Differential Proteomics Based on TMT and PRM Reveal the Resistance Response of Bambusa pervariabilis × Dendrocalamopisis grandis Induced by AP-Toxin


Abstract
:1. Introduction
2. Results
2.1. Symptoms of B. Pervariabilis × D. Grandis after AP-Toxin Induction
2.2. Quantitative Proteomic Response to Inducer
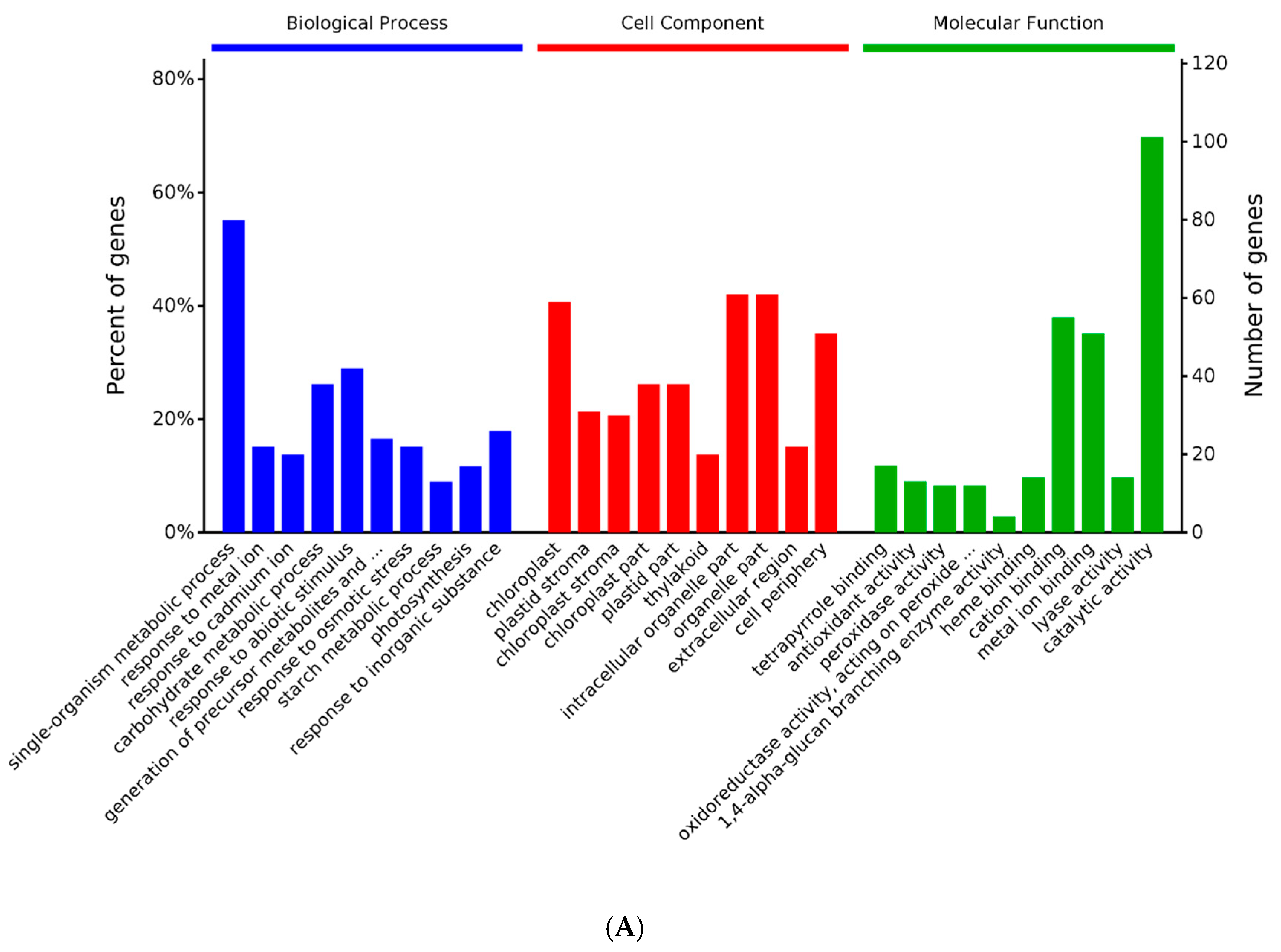
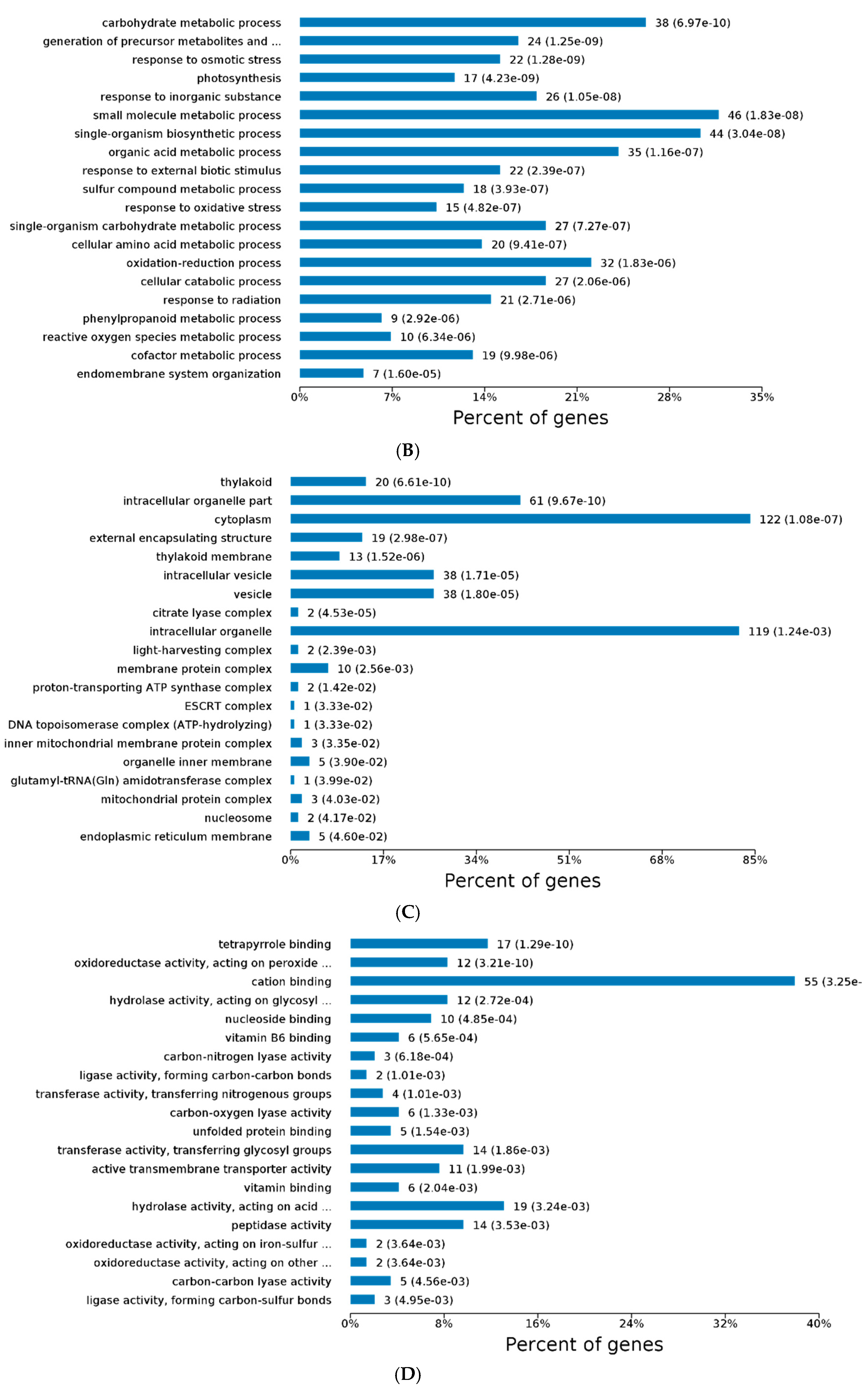
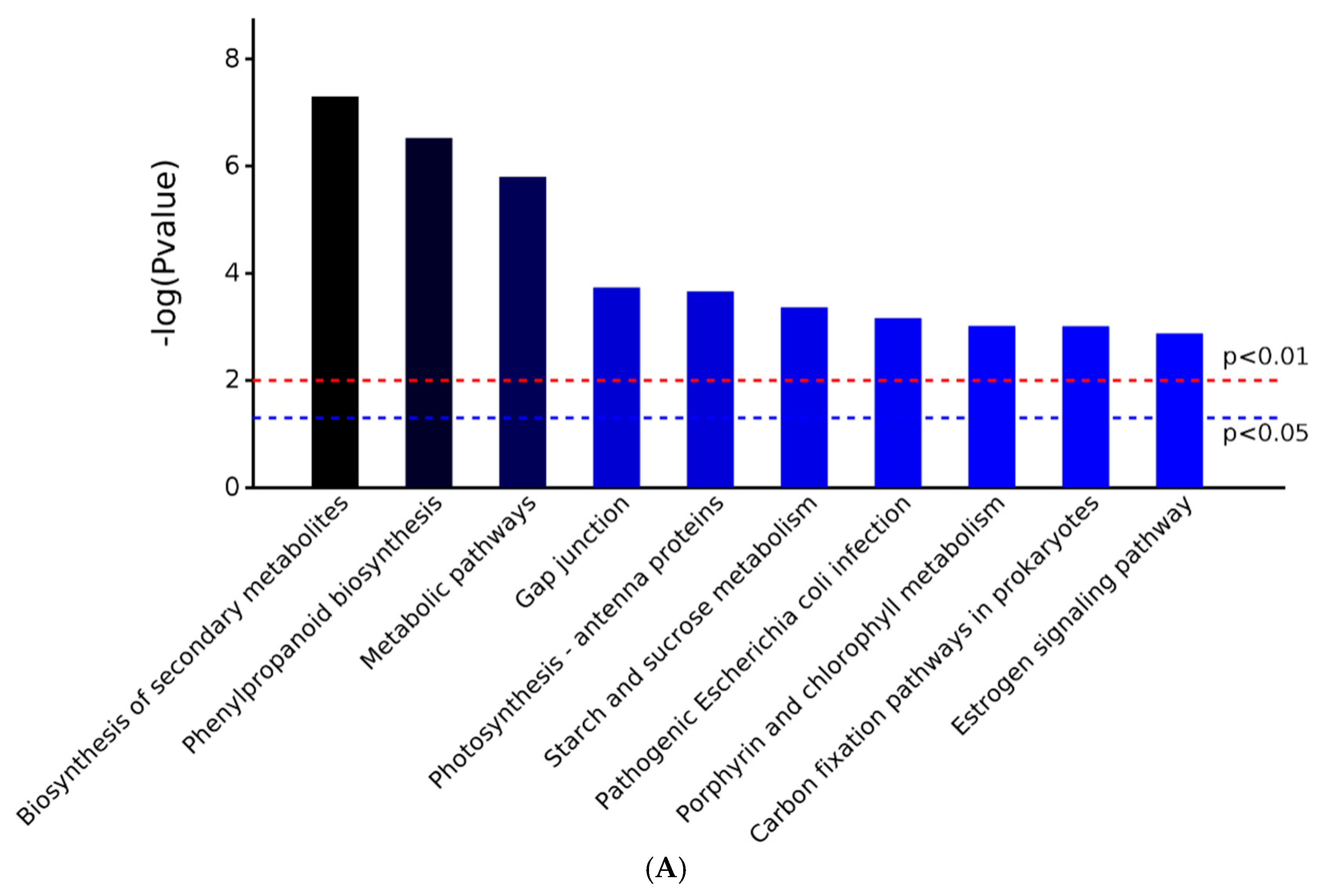
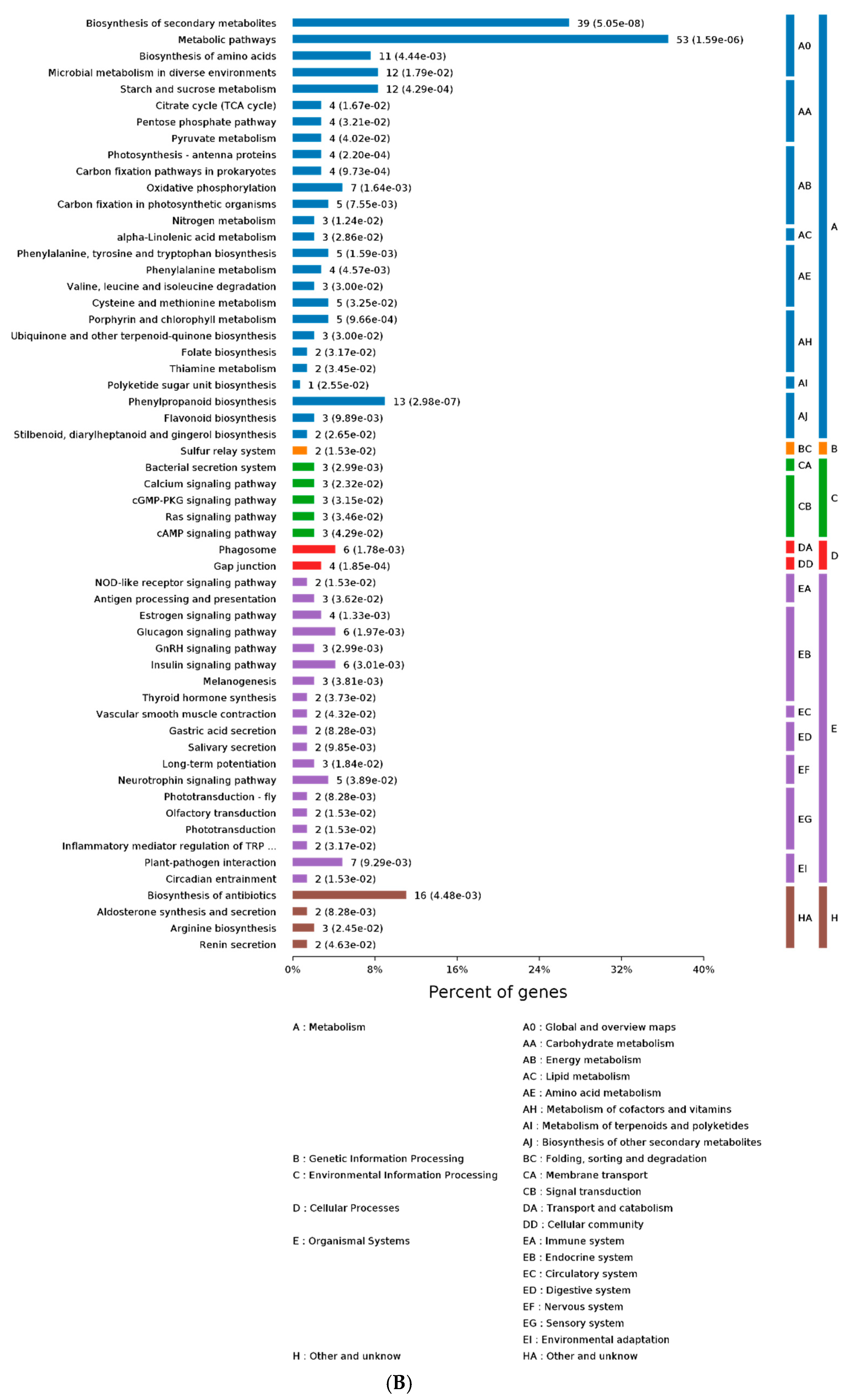
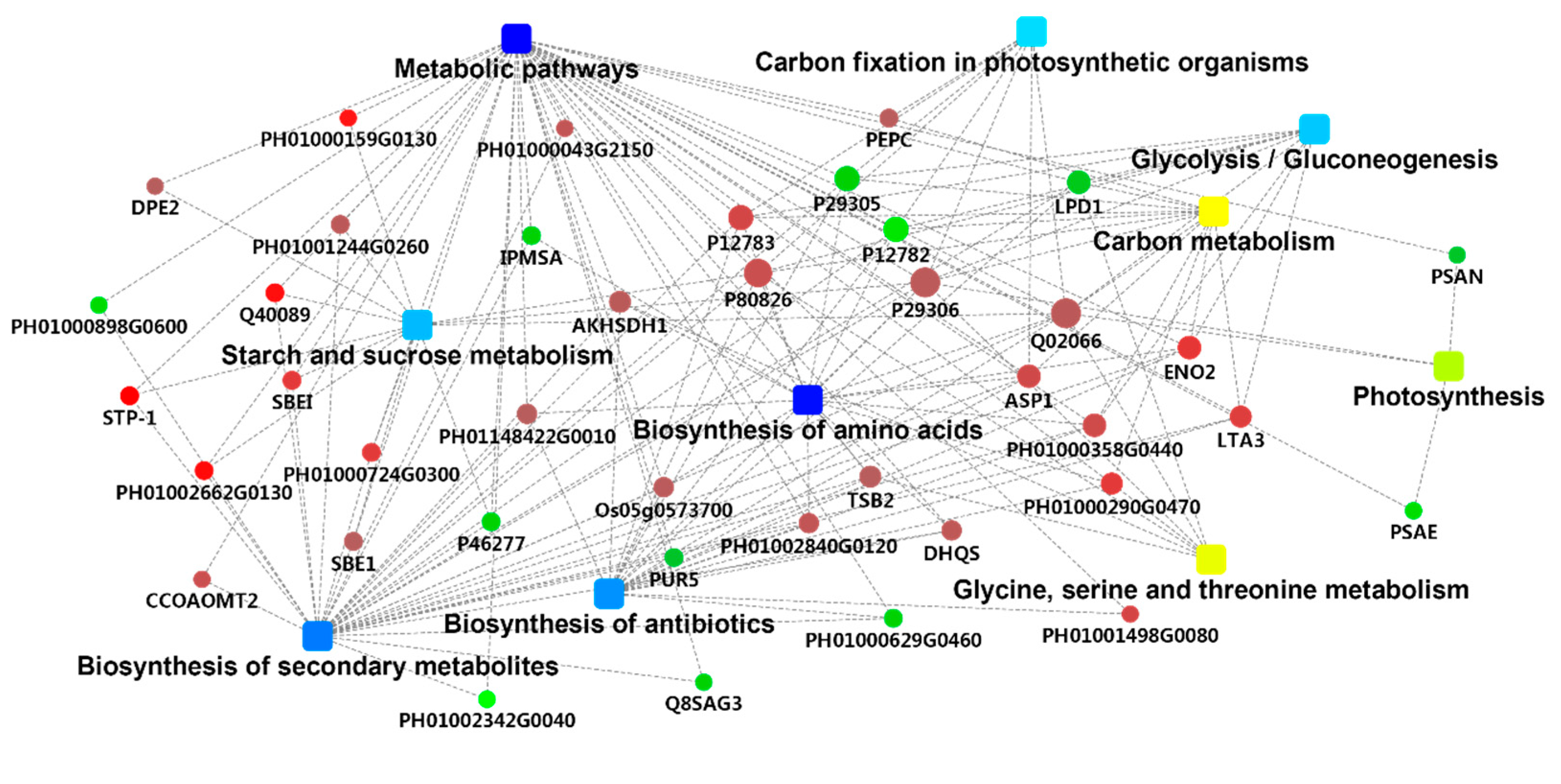
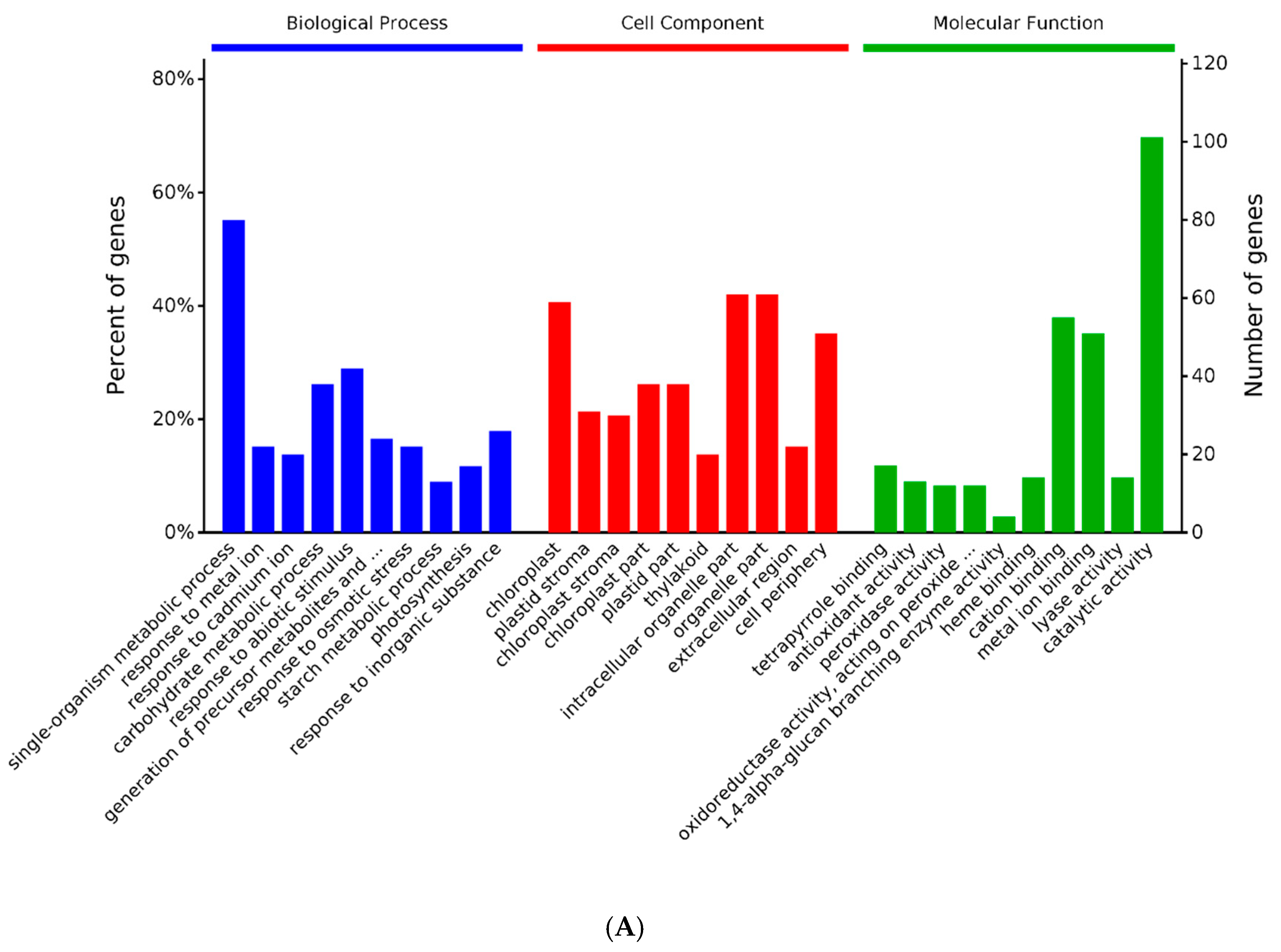
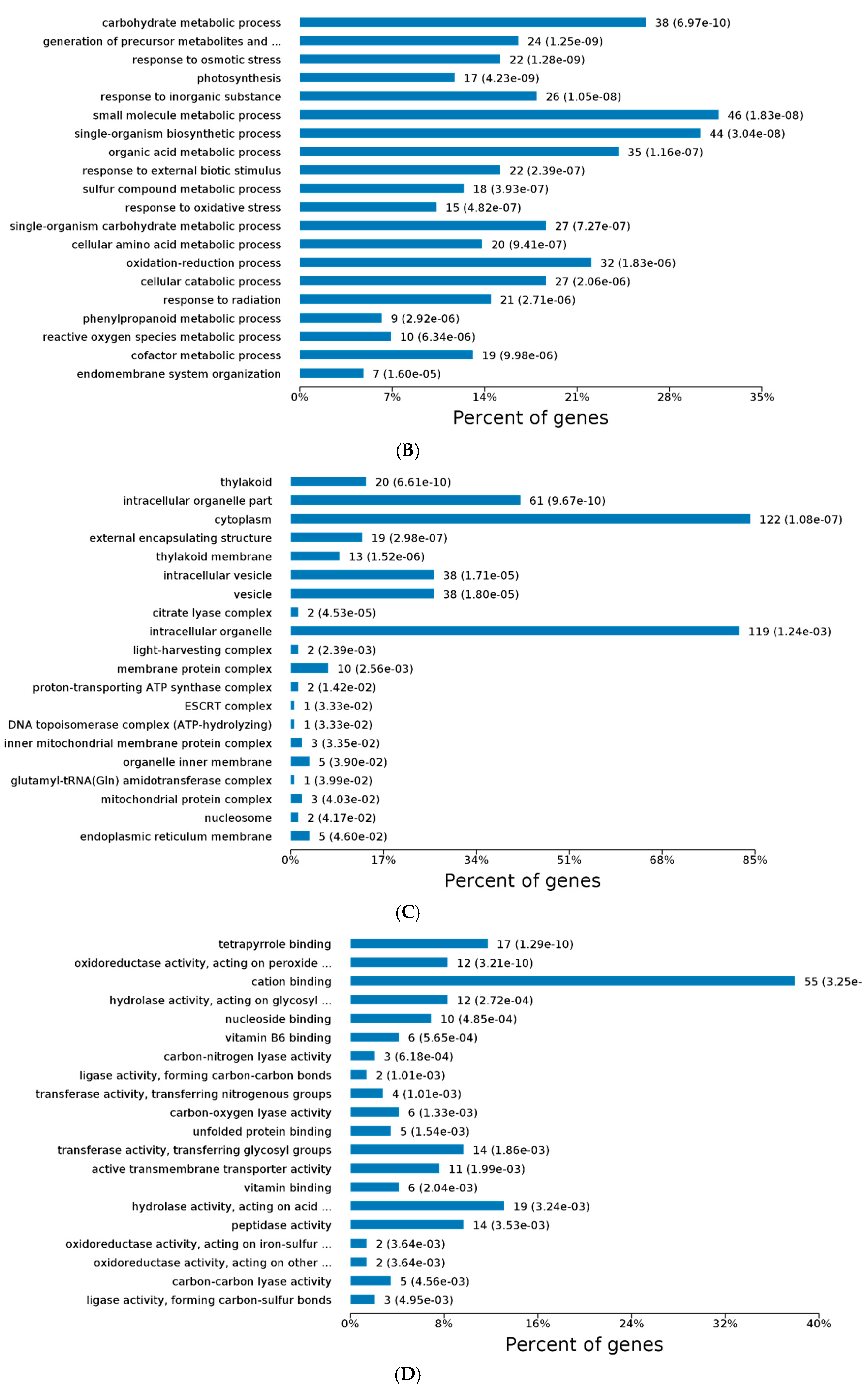
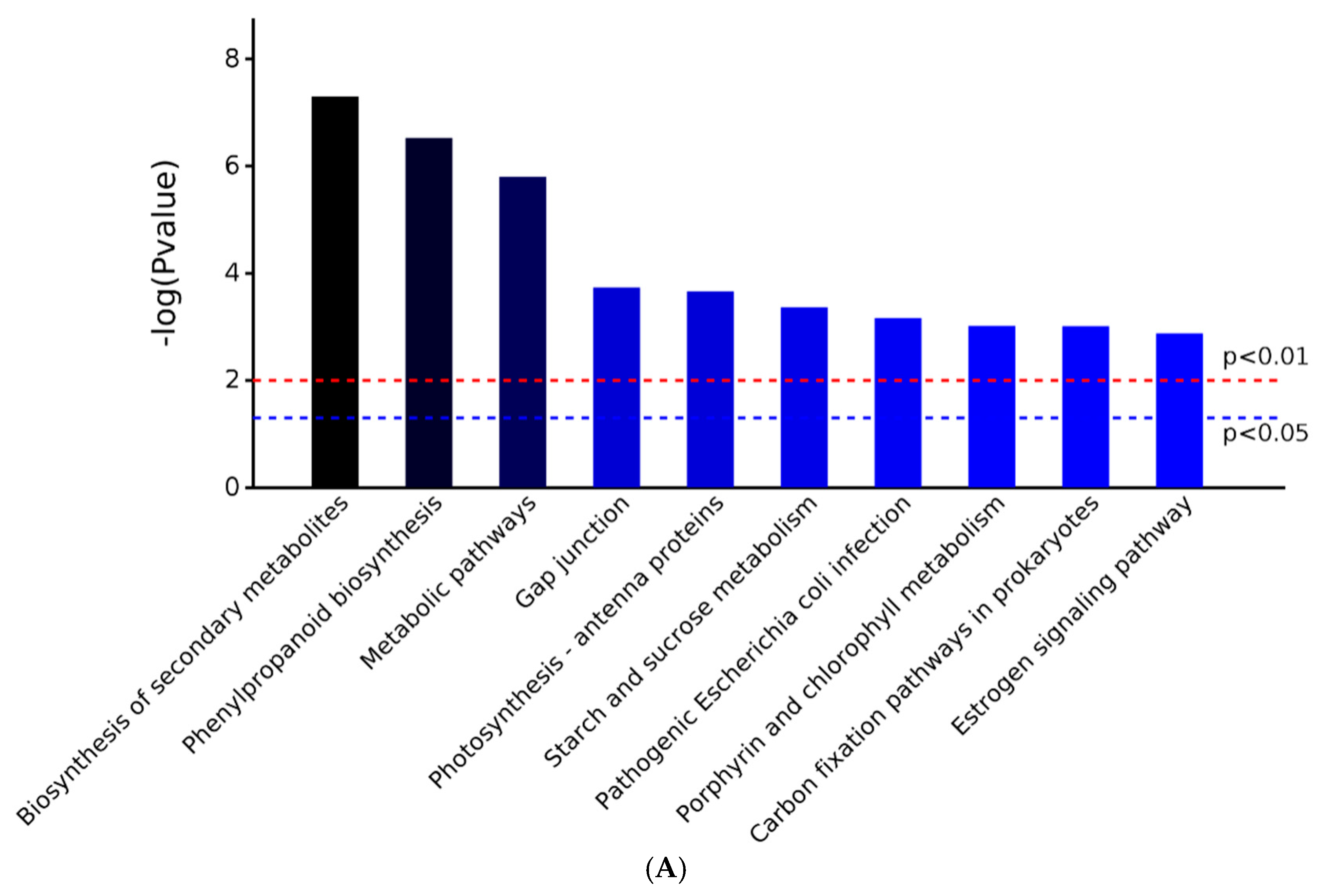
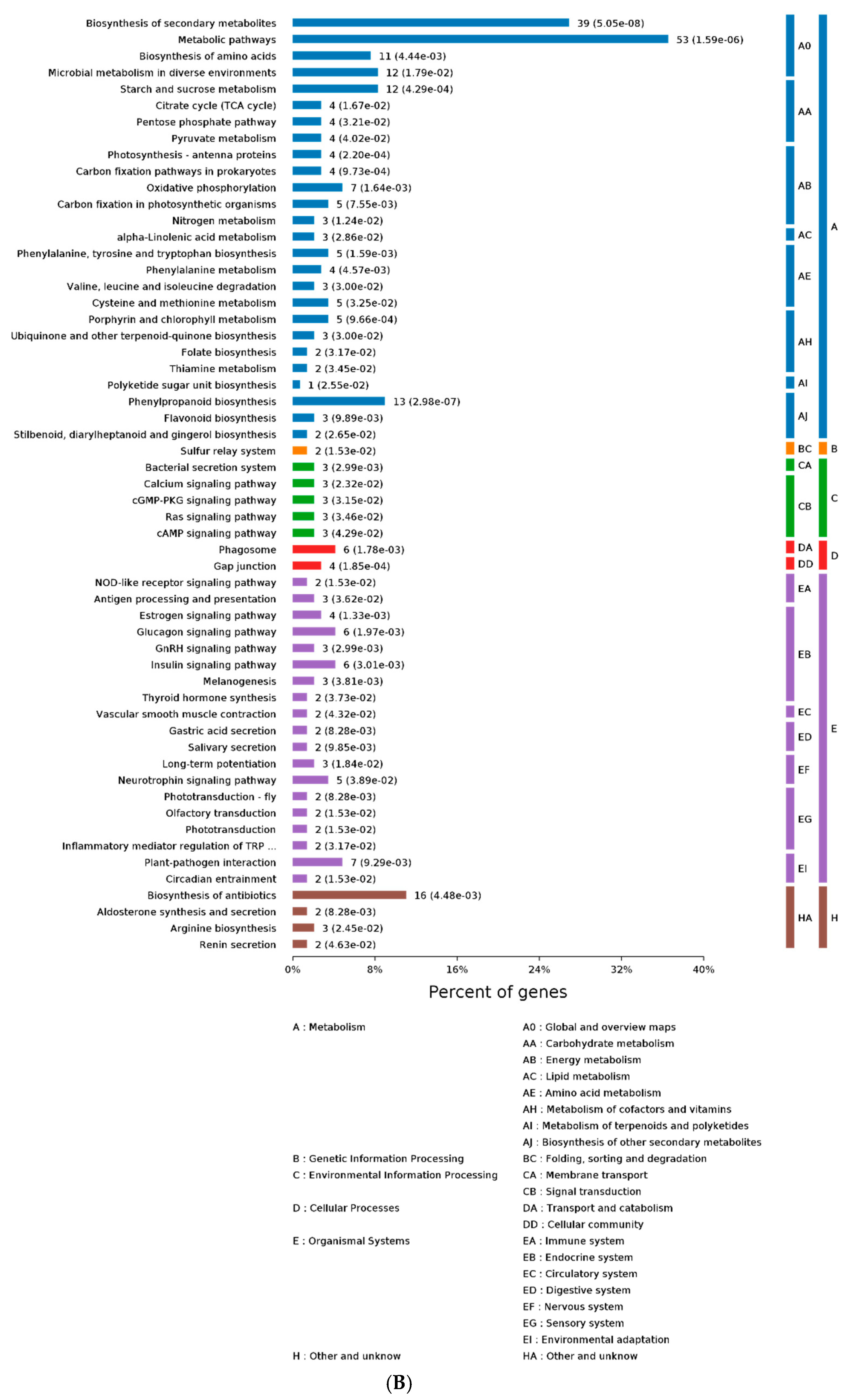
2.3. GO and KEGG Analysis of Differentially Expressed Proteins
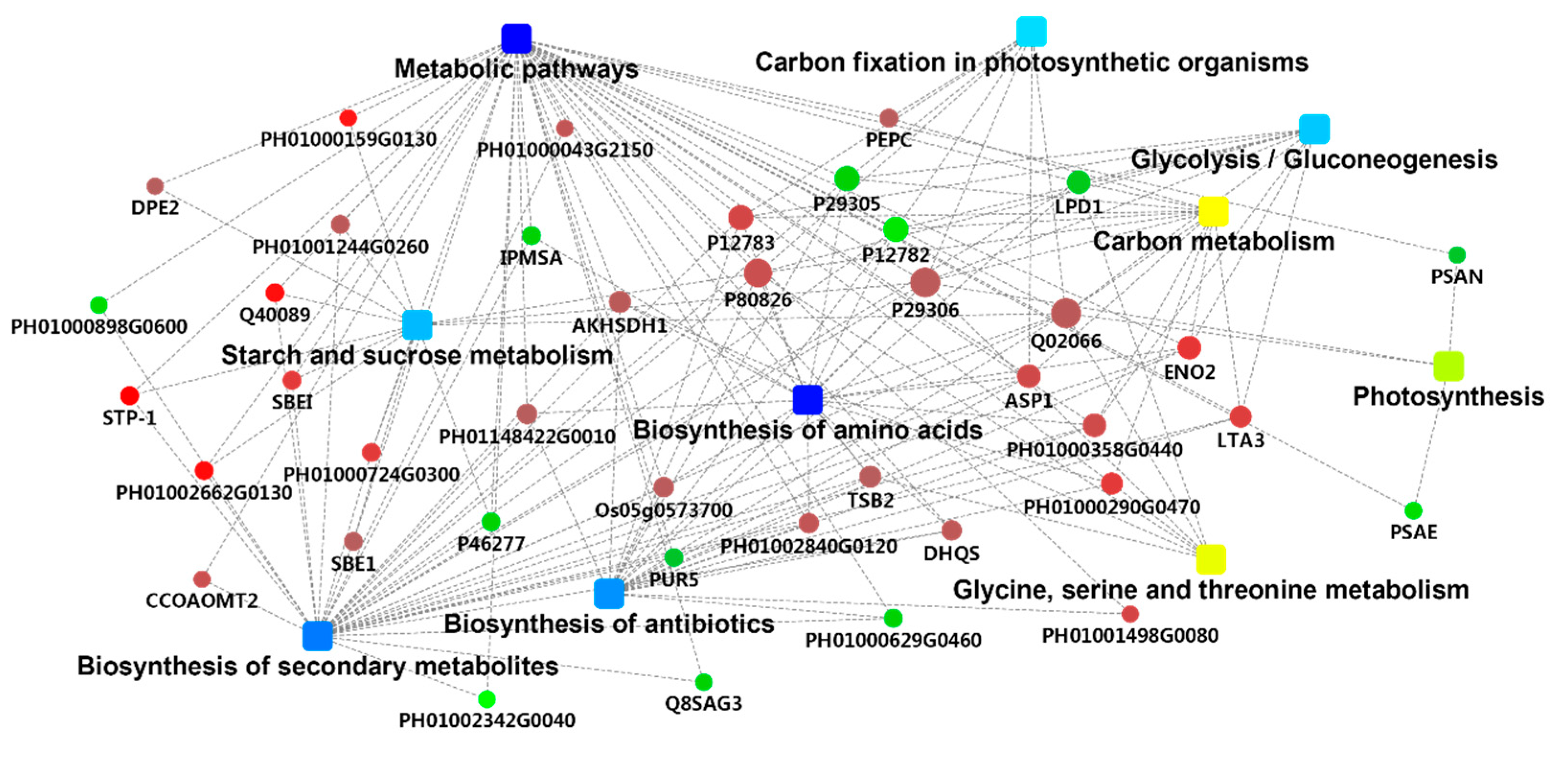
2.4. PPI Analysis
2.5. Mass Spectrometry Analysis of PRM Validation Candidate Peptides
2.6. PRM Verification of the Top Candidates for Differentially Expressed Proteins
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Plant Sample Processing
4.3. Protein Extraction, Enzymatic Hydrolysis and Peptide Quantification
4.4. TMT Peptide Labeling and Peptide Grading
4.5. Liquid Chromatography Mass Spectrometry (LC-MS/MS) Analysis
4.6. Mass Spectrometry Data Analysis
4.7. Bioinformatics Analysis
4.8. PRM Verification
4.9. Data Analysis Software
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bronwyn, J.B.; Thelma, C.C.; Jose, L.D.; Andrea, M.; Hans, P.M.; Francisco, P.A.; Ghasem, H.S.; Katja, W.; Christian, Z. Elucidation of salt stress defense and tolerance mechanisms of crop plants using proteomics-current achievements and perspectives. Proteomics 2013, 13, 1885–1900. [Google Scholar]
- Wang, Q.G.; Li, Z.; Liu, W.; Pan, J. Studies on drought-responsive proteins of foxtail millet by applying quantitative proteomic. Shandong Agric. Sci. 2018, 50, 1–8. [Google Scholar]
- Zhong, Y. Research of Transcription Genomics and Proteomics of Citrus Induced by Candidatus Liberibacter Asiaticus. Ph.D. Thesis, Hunan Agricultural University, Changsha, China, 2012. [Google Scholar]
- Chen, Q.Z.; Guo, W.S.; Feng, L.Z.; Miao, S.H.; Lin, J.S. Pathogenic Identification of Cylindro Cladium Leaf Blight Collected from Fujian Province and Studies on Transcriptomics and Proteomics of Eucalyptus Induced by Calonectria Pseudoreteaudii. Ph.D. Thesis, Fujian Agriculture and Forestry University, Fuzhou, China, 2013. [Google Scholar]
- González-Fernández, R.; Prats, E.; Jorrín-Novo, V.J. Proteomics of plant pathogenic fungi. J. Biomed. Biotechnol. 2010, 2010, 932527. [Google Scholar] [CrossRef] [PubMed]
- Rustagi, A.; Singh, G.; Agrawal, S.; Gupta, P.K. Proteomic studies revealing enigma of plant–pathogen interaction. In Molecular Aspects of Plant-Pathogen Interaction; Springer: Berlin, Germany, 2018; pp. 239–264. [Google Scholar]
- Yang, Y.L.; Xiao, L.T.; Hu, X.Q. Study on the relationship between the toxin of Phytophthora infestans (Mont.) de bary and resistance of potato. Agric. Sci. China 2011, 10, 238–245. [Google Scholar] [CrossRef]
- Hui, Q.; Liu, H.P.; Han, J.C. Induce of crude toxin from Cladosporium fulvum on defense enzyme and activity oxygen of tomato seedlings. North China Agric. J. 2009, 24, 215–218. [Google Scholar]
- Ma, C.H.; Zhai, C.X.; Wang, L.A.; Chen, X.; Li, Y.C.; Guo, X.L.; Cui, S.P.; Li, G.M. Induced resistance by the toxin filtrate of Bipolaris maydis race T cultivation. Agric. Sci. China 2006, 5, 678–684. [Google Scholar] [CrossRef]
- Conrath, U.; Thulke, O.; Katz, V.; Schwindling, S.; Kohler, A. Priming as a mechanism in induced systemic resistance of plant. Eur. J. Plant Pathol. 2001, 107, 113–119. [Google Scholar] [CrossRef]
- Zhu, T.H.; Huang, Z.C.; Gao, Q.Z.; Mou, Y.M.; Luo, Y.Q.; Mao, X.T.; Li, X.D. Research on survey and space distribution of Bambusa pervariabilis × Dendrocalamopsis daii blight. J. Sichuan For. Sci. Technol. 2009, 30, 29–31, 37. [Google Scholar]
- Zhu, J.H. Preliminarily approaching the relationship between the universal incidence and gravity of Ceratosphaeria phyllostachydis. J. Fujian For. Sci. Tech. 1997, 24, 43–46. [Google Scholar]
- Ma, G.L.; Hu, G.B.; Yu, C.Z.; Wu, J.L.; Xu, B.C. Phyllostachys prominens plum shoot wilt pathogenic fungoid and its biological characteristics. J. Zhejiang For. Coll. 2003, 20, 44–48. [Google Scholar]
- Li, S.J. Protein toxin produced from Arthrinium Phaeospermum Causing Blight in Bambusa Pervariabilis × Dendrocalamopsis Grandis and Its Accurate Pathogenic Mechanism. Ph.D. Thesis, Sichuan Agricultural University, Chengdu, China, July 2013. [Google Scholar]
- Li, S.J.; Zhu, T.H. Purification of the toxin protein Pc from Arthrinium phaeospermum and its effect on the defence enzymes of Bambusa pervariabilis × Dendrocalamopsis grandis varieties. For. Pathol. 2014, 44, 96–106. [Google Scholar] [CrossRef]
- Li, S.J.; Zhu, T.H.; Zhu, H.M.Y.; Liang, M.; Qiao, T.M.; Han, S.; Che, G.N. Purification of protein AP-toxin from Arthrinium phaeospermum causing blight in Bambusa pervariabilis × Dendrocalamopsis grandis and its metabolic effects on four bamboo varieties. Phytopathology 2013, 103, 135–145. [Google Scholar] [CrossRef]
- He, Q.Q.; Liu, Y.X.; Fang, X.M.; Zhu, T.H.; Qiao, T.M.; Han, S.; Li, S.J. Screening of induced factor of Bambusa pervariabilis × Dendrocalamopisis grandis against Arthrinium phaeospermum and its duration. Plant Prot. 2019, 45, 81–89. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon J, J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteom. 2012, 11, 1475–1488. [Google Scholar] [CrossRef]
- Overall, C.M. Can proteomics fill the gap between genomics and phenotypes? J. Proteom. 2014, 100, 1–2. [Google Scholar] [CrossRef]
- Shoresh, M.; Harman, G.E.; Mastouri, F. Induced systemic resistance and plant responses to fungal biocontrol agents. Annu. Rev. Phytopathol. 2010, 48, 21–43. [Google Scholar] [CrossRef]
- Yu, Z.M.; Li, J.Y.; Lin, Z.H.; Zhang, Q.; He, H.B. Research advances on induced-defense of plant resistance. J. Agric. Sci. 2013, 34, 69–76. [Google Scholar]
- Lan, P.; Li, W.; Wen, T.N.; Shiay, J.Y.; Wu, Y.C.; Lin, W.; Schmidt, W. iTRAQ protein profile analysis of Arabidopsis roots reveals new aspects critical for iron homeostasis. Plant Physiol. 2011, 155, 821–834. [Google Scholar] [CrossRef]
- Fan, J.; Chen, C.; Yu, Q.; Brlansky, R.H.; Li, Z.J.; Gmitter, G.G.J. Comparative iTRAQ proteome and transcriptome analyses of sweet orange infected by “Candidatus liberibacter asiaticus”. Physiol. Plantarum 2011, 143, 235–245. [Google Scholar] [CrossRef]
- Lippert, D.N.; Ralph, S.G.; Phillips, M.; Phillips, M.; White, R.; Smith, D.; Hardie, D.; Gershenzon, J.; Ritland, K.; Borchers, C.H.; et al. Quantitative iTRAQ proteome and comparative transcriptome analysis of elicitor-induced Norway spruce (Picea abies) cells reveals elements of calcium signaling in the early conifer defense response. Proteomics 2009, 9, 350–367. [Google Scholar] [CrossRef]
- Duffey, S.; Stout, M. Antinutritive and toxic components of plant defense against insects. Arch. Insect Biochem. Physiol. 1996, 32, 3–37. [Google Scholar] [CrossRef]
- Felton, G.W. Nutritive quality of plant protein; sources of variation and insect herbivore responses. Arch. Insect Biochem. Physiol. 1996, 32, 107–130. [Google Scholar] [CrossRef]
- Wang, J.; Constabel, C. Polyphenol oxidase overexpression in transgenic Populus enhances resistance to herbivory by forest tent caterpillar (Malacosoma disstria). Planta 2004, 220, 87–96. [Google Scholar] [CrossRef]
- Henrik, U.S.; Barry, R.P.; Jürgen, K.; Kerstin, W.; Jacqueline, F.; Antje, B.; Thomas, M.O. Induced plant defense responses against chewing insects. Ethylene signaling reduces resistance of Arabidopsis against Egyptian cotton worm but not diamondback moth. Plant Physiol. 2000, 124, 1007–1017. [Google Scholar]
- Timpa, J.D.; Burke, J.J.; Quiseberry, J.E.; Wendt, C.W. Effects of water stress on the organic acid and carbohydrate compositions of cotton plants. Plant Physiol. 1986, 3, 724–728. [Google Scholar] [CrossRef]
- Francoise, F.; Daniel, L.R.; John, G.S. Effects of salt stress on amino acid, organic acid, and carbohydrate composition of roots, bacteroids and cytosol of alfalfa (Medicago sativa L.). Plant Physiol. 1991, 96, 1228–1236. [Google Scholar]
- Ghosh, D.; Xu, J. Abiotic stress responses in plant roots: A proteomics perspective. Front. Plant Sci. 2014, 5, 6. [Google Scholar] [CrossRef]
- Wang, X.; Fan, P.; Song, H.; Chen, X.; Li, X.; Li, Y. Comparative proteomic analysis of differentially expressed proteins in shoots of Salicornia europaea under different salinity. J. Proteome Res. 2009, 8, 3331–3345. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, H.; Wang, T.; Chen, S.X.; Dai, S.J. Proteomics-based investigation of salt-responsive mechanisms in plant roots. J. Proteomics 2013, 82, 230–253. [Google Scholar] [CrossRef]
- Waduwara-Jayabahu, I.; Oppermann, Y.; Wirtz, M.; Hull, Z.T.; Schoor, S.; Plotnikov, A.N.; Hell, R.; Sauter, M.; Moffatt, B.A. Recycling of methylthioadenosine is essential for normal vascular development and reproduction in Arabidopsis. Plant Physiol. 2012, 158, 1728–1744. [Google Scholar] [CrossRef]
- Zheng, B.B.; Fang, Y.N.; Pan, Z.Y.; Sun, L.; Deng, X.X.; Grosser, J.W.; Guo, W.W. iTRAQ-based quantitative proteomics analysis revealed alterations of carbohydrate metabolism pathways and mitochondrial proteins in a male sterile cybrid pummelo. J. Proteome Res. 2014, 13, 2998–3015. [Google Scholar] [CrossRef]
- Coleman, H.D.; Yan, J.; Mansfield, S.D. Sucrose synthase affects carbon partitioning to increase cellulose production and altered cell wall ultrastructure. Proc. Natl. Acad. Sci. USA 2009, 106, 13118–13123. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.L. Sucrose metabolism: Gateway to diverse car-bon use and sugar signaling. Annu. Rev. Plant. Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef]
- Xu, S.M.; Brill, E.; Llewellyn, D.J.; Llewellyn, D.J.; Furbank, R.T.; Ruan, Y.L. Overexpression of a potato sucrose synthase gene in cotton accelerates leaf expansion, reduces seed abortion, and enhances fiber production. Mol. Plant 2012, 5, 430–441. [Google Scholar] [CrossRef]
- Shewmaker, C.K.; Boyer, C.D.; Wiesenborn, D.P.; Thompson, D.B.; Boersig, M.R.; Oakes, J.V.; Stalker, D.M. Expression of Escherichia coli glycogen synthase in the tubers of transgenic potatoes (Solanum tuberosum) results in a highly branched starch. Plant Physiol. 1994, 104, 1159–1166. [Google Scholar] [CrossRef]
- Li, Y.Q.; Zhao, S.B.; Ni, K.C.; Chen, W.R.; Xin, D.D.; Guo, W.D. Studies on differentially expressed genes in flower bud during dormancy release in Prunus pseudocerasus Lindl. ‘duanbing’. Acta Hortic. Sin. 2012, 39, 1253–1262. [Google Scholar]
- Schmidtmann, E.; Konig, A.C.; Orwat, A.; Leister, D.; Hartl, M.; Finkemeier, I. Redox regulation of Arabidopsis mitochondrial citrate synthase. Mol. Plant 2014, 7, 156–169. [Google Scholar] [CrossRef]
- Yang, J.L. Effect of l-Glutamate on Inhibiting Postharvest Diseases by Inducing Resistance in Fruit and the Possible Defense Mechanisms Involved. Ph.D. Thesis, Zhejiang Univercity, Hangzhou, China, 2017. [Google Scholar]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Van Breusegem, F. Reactive oxygen gene network of plants. Trends. Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef]
- Sun, F. Effects of Chilling Stress on Physiological Metabolism Protein Expression in Chloroplasts from Settlings of Sugarcane. Ph.D. Thesis, Guangxi university, Nanning, China, 2011. [Google Scholar]
- Song, L.T. Study on the relations of esterase, peroxidase and cold hardiness of winter rape (B. napu L.). J. Anhui Agric. Sci. 1995, 23, 333–336. [Google Scholar]
- Ma, X.J.; Zhu, D.H. Functional roles of the plant superoxide dismutase. Hereditas 2003, 25, 225–231. [Google Scholar]
- Avdiushko, S.A.; Ye, X.; Kuc, J. Detection of several enzymatic activities in leaf prints of cucumber plants. Physiol. Mol. Plant P 1993, 42, 441–454. [Google Scholar] [CrossRef]
- Zhu, J.X.; Sun, J.J.; Yuan, X.L.; Wang, J.; Yang, Y.M.; Wang, Y. Cloning and Sequence Analysis of Cinnamoyl Alcohol Dehydrogenase Gene from Guadua paraguayana. J. West China For. Sci. 2017, 1, 6–11. [Google Scholar]
- Whetten, R.W.; Mackay, J.J.; Sederoff, R.R. Recent advances in understanding lignin biosynthesis. Annu. Rev. Plant Physiol. Plant Mol Biol. 1998, 1, 585–609. [Google Scholar] [CrossRef]
- Frey, M.; Chomet, P.; Glawischnig, E.; Stettner, C.; Grun, S.; Winklmair, A.; Eisenreich, W.; Bacher, A.; Meeley, R.B.; Briggs, S.P.; et al. Analysis of a chemical plant defense mechanism in grasses. Science 1997, 277, 696–699. [Google Scholar] [CrossRef]
- Yoda, H.; Fujimura, K.; Takahashi, H.; Munemura, I.; Uchimiya, H.; Sano, H. Polyamines as a common source of hydrogen peroxide in host-and nonhost hypersensitive response during pathogen infection. Plant Mol. Biol. 2009, 70, 103–112. [Google Scholar] [CrossRef]
- Bell, A.A. Biochemical mechanism of disease resistance. Ann. Rev. Plant Physiol. 1981, 32, 21–81. [Google Scholar] [CrossRef]
- Qiu, Y.X.; Ke, Y.Q.; Dai, H.J.; Pan, T.G. Phenolics metabolism in sweet potato infected by Fusarium oxysporum Schlecht. f. sp. Chin. J. Eco Agric. 2007, 15, 167–170. [Google Scholar]
- Ritter, H.; Schulz, G.E. Structural basis for the entrance into the phenylpropanoid metabolism catalyzed by phenylalanine ammonia-lyase. Plant Cell 2004, 16, 3426–3436. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.F.; Yang, C.Y.; Bai, M.Z.; Shu, K.X.; Li, K.L.; Zhang, G.Q.; Jin, Z.; He, F.C.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2018, 47, D1211–D1217. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of largescale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Result | Unique Peptides | Quantified Proteins | Up-Regulated | Down-Regulated | Significantly Different Proteins |
|---|---|---|---|---|---|
| Total | 3320 | 1791 | 75 | 88 | 163 |
| Protein ID | Description | KEGG Pathways |
|---|---|---|
| PH01000123G1100 | porphobilinogen deaminase | Biosynthesis of secondary metabolites |
| Metabolic pathways | ||
| PH01000159G0130 | sucrose synthase | Starch and sucrose metabolism |
| PH01000713G0340 | Adenosine triphosphate (ATP)-citrate synthase beta chain protein 1 | Citrate cycle (Tricarboxylic acid (TCA) cycle) |
| Energy production and conversion | ||
| PH01000761G0570 | Peroxidase | Phenylpropanoid biosynthesis |
| Carbohydrate transport and metabolism | ||
| PH01003309G0170 | Phenylalanine ammonia-lyase | Phenylalanine metabolism |
| Phenylpropanoid biosynthesis | ||
| Secondary metabolites biosynthesis, transport and catabolism |
| Protein Name | Peptide Sequence | AP-Toxin + A. phaeospermum | Sterile Water + A. phaeospermum | PRM Fold Change | TMT Fold Change | Consistency between PRM and TMT |
|---|---|---|---|---|---|---|
| PH01000087G1730 | AVAHQPVSVAIEAGGR | 1,756,917.422 | 1,409,646.38 | 1.25 | 1.52 | Yes |
| PH01000123G1100 | TTGDMILDKPLADIGGK | 3,128,973.576 | 3,315,437.047 | 0.94 | 1.62 | No |
| PH01000133G0340 | LVENDEVVR | 6,132,972.898 | 4710,029.37 | 1.30 | 2.01 | Yes |
| PH01000159G0130 | QQGLNITPR | 30,652,687.98 | 5,578,763.154 | 5.49 | 3.27 | Yes |
| PH01000713G0340 | FGGAIDDAAR | 21,475,599.31 | 8,257,753.841 | 2.60 | 1.63 | Yes |
| PH01000761G0570 | NNPSDIDPSLNPSYAK | 4,349,530.158 | 1,633,319.523 | 2.66 | 1.59 | Yes |
| PH01000860G0570 | DVDLSTYK | 2,738,630.087 | 1,653,255.404 | 1.66 | 1.54 | Yes |
| PH01000898G0600 | MGNINPLTGTAGQIR | 66,407,505.35 | 444,831,682.9 | 0.15 | 0.35 | Yes |
| PH01001064G0040 | EHLIAGWAPK | 7,994,610.908 | 4,179,165.695 | 1.91 | 1.44 | Yes |
| PH01001724G0160 | VNVYYNEASCGR | 4,996,071.907 | 3,097,515.346 | 1.61 | 1.57 | Yes |
| PH01001918G0090 | APDFEAEAVFDQEFIK | 1,768,290.264 | 3,727,484.2 | 0.47 | 0.64 | Yes |
| PH01002295G0170 | VVVSSCGHDGPFGATGVK | 11,037,762.06 | 6,282,695.56 | 1.76 | 1.45 | Yes |
| PH01003309G0170 | VGQVAAVAQAK | 63,790,370.98 | 19,648,147.32 | 3.25 | 2.24 | Yes |
| PH01100083G0010 | YFSAAASQALDTAER | 2,771,584.723 | 16,486,693.42 | 0.17 | 0.46 | Yes |
| Item | Value |
|---|---|
| Type | Reporter ion MS2 |
| Isobaric labels | TMT 6plex |
| Enzyme | Trypsin |
| Reporter mass tolerance | 0.005 Da |
| Max missed cleavages | 2 |
| Main search peptide tolerance | 4.5 ppm |
| First search peptide tolerance | 20 ppm |
| Mass spectrometry (MS) /MS tolerance | 20 ppm |
| Fixed modifications | Carbamidomethyl (C) |
| Variable modifications | Oxidation (M), Acetyl (Protein N-term) |
| Database | Bamboo.fasta |
| Database pattern | Target-Reverse |
| Peptide spectrum matches false discovery rate (PSM FDR) | ≤0.01 |
| Protein FDR | ≤0.01 |
| Protein quantification | Razor and unique peptides were used for protein quantification. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Q.; Fang, X.; Zhu, T.; Han, S.; Zhu, H.; Li, S. Differential Proteomics Based on TMT and PRM Reveal the Resistance Response of Bambusa pervariabilis × Dendrocalamopisis grandis Induced by AP-Toxin. Metabolites 2019, 9, 166. https://doi.org/10.3390/metabo9080166
He Q, Fang X, Zhu T, Han S, Zhu H, Li S. Differential Proteomics Based on TMT and PRM Reveal the Resistance Response of Bambusa pervariabilis × Dendrocalamopisis grandis Induced by AP-Toxin. Metabolites. 2019; 9(8):166. https://doi.org/10.3390/metabo9080166
Chicago/Turabian StyleHe, Qianqian, Xinmei Fang, Tianhui Zhu, Shan Han, Hanmingyue Zhu, and Shujiang Li. 2019. "Differential Proteomics Based on TMT and PRM Reveal the Resistance Response of Bambusa pervariabilis × Dendrocalamopisis grandis Induced by AP-Toxin" Metabolites 9, no. 8: 166. https://doi.org/10.3390/metabo9080166
APA StyleHe, Q., Fang, X., Zhu, T., Han, S., Zhu, H., & Li, S. (2019). Differential Proteomics Based on TMT and PRM Reveal the Resistance Response of Bambusa pervariabilis × Dendrocalamopisis grandis Induced by AP-Toxin. Metabolites, 9(8), 166. https://doi.org/10.3390/metabo9080166