N.N.; Scherschligt, J. Nuclear-Spin Dependent Parity Violation in Optically Trapped Polyatomic Molecules. arXiv, 2018; arXiv:1812.00064. [Google Scholar]
- Kozyryev, I.; Hutzler, N. Precision measurement of time-reversal symmetry violation with laser-cooled polyatomic molecules. Phys. Rev. Lett. 2017, 119, 133002. [Google Scholar] [CrossRef] [PubMed]
- Shafer-Ray, N.E. Possibility of 0-g- factor paramagnetic molecules for measurement of the electron’s electric dipole moment. Phys. Rev. A 2006, 73, 034102. [Google Scholar] [CrossRef]
- DeMille, D. CeNTREX Experiment (DAMOP 2017 Report). 2017. Available online: http://meetings.aps.org/Meeting/DAMOP17/Session/B2.3 (accessed on 29 April 2019).
- Norrgard, E.B.; Edwards, E.R.; McCarron, D.J.; Steinecker, M.H.; DeMille, D.; Alam, S.S.; Peck, S.K.; Wadia, N.S.; Hunter, L.R. Hyperfine structure of the B3Π1 state and predictions of optical cycling behavior in the x→B transition of TlF. Phys. Rev. A 2017, 95, 062506. [Google Scholar] [CrossRef]
- Lanzisera, D.V.; Andrews, L. Reactions of Laser-Ablated Al, Ga, In, and Tl Atoms with Hydrogen Cyanide in Excess Argon. Matrix Infrared Spectra and Density Functional Theory Calculations on New Cyanide and Isocyanide Products. J. Phys. Chem. A 1997, 101, 9660. [Google Scholar] [CrossRef]
- Essers, R.; Tennyson, J.; Wormer, P. An SCF potential energy surface for Lithium Cyanide. Chem. Phys. Lett. 1982, 89, 223. [Google Scholar] [CrossRef]
- Van Vaals, J.; Meerts, W.L.; Dymanus, A. Structure of sodium cyanide by molecular beam electric resonance spectroscopy. J. Chem. Phys. 1982, 77, 5245. [Google Scholar] [CrossRef]
- Marsden, C. Ab initio correlated potential energy surfaces for monomeric sodium and potassium cyanides. J. Chem. Phys. 1982, 76, 6451. [Google Scholar] [CrossRef]
- Lee, D.-K.; Lim, I.S.; Lee, Y.S.; Hagebaum-Reignier, D.; Jeung, G.-H. Molecular properties and potential energy surfaces of the cyanides of the groups 1 and 11 metal atoms. J. Chem. Phys. 2007, 126, 244313. [Google Scholar] [CrossRef]
- Van Vaals, J.; Meerts, W.L.; Dymanus, A. Molecular beam electric resonance study of KCN, K13CN and KC15N. J. Mol. Spectrosc. 1984, 106, 280. [Google Scholar] [CrossRef]
- Törring, T.; Bekooy, J.; Meerts, W.L.; Hoeft, J.; Tiemann, E.; Dymanus, A. Rotational spectrum and structure of KCN. J. Chem. Phys. 1980, 73, 4875. [Google Scholar] [CrossRef]
- Petrie, S. Trends in M (CN) isomerism: A computational study of monocyanides of the main-group third row atoms. Phys. Chem. Chem. Phys. 1999, 1, 2897. [Google Scholar] [CrossRef]
- Hinds, E.A.; Sandars, P.G.H. Electric dipole hyperfine structure of TIF. Phys. Rev. A 1980, 21, 471. [Google Scholar] [CrossRef]
- Available online: http://www.qchem.pnpi.spb.ru (accessed on 29 April 2019).
- Vissher, L.; Lee, T.J.; Dyall, K.G. Formulation and implementation of a relativistic unrestricted coupled-cluster method including noniterative connected triples. J. Chem. Phys. 1996, 105, 8769. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Zaitsevskii, A.; Eliav, E. Padé´ extrapolated effective Hamiltonians in the Fock space relativistic coupled cluster method. Int. J. Quantum Chem. 2018, 118, e25772. [Google Scholar] [CrossRef]
- Petersson, G.A.; Frisch, M.J. A journey from generalized valence bond theory to the full CI complete basis set limit. J. Phys. Chem. A 2000, 104, 2183. [Google Scholar] [CrossRef]
- Mourik, V.T.; Wilson, A.K.; Peterson, K.A.; Woon, D.E.; Dunning, J.T.H. Advances in Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 1998; Volume 31, pp. 105–135. [Google Scholar]
- van Wüllen, C. A quasirelativistic two-component density functional and hartree-fock program. Zeitschrift für Physikalische Chemie 2010, 224, 413–426. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560. [Google Scholar] [CrossRef]
- Byrd, R.H.; Lu, P.; Nocedal, J.; Zhu, C. A limited memory algorithm for bound constrained optimization. SIAM J. Sci. Comput. 1995, 16, 1190. [Google Scholar] [CrossRef]
- Sushkov, O.P.; Flambaum, V.V.; Khriplovich, I.B. Possibility of investigating P-and T-odd nuclear forces in atomic and molecular experiments. Sov. Phys. JETP 1984, 87, 1521. [Google Scholar]
- Petrov, A.N.; Mosyagin, N.S.; Isaev, T.A.; Titov, A.V.; Ezhov, V.F.; Eliav, E.; Kaldor, U. Calculation of P,T-Odd Effects in 205TIF Including Electron Correlation. Phys. Rev. Lett. 2002, 88, 073001. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.V.; Mosyagin, N.S.; Petrov, A.N.; Isaev, T.A.; DeMille, D.P. P, T-parity violation effects in polar heavy-atom molecules. Prog. Theor. Chem. Phys. 2006, 15, 253. [Google Scholar]
- Skripnikov, L.V.; Titov, A.V. Theoretical study of ThF+ in the search for T,P-violation effects: Effective state of a Th atom in ThF+ and ThO compounds. Phys. Rev. A 2015, 91, 042504. [Google Scholar] [CrossRef]
- Skripnikov, L.V.; Titov, A.V. LCAO-based theoretical study of PbTiO3 crystal to search for parity and time reversal violating interaction in solids. J. Chem. Phys. 2016, 145, 054115. [Google Scholar] [CrossRef] [PubMed]
- Skripnikov, L.V.; Titov, A.V.; Petrov, A.N.; Mosyagin, N.S.; Sushkov, O.P. Enhancement of the electron electric dipole moment in Eu2+. Phys. Rev. A 2011, 84, 022505. [Google Scholar] [CrossRef]
- Titov, A.V.; Mosyagin, N.S. Generalized relativistic effective core potential: Theoretical grounds. Int. J. Quantum Chem. 1999, 71, 359. [Google Scholar] [CrossRef]
- Mosyagin, N.S.; Zaitsevskii, A.V.; Titov, A.V. Shape-consistent relativistic effective potentials of small atomic cores. Rev. At. Mol. Phys. 2010, 1, 63. [Google Scholar]
- Mosyagin, N.S.; Zaitsevskii, A.V.; Skripnikov, L.V.; Titov, A.V. Generalized relativistic effective core potentials for actinides. Int. J. Quantum Chem. 2016, 116, 301. [Google Scholar] [CrossRef]
- Skripnikov, L.V. Combined 4-component and relativistic pseudopotential study of ThO for the electron electric dipole moment search. J. Chem. Phys. 2016, 145, 214301. [Google Scholar] [CrossRef] [PubMed]
- Skripnikov, L.V.; Mosyagin, N.S.; Titov, A.V. Relativistic coupled-cluster calculations of spectroscopic and chemical properties for element 120. Chem. Phys. Lett. 2013, 555, 79. [Google Scholar] [CrossRef]
- Visscher, L.; Jensen, H.J.A.; Bast, R.; Saue, T.; Bakken, V.; Dyall, K.G.; Dubillard, S.; Ekström, U.; Eliav, E.; Enevoldsen, T.; et al. DIRAC, a Relativistic Ab Initio Electronic Structure Program, Release DIRAC17 (2017). Available online: http://www.diracprogram.org (accessed on 29 April 2019).
- Kállay, M.; Rolik, Z.; Ladjánszki, I.; Szegedy, L.; Ladóczki, B.; Csontos, J.; Kornis, B. mrcc, a quantum chemical program suite. Available online: www.mrcc.hu (accessed on 29 April 2019).
- Rolik, Z.; Kállay, M. A general-order local coupled-cluster method based on the cluster-in-molecule approach. J. Chem. Phys. 2011, 135, 104111. [Google Scholar] [CrossRef]
- Ma, B.; Yamaguchi, Y.; Schaefer, H.F., III. Spectroscopic constants and potential energy surfaces for the possible interstellar molecules A1NC and A1CN. Mol. Phys. 1995, 86, 1331. [Google Scholar] [CrossRef]
- Fukushima, M. Laser induced fluorescence spectroscopy of AlNC/AlCN in supersonic free expansions. Chem. Phys. Lett. 1998, 283, 337. [Google Scholar] [CrossRef]
- Walker, K.A.; Evans, C.J.; Suh, S.-H.K.; Gerry, M.C.; Watson, J.K. Fourier Transform Microwave Spectroscopy of Cyanides and Isocyanides of Al, Ga, and In. J. Mol. Spectrosc. 2001, 209, 178. [Google Scholar] [CrossRef]
- Robinson, J.; Apponi, A.; Ziurys, L. The millimeter-wave spectrum of AlNC: chemical trends in metal isocyanide molecules. Chem. Phys. Lett. 1997, 278, 1. [Google Scholar] [CrossRef]
- Gerasimov, I.; Yang, X.; Dagdigian, P.J. Laser fluorescence excitation spectra of the AlNC and AlCN isomers. J. Chem. Phys. 1999, 110, 220. [Google Scholar] [CrossRef]
- Ismail, Z.K.; Hauge, R.H.; Margrave, J.L. Infrared spectra of matrix-isolated sodium and potassium cyanides. J. Mol. Spectrosc. 1973, 45, 304. [Google Scholar] [CrossRef]
- Kudashov, A.D.; Petrov, A.N.; Skripnikov, L.V.; Mosyagin, N.S.; Titov, A.V.; Flambaum, V.V. Calculation of the parity- and time-reversal-violating interaction in 225RaO. Phys. Rev. A 2013, 87, 020102(R). [Google Scholar] [CrossRef]
- Kudashov, A.D.; Petrov, A.N.; Skripnikov, L.V.; Mosyagin, N.S.; Isaev, T.A.; Berger, R.; Titov, A.V. Ab initio study of radium monofluoride (RaF) as a candidate to search for parity- and time-and-parity-violation effects. Phys. Rev. A 2014, 90, 052513. [Google Scholar] [CrossRef]
- Isaev, T.A.; Petrov, A.N.; Mosyagin, N.S.; Titov, A.V. Search for the nuclear Schiff moment in liquid xenon. Phys. Rev. A 2007, 75, 032515. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).