Polar Lipids in Starch-Rich Commodities to be Analyzed with LC-MS-Based Metabolomics—Optimization of Ionization Parameters and High-Throughput Extraction Protocols


Abstract
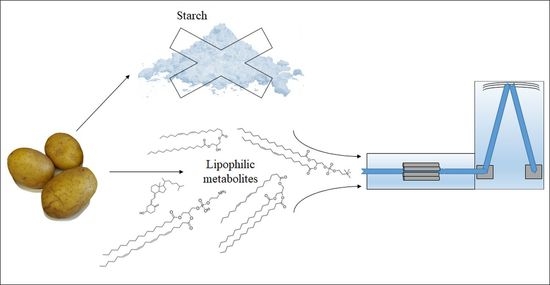
1. Introduction
2. Results
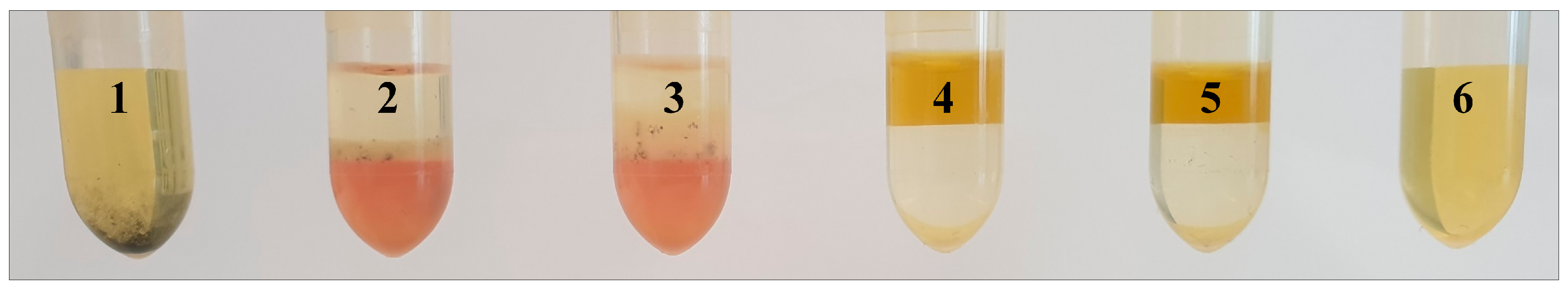
2.1. Comparison of Extraction Procedures
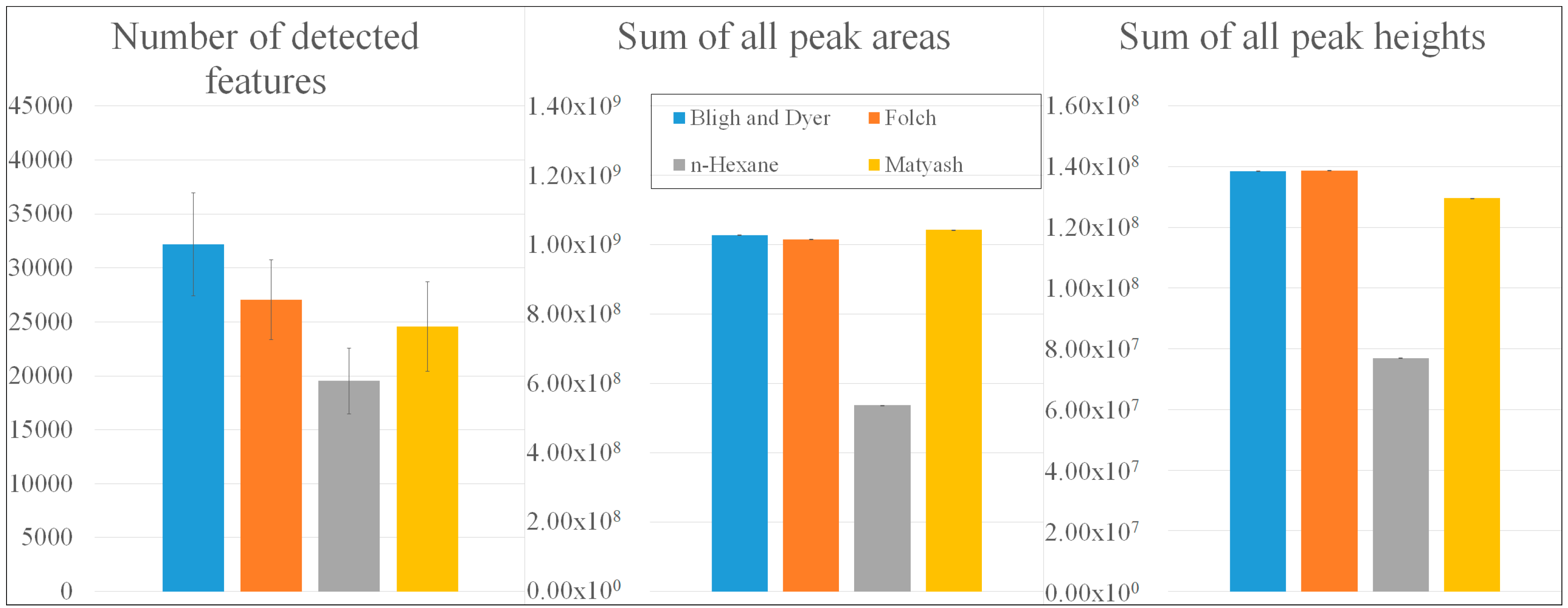
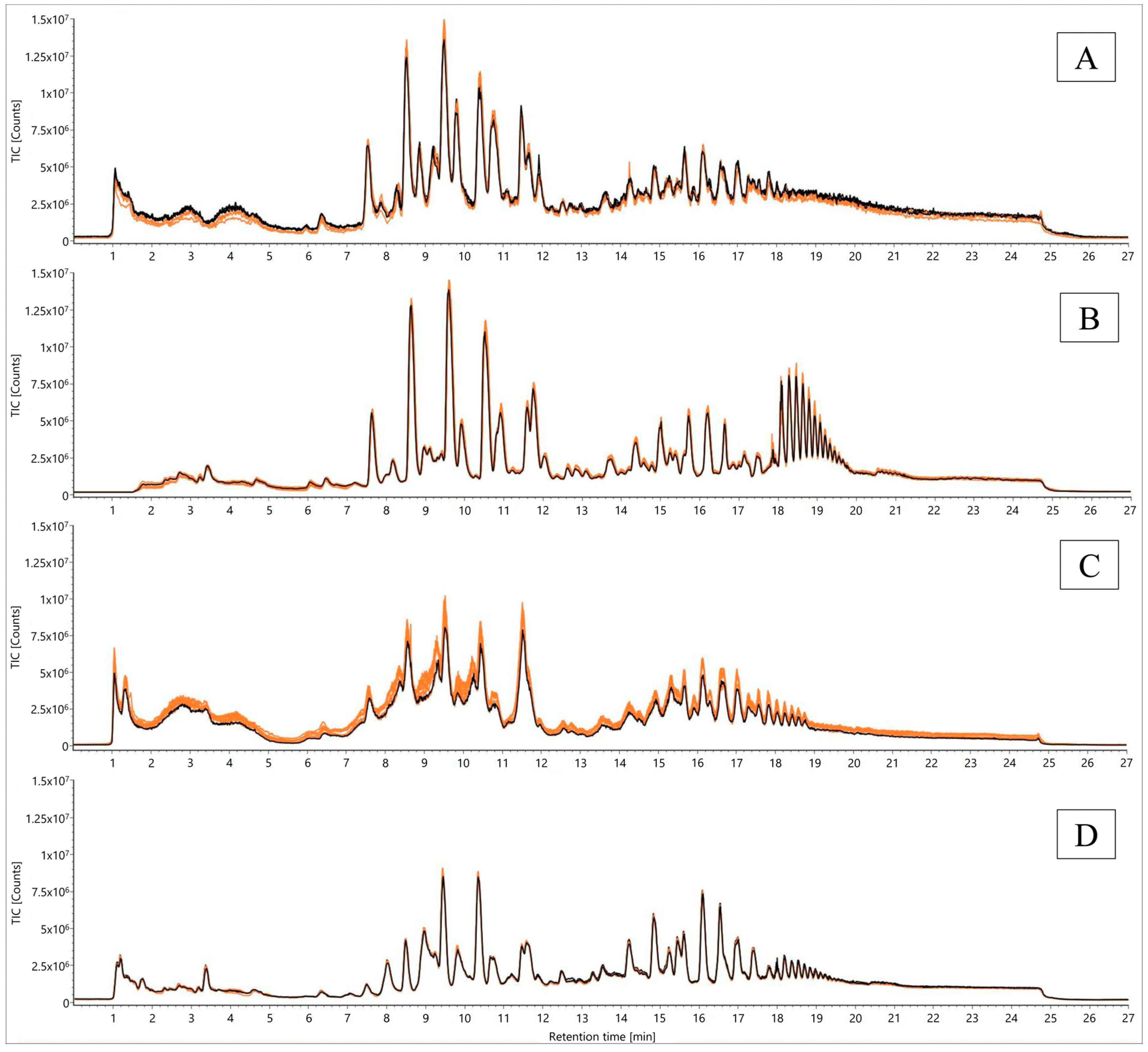
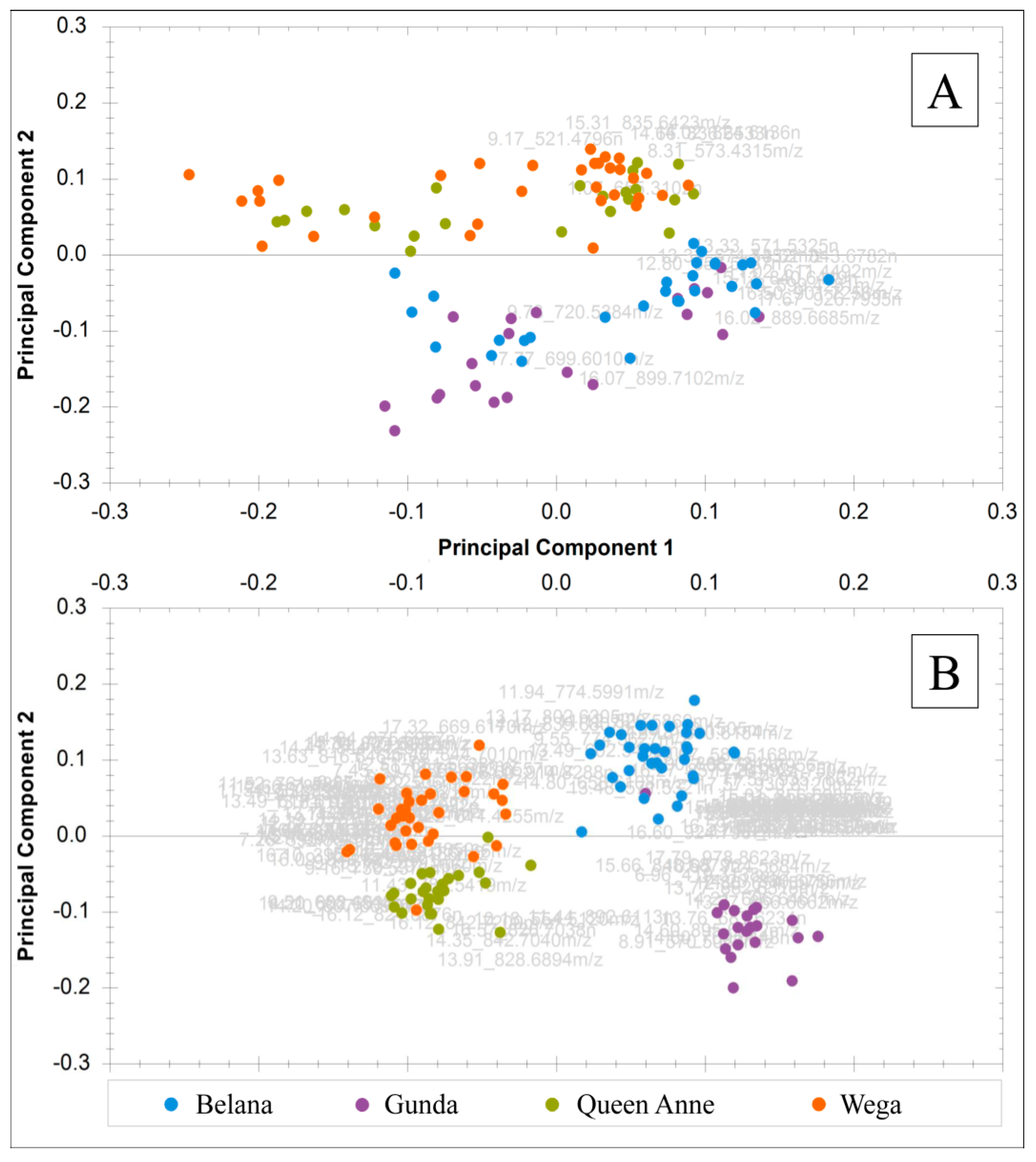
2.1.1. Non-Targeted Evaluation
2.1.2. Targeted Evaluation
2.1.3. Upscaling to Large Metabolomic Datasets
2.2. Comparison of Ionization Parameters Regarding the Removal of Starch Residues
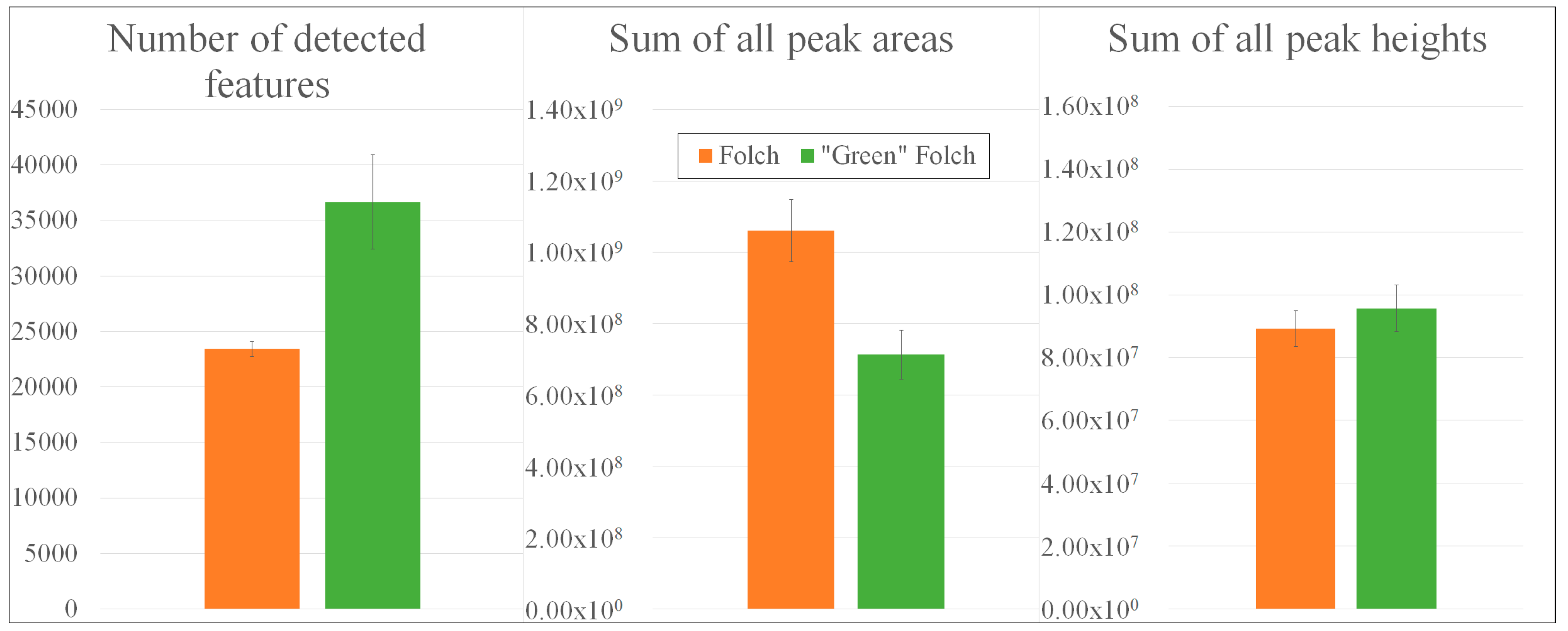
2.3. Enhancement of the Extraction Method According to Folch
2.3.1. Non-Targeted Evaluation
2.3.2. Targeted Evaluation
2.3.3. Detection of Starch Residues
2.4. Sustainability of Signal Intensities and Analysis of Authentic Potato Samples
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Potato Samples
4.3. Sample Preparation and Extraction
4.4. UPLC-IMS-QToF Analysis
4.5. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harrigan, G.G.; Goodacre, R. Metabolic Profiling: Its Role in Biomarker Discovery and Gene Function Analysis; Springer Science & Business Media: Luxembourg, 2003. [Google Scholar]
- Monton, M.R.; Soga, T. Metabolome analysis by capillary electrophoresis-mass spectrometry. J. Chromatogr. A 2007, 1168, 237–246. [Google Scholar] [CrossRef] [PubMed]
- De Livera, A.M.; Dias, D.A.; De Souza, D.; Rupasinghe, T.; Pyke, J.; Tull, D.; Roessner, U.; McConville, M.; Speed, T.P. Normalizing and integrating metabolomics data. Anal. Chem. 2012, 84, 10768–10776. [Google Scholar] [CrossRef] [PubMed]
- Deutsche Forschungsanstalt für Lebensmittelchemie. Der kleine Souci, Fachmann, Kraut—Lebensmitteltabelle für die Praxis; Andersen, G., Soyka, K., Eds.; Wissenschaftliche Verlagsgesellschaft mbH (WVG): Stuttgart, Germany, 2011. [Google Scholar]
- Malachová, A.; Sulyok, M.; Beltrán, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography–tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chromatogr. A 2014, 1362, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Niessen, W.; Manini, P.; Andreoli, R. Matrix effects in quantitative pesticide analysis using liquid chromatography–mass spectrometry. Mass Spectrom. Rev. 2006, 25, 881–899. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003. [Google Scholar]
- De Vos, R.C.; Moco, S.; Lommen, A.; Keurentjes, J.J.; Bino, R.J.; Hall, R.D. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2007, 2, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Creydt, M.; Arndt, M.; Hudzik, D.; Fischer, M. Plant Metabolomics: Evaluation of Different Extraction Parameters for Nontargeted UPLC-ESI-QTOF-Mass Spectrometry at the Example of White Asparagus officinalis. J. Agric. Food Chem. 2018, 66, 12876–12887. [Google Scholar] [CrossRef] [PubMed]
- Arvanitoyannis, I.S.; Vaitsi, O.; Mavromatis, A. Potato: A comparative study of the effect of cultivars and cultivation conditions and genetic modification on the physico-chemical properties of potato tubers in conjunction with multivariate analysis towards authenticity. Crit. Rev. Food Sci. Nutr. 2008, 48, 799–823. [Google Scholar] [CrossRef]
- Casañas, R.; González, M.; Rodríguez, E.; Marrero, A.; Díaz, C. Chemometric studies of chemical compounds in five cultivars of potatoes from Tenerife. J. Agric. Food Chem. 2002, 50, 2076–2082. [Google Scholar] [CrossRef]
- Chatterjee, C.; Gopal, R.; Dube, B. Impact of iron stress on biomass, yield, metabolism and quality of potato (Solanum tuberosum L.). Sci. Hortic. 2006, 108, 1–6. [Google Scholar] [CrossRef]
- Amrein, T.M.; Bachmann, S.; Noti, A.; Biedermann, M.; Barbosa, M.F.; Biedermann-Brem, S.; Grob, K.; Keiser, A.; Realini, P.; Escher, F.; et al. Potential of Acrylamide Formation, Sugars, and Free Asparagine in Potatoes: A Comparison of Cultivars and Farming Systems. J. Agric. Food Chem. 2003, 51, 5556–5560. [Google Scholar] [CrossRef]
- Lehesranta, S.J.; Koistinen, K.M.; Massat, N.; Davies, H.V.; Shepherd, L.V.; McNicol, J.W.; Cakmak, I.; Cooper, J.; Luck, L.; Karenlampi, S.O.; et al. Effects of agricultural production systems and their components on protein profiles of potato tubers. Proteomics 2007, 7, 597–604. [Google Scholar] [CrossRef]
- Jandrić, Z.; Cannavan, A. An investigative study on differentiation of citrus fruit/fruit juices by UPLC-QToF MS and chemometrics. Food Control 2017, 72, 173–180. [Google Scholar] [CrossRef]
- Díaz, R.; Pozo, O.J.; Sancho, J.V.; Hernández, F. Metabolomic approaches for orange origin discrimination by ultra-high performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry. Food Chem. 2014, 157, 84–93. [Google Scholar] [CrossRef]
- Vallverdú-Queralt, A.; Medina-Remón, A.; Casals-Ribes, I.; Amat, M.; Lamuela-Raventós, R.M. A metabolomic approach differentiates between conventional and organic ketchups. J. Agric. Food Chem. 2011, 59, 11703–11710. [Google Scholar] [CrossRef]
- Hubbard, W.; Sheppard, A.; Newkirk, D.; Prosser, A.; Osgood, T. Comparison of various methods for the extraction of total lipids, fatty acids, cholesterol, and other sterols from food products. J. Am. Oil Chem. Soc. 1977, 54, 81–83. [Google Scholar] [CrossRef]
- Galliard, T. Lipids of potato tubers. 1. Lipid and fatty acid composition of tubers from different varieties of potato. J. Sci. Food Agric. 1973, 24, 617–622. [Google Scholar] [CrossRef]
- Lepage, M. The lipid components of white potato tubers (Solanum tuberosum). Lipids 1968, 3, 477–481. [Google Scholar] [CrossRef]
- Hassanien, M.F.R.; Elsanhoty, R.M. Lipid classes, fatty acids and bioactive lipids of genetically modified potato Spunta with Cry V gene. Food Chem. 2012, 133, 1169–1176. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef]
- Reis, A.; Rudnitskaya, A.; Blackburn, G.J.; Mohd Fauzi, N.; Pitt, A.R.; Spickett, C.M. A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J. Lipid Res. 2013, 54, 1812–1824. [Google Scholar] [CrossRef]
- Vorkas, P.A.; Isaac, G.; Anwar, M.A.; Davies, A.H.; Want, E.J.; Nicholson, J.K.; Holmes, E. Untargeted UPLC-MS profiling pipeline to expand tissue metabolome coverage: Application to cardiovascular disease. Anal. Chem. 2015, 87, 4184–4193. [Google Scholar] [CrossRef]
- Breil, C.; Abert Vian, M.; Zemb, T.; Kunz, W.; Chemat, F. “Bligh and Dyer” and Folch Methods for Solid-Liquid-Liquid Extraction of Lipids from Microorganisms. Comprehension of Solvatation Mechanisms and towards Substitution with Alternative Solvents. Int. J. Mol. Sci. 2017, 18, 708. [Google Scholar] [CrossRef]
- Magel, E. Qualitative and quantitative determination of starch by a colorimetric method. Starch 1991, 43, 384–387. [Google Scholar] [CrossRef]
- Klockmann, S.; Reiner, E.; Bachmann, R.; Hackl, T.; Fischer, M. Food Fingerprinting: Metabolomic Approaches for Geographical Origin Discrimination of Hazelnuts (Corylus avellana) by UPLC-QTOF-MS. J. Agric. Food Chem. 2016, 64, 9253–9262. [Google Scholar] [CrossRef]
- Claassen, C.; Kuballa, J.; Rohn, S. Metabolomics-Based Approach for the Discrimination of Potato Varieties (Solanum tuberosum) using UPLC-IMS-QToF. J. Agric. Food Chem. 2019, 67, 5700–5709. [Google Scholar] [CrossRef]
- Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues and Analysis in Food and Feed. SANTE/11813/2017. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf (accessed on 29 May 2019).
- European Commission. Commission directive 2002/63/EC of 11 July 2002-Establishing community methods of sampling for the official control of pesticide residues in and on products of plant and animal origin and repealing directive 79/700. Off. J. Eur. Commun. 2002, L 187/30, 30–43. [Google Scholar]
- Zhang, R.; Brennan, M.L.; Shen, Z.; MacPherson, J.C.; Schmitt, D.; Molenda, C.E.; Hazen, S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem. 2002, 277, 46116–46122. [Google Scholar] [CrossRef]
- Hara, A.; Radin, N.S. Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem. 1978, 90, 420–426. [Google Scholar] [CrossRef]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated Platform of Small Molecules and Biological Activities. In Annual Repports Computational Chemistry; Wheeler, R.A., Spellmeyer, D.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 4, pp. 217–241, Chapter 12. [Google Scholar]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences; Routledge: Abingdon, UK, 2013. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bligh and Dyer [22] | Folch [23] | n-Hexane [25] | Matyash [24] | “Green” Folch [27] |
|---|---|---|---|---|
| 1.0 g lyophilisat | 1.0 g lyophilisat | 1.0 g lyophilisat | 0.5 g lyophilisat | 1.0 g lyophilisat |
| +6 glass beads | ||||
| +5 mL TCM +10 mL MeOH | +10 mL TCM +5 mL MeOH | +25 mL n-hexane/2-propanol (v/v; 3/2) | +6.3 mL MeOH | +8.3 mL EtOAc/MeOH (v/v; 70/30) |
| 5 min shaking | ||||
| +5 mL water | +4.7 mL water | +6.3 mL n-hexane/2-propanol (v/v; 3/2) | +7.8 mL TBME | +8.3 mL EtOAc/MeOH (v/v; 70/30) |
| 5 min shaking | ||||
| +5 mL TCM | +7.8 mL TCM/MeOH (v/v; 2/1) | +5 mL MeOH +5 mL n-hexane | +7.8 mL water | +9.1 mL water +1.5 mL EtoAc |
| 5 min shaking | Two times 1 min shaking, releasing pressure every minute | 5 min shaking | ||
| +4 mL water | +5 mL water | +3.1 mL TBME | ||
| 5 min shaking | 5 min shaking | 4 min shaking, releasing pressure every two minutes | ||
| +4.7 mL water | ||||
| 3 min shaking | ||||
| Centrifugation for 10 min at 3846 g | Centrifugation for 15 min at 3846 g | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claassen, C.; Kuballa, J.; Rohn, S. Polar Lipids in Starch-Rich Commodities to be Analyzed with LC-MS-Based Metabolomics—Optimization of Ionization Parameters and High-Throughput Extraction Protocols. Metabolites 2019, 9, 167. https://doi.org/10.3390/metabo9080167
Claassen C, Kuballa J, Rohn S. Polar Lipids in Starch-Rich Commodities to be Analyzed with LC-MS-Based Metabolomics—Optimization of Ionization Parameters and High-Throughput Extraction Protocols. Metabolites. 2019; 9(8):167. https://doi.org/10.3390/metabo9080167
Chicago/Turabian StyleClaassen, Christin, Jürgen Kuballa, and Sascha Rohn. 2019. "Polar Lipids in Starch-Rich Commodities to be Analyzed with LC-MS-Based Metabolomics—Optimization of Ionization Parameters and High-Throughput Extraction Protocols" Metabolites 9, no. 8: 167. https://doi.org/10.3390/metabo9080167
APA StyleClaassen, C., Kuballa, J., & Rohn, S. (2019). Polar Lipids in Starch-Rich Commodities to be Analyzed with LC-MS-Based Metabolomics—Optimization of Ionization Parameters and High-Throughput Extraction Protocols. Metabolites, 9(8), 167. https://doi.org/10.3390/metabo9080167