Biotransformation of the Mycotoxin Enniatin B1 by CYP P450 3A4 and Potential for Drug-Drug Interactions

Abstract
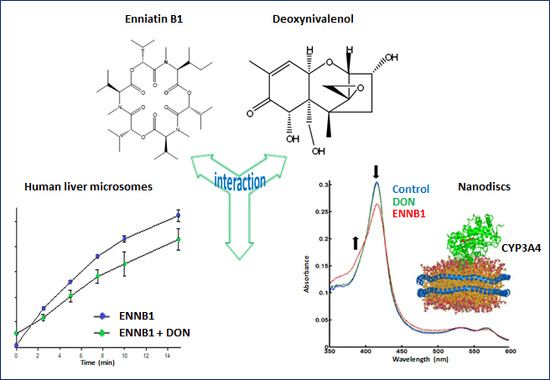
1. Introduction
2. Results
2.1. Determination of Kinetic Parameters of ENNB1 Depletion in HLM
2.2. Biotransformation Products of ENNB1 in HLM
2.3. Role of CYP3A4/5 in ENNB1 Biotransformation
2.3.1. Decrease of ENNB1 Metabolism with CYP3A4/5-Specific Inhibitor
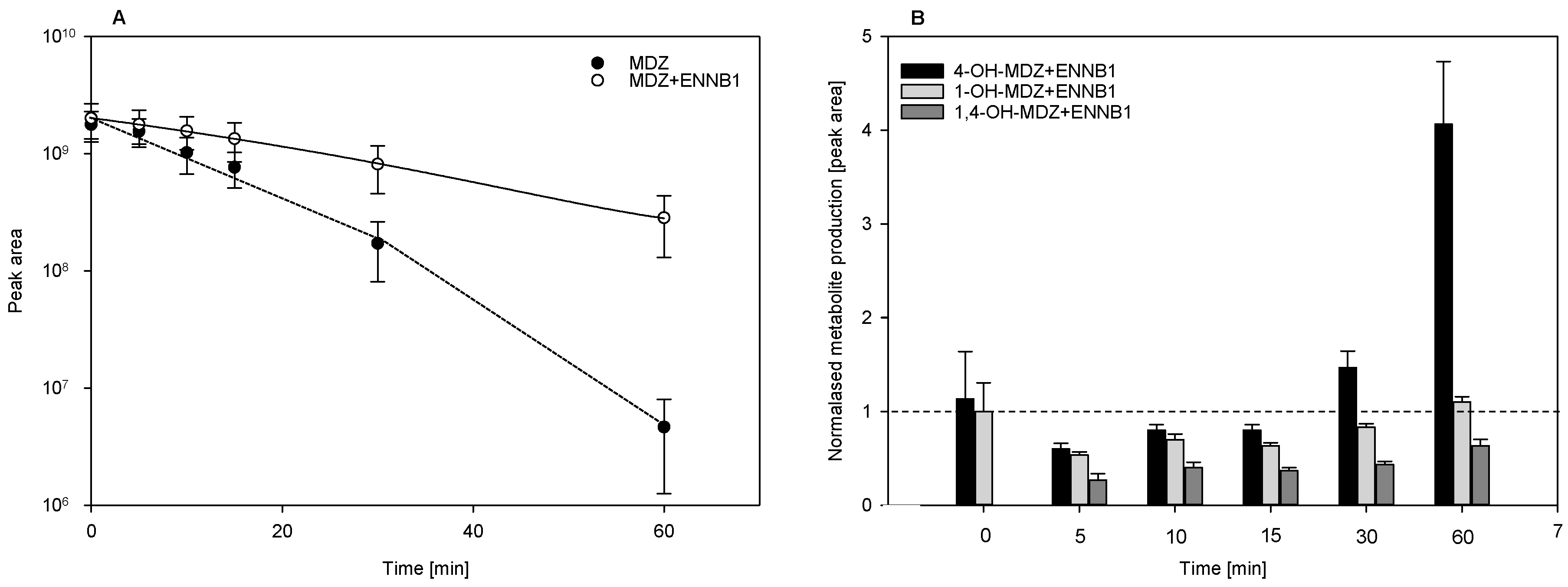
2.3.2. ENNB1 as Inhibitor of Midazolam Biotransformation
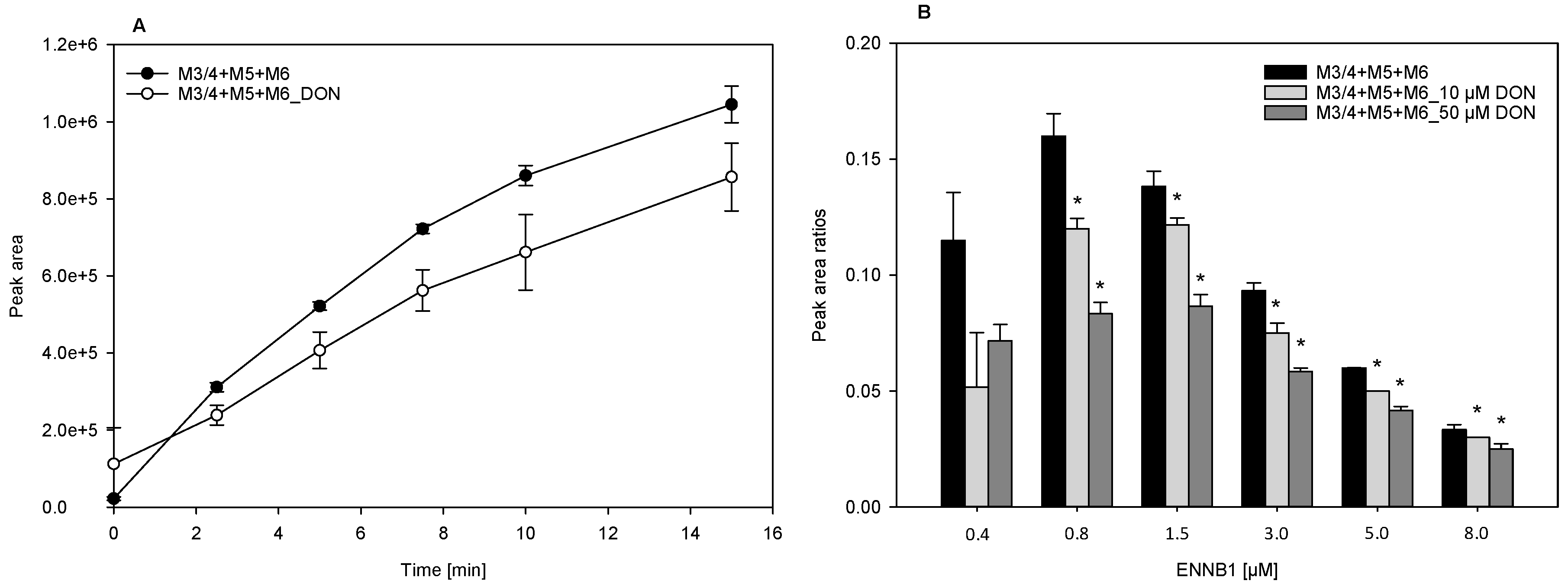
2.4. DON Decreases the Formation of ENNB1 Metabolites in HLM and ND
2.4.1. DON Decreases the Hydroxylation Efficiency of Progesterone and Atorvastatin Lactone in ND
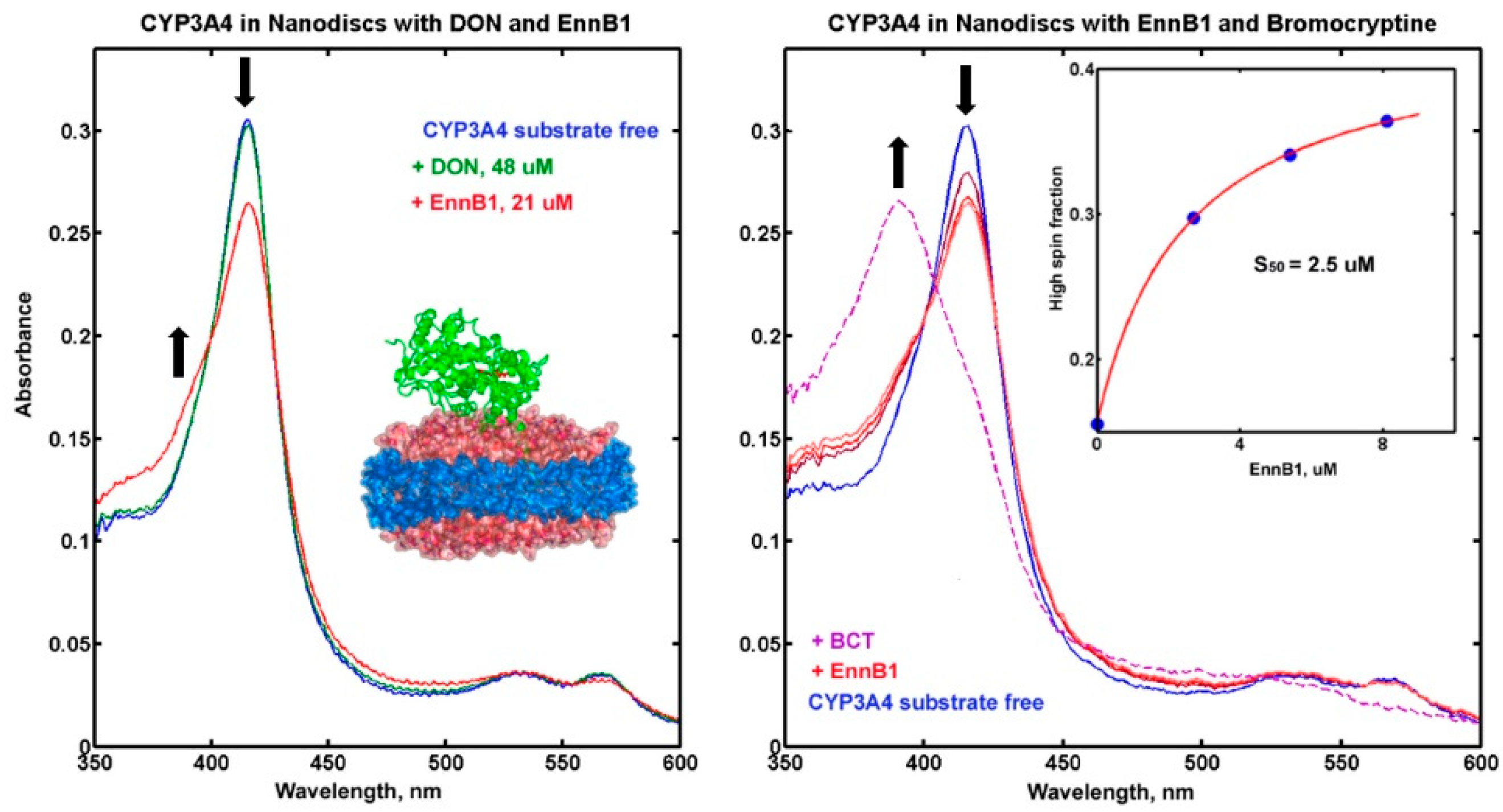
2.4.2. Binding of DON and ENNB1 to CYP3A4 in Nanodiscs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Microsomal Incubations
4.2.1. Performance of Metabolism Experiments
4.2.2. In Vitro-to-In Vivo Extrapolation of Kinetic Parameters
4.3. Incubations in Nanodiscs
4.3.1. Protein Expression and Purification of Functional Nanodiscs
4.3.2. Titration of Substrate Binding to CYP3A4/CPR-ND
4.3.3. Interference of DON with the Metabolism of Typical CYP3A4-s Substrates in Functional ND
4.4. Liquid Chromatography-Iontrap Mass Spectrometry (LC-ITMS)
4.5. Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS)
4.6. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging mycotoxins: Beyond traditionally determined food contaminants. J. Agric. Food Chem. 2017, 65, 7052–7070. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.; Meca, G.; Uhlig, S.; Ritieni, A. Fusaproliferin, beauvericin and enniatins: Occurrence in food—A review. World Mycotoxin J. 2012, 5, 71–81. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA); EFSA Panel on Contaminants in Food Chain (CONTAM). Scientific opinion on the risk to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J. 2014, 12, 3802–3976. [Google Scholar] [CrossRef]
- Prosperini, A.; Berrada, H.; Ruiz, M.J.; Caloni, F.; Coccini, T.; Spicer, L.J.; Perego, M.C.; Lafranconi, A. A Review of the Mycotoxin Enniatin B. Front. Public Health 2017, 5, article 304. [Google Scholar] [CrossRef] [PubMed]
- Maranghi, F.; Tassinari, R.; Narciso, L.; Tait, S.; La Rocca, C.; Di Felice, G.; Butteroni, C.; Corinti, S.; Barletta, B.; Cordelli, E.; et al. In vivo toxicity and genotoxicity of beauvericin and enniatins. Combined approach to study in vivo toxicity and genotoxicity of mycotoxins beauvericin (BEA) and enniatin B (ENNB). EFSA Support. Publ. 2018, 15, EN-1406. [Google Scholar] [CrossRef]
- Dornetshuber, R.; Heffeter, P.; Lemmens-Gruber, R.; Elbling, L.; Marko, D.; Micksche, M.; Berger, W. Oxidative stress and DNA interactions are not involved in Enniatin- and Beauvericin-mediated apoptosis induction. Mol. Nutr. Food Res. 2009, 53, 1112–1122. [Google Scholar] [CrossRef]
- Ivanova, L.; Egge-Jacobsen, W.M.; Solhaug, A.; Thoen, E.; Fæste, C.K. Lysosomes as a Possible Target of Enniatin B-Induced Toxicity in Caco-2 Cells. Chem. Res. Toxicol. 2012, 25, 1662–1674. [Google Scholar] [CrossRef]
- Prosperini, A.; Juan-Garcia, A.; Font, G.; Ruiz, M.J. Reactive oxygen species involvement in apoptosis and mitochondrial damage in Caco-2 cells induced by enniatins A, A1, B and B1. Toxicol. Lett. 2013, 222, 36–44. [Google Scholar] [CrossRef]
- Behm, C.; Degen, G.H.; Föllmann, W. The Fusarium toxin enniatin B exerts no genotoxic activity, but pronounced cytotoxicity in vitro. Mol. Nutr. Food Res. 2009, 53, 423–430. [Google Scholar] [CrossRef]
- Bertero, A.; Moretti, A.; Spicer, L.J.; Caloni, F. Fusarium Molds and Mycotoxins: Potential Species-Specific Effects. Toxins 2018, 10, 244–271. [Google Scholar] [CrossRef]
- Smith, M.-C.; Madec, S.; Coton, E.; Hymery, N. Natural Co-Occurrence of Mycotoxins in Foods and Feeds and Their in vitro Combined Toxicological Effects. Toxins 2016, 8, 94–130. [Google Scholar] [CrossRef] [PubMed]
- Gutleb, A.C.; Morrison, E.; Murk, A.J. Cytotoxicity assays for mycotoxins produced by Fusarium strains: A review. Environ. Toxicol. Pharmacol. 2002, 11, 309–320. [Google Scholar] [CrossRef]
- Fernández-Blanco, C.; Font, G.; Ruiz, M.-J. Interaction effects of enniatin B, deoxinivalenol and alternariol in Caco-2 cells. Toxicol. Lett. 2016, 241, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.J.; Franzova, P.; Juan-Garcia, A.; Font, G. Toxicological interactions between the mycotoxins beauvericin, deoxynivalenol and T-2 toxin in CHO-K1 cells in vitro. Toxicon 2011, 58, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Fæste, C.K.; Uhlig, S. In vitro phase I metabolism of the depsipeptide enniatin B. Anal. Bioanal. Chem. 2011, 400, 2889–2901. [Google Scholar] [CrossRef]
- Ivanova, L.; Uhlig, S.; Devreese, M.; Croubels, S.; Fæste, C.K. Biotransformation of the mycotoxin enniatin B1 in pigs: A comparative in vitro and in vivo approach. Food Chem. Toxicol. 2017, 105, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Fæste, C.K.; Uhlig, S. Enzyme-Assisted Synthesis and Structural Characterization of the 3-, 8-, and 15-Glucuronides of Deoxynivalenol. J. Agric. Food Chem. 2013, 61, 2006–2012. [Google Scholar]
- Ivanova, L.; Uhlig, S.; Eriksen, G.S.; Johannessen, L.E. Enniatin B1 is a substrate of intestinal P-glycoprotein, multidrug resistance-associated protein 2 and breast cancer resistance protein. World Mycotoxin J. 2010, 3, 271–281. [Google Scholar] [CrossRef]
- Solhaug, A.; Stanic, A.; Uhlig, S.; Rise, F.; Wilkins, A.L.; Miles, C.O. Nucleophilic Addition of Thiols to Deoxynivalenol. J. Agric. Food Chem. 2015, 63, 7556–7566. [Google Scholar]
- Denisov, I.G.; Grinkova, Y.V.; Baylon, J.L.; Tajkhorshid, E.; Sligar, S.G. Mechanism of Drug-Drug Interactions Mediated by Human Cytochrome P450 CYP3A4 Monomer. Biochem. 2015, 54, 2227–2239. [Google Scholar] [CrossRef]
- Denisov, I.G.; Baas, B.J.; Grinkova, Y.V.; Sligar, S.G. Cooperativity in Cytochrome P450 3A4: Linkages in Substrate Binding, Spin State, Uncoupling, and Product Formation. J. Boil. Chem. 2007, 282, 7066–7076. [Google Scholar] [CrossRef] [PubMed]
- Luthra, A.; Gregory, M.; Grinkova, Y.V.; Denisov, I.G.; Sligar, S.G. Nanodiscs in the studies of membrane-bound cytochrome P450 enzymes. Bioconjugation 2013, 987, 115–127. [Google Scholar]
- Denisov, I.G.; Baylon, J.L.; Grinkova, Y.V.; Tajkhorshid, E.; Sligar, S.G. Drug-drug interactions between atorvastatin and dronedarone mediated by monomeric CYP3A4. Biochemistry 2018, 57, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Faeste, C.K.; Ivanova, L.; Uhlig, S.; Hosoda, K.; Furuta, T.; Ishii, K. In Vitro Metabolism of the Mycotoxin Enniatin B in Different Species and Cytochrome P450 Enzyme Phenotyping by Chemical Inhibitors. Drug Metab. Dispos. 2011, 39, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.C.; Hall, S.D.; Jones, D.R.; Vandenbranden, M. Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem. Pharmacol. 1994, 47, 1643–1653. [Google Scholar] [CrossRef]
- Wandel, C.; Böcker, R.; Böhrer, H.; Browne, A.; Rügheimer, E.; Martin, E. Midazolam is metabolized by at least three different cytochrome P450 enzymes. Br. J. Anaesth. 1994, 73, 658–661. [Google Scholar] [CrossRef]
- Patki, K.C. In vitro Metabolism of Midazolam, Triazolam, Nifedipine, and Testosterone by Human Liver Microsomes and Recombinant Cytochromes p450: Role of cyp3a4 and cyp3a5. Drug Metab. Dispos. 2003, 31, 938–944. [Google Scholar] [CrossRef]
- Nguyen, H.Q.; Kimoto, E.; Callegari, E.; Obach, R.S. Mechanistic Modeling to Predict Midazolam Metabolite Exposure from In vitro Data. Drug Metab. Dispos. 2016, 44, 781–791. [Google Scholar] [CrossRef]
- Denisov, I.G.; Sligar, S.G. Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Boil. 2016, 23, 481–486. [Google Scholar] [CrossRef]
- Denisov, I.G.; Sligar, S.G. Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev. 2017, 117, 4669–4713. [Google Scholar] [CrossRef]
- Devreese, M.; Broekaert, N.; De Mil, T.; Fraeyman, S.; De Backer, P.; Croubels, S. Pilot toxicokinetic study and absolute oral bioavailability of the Fusarium mycotoxin enniatin B1 in pigs. Food Chem. Toxicol. 2014, 63, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; De Baere, S.; De Backer, P.; Croubels, S. Quantitative determination of the Fusarium mycotoxins beauvericin, enniatin A, A1, B and B1 in pig plasma using high performance liquid chromatography–tandem mass spectrometry. Talanta 2013, 106, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; Antonissen, G.; Fraeyman, S.; De Baere, S.; Rychlik, M.; Croubels, S. Comparative Oral Bioavailability, Toxicokinetics, and Biotransformation of Enniatin B1 and Enniatin B in Broiler Chickens. J. Agric. Food Chem. 2016, 64, 7259–7264. [Google Scholar]
- Sevrioukova, I.F.; Poulos, T.L. Current Approaches for Investigating and Predicting Cytochrome P450 3A4-Ligand Interactions. Results Probl. Cell Differ. 2015, 851, 83–105. [Google Scholar]
- Sevrioukova, I.F.; Poulos, T.L. Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2017, 114, 486–491. [Google Scholar] [CrossRef]
- Schelstraete, W.; Devreese, M.; Croubels, S. Impact of Subacute Exposure to T-2 Toxin and Zearalenone on the Pharmacokinetics of Midazolam as CYP3A Probe Drug in a Porcine Animal Model: A Pilot Study. Front. Pharmacol. 2019, 10, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Waterman, M.R.; Egli, M. Recent Structural Insights into Cytochrome P450 Function. Trends Pharmacol. Sci. 2016, 37, 625–640. [Google Scholar] [CrossRef]
- Yamazaki, H.; Shimada, T. Progesterone and Testosterone Hydroxylation by Cytochromes P450 2C19, 2C9, and 3A4 in Human Liver Microsomes. Arch. Biochem. Biophys. 1997, 346, 161–169. [Google Scholar] [CrossRef]
- Isin, E.M.; Guengerich, F.P. Kinetics and Thermodynamics of Ligand Binding by Cytochrome P450 3A4. J. Boil. Chem. 2006, 281, 9127–9136. [Google Scholar] [CrossRef]
- Obach, R.S.; Reed-Hagen, A.E. Measurement of Michaelis Constants for Cytochrome P450-Mediated Biotransformation Reactions Using a Substrate Depletion Approach. Drug Metab. Dispos. 2002, 30, 831–837. [Google Scholar] [CrossRef]
- Ito, K.; Houston, J.B. Prediction of Human Drug Clearance from in Vitro and Preclinical Data Using Physiologically Based and Empirical Approaches. Pharm. Res. 2005, 22, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Baas, B.J.; Denisov, I.G.; Sligar, S.G. Homotropic cooperativity of monomeric cytochrome P450 3A4 in a nanoscale native bilayer environment. Arch. Biochem. Biophys. 2004, 430, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Grinkova, Y.V.; Lazarides, A.A.; Sligar, S.G. Directed Self-Assembly of Monodisperse Phospholipid Bilayer Nanodiscs with Controlled Size. J. Am. Chem. Soc. 2004, 126, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Grinkova, Y.V.; Baas, B.J.; Sligar, S.G. The Ferrous-Dioxygen Intermediate in Human Cytochrome P450 3A4: Substrate Dependence of Formation and Decay Kinetics. J. Boil. Chem. 2006, 281, 23313–23318. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Grinkova, Y.V.; McLean, M.A.; Sligar, S.G. The One-electron Autoxidation of Human Cytochrome P450 3A4. J. Boil. Chem. 2007, 282, 26865–26873. [Google Scholar] [CrossRef] [PubMed]
- Grinkova, Y.V.; Denisov, I.G.; Sligar, S.G. Functional reconstitution of monomeric CYP3A4 with multiple cytochrome P450 reductase molecules in Nanodiscs. Biochem. Biophys. Res. Commun. 2010, 398, 194–198. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | HLM * |
|---|---|
| ke [min−1] a | 0.065 |
| t1/2, assay [min] | 11 |
| KM, assay [µM] b | 10.1 |
| CLint, assay [mL/min] | 0.07 |
| CLint [L/(h×kg×BW)] | 1.72 |
| CLb [L/(h×kg×BW)] | 0.77 |
| fmax (%) | 45 |
| Substrate (µM) | Without DON 1 | DON 1, 10 μM | DON 1, 49 μM |
|---|---|---|---|
| nmol/(min×nmol CYP3A4) | nmol/(min×nmol CYP3A4) | nmol/(min×nmol CYP3A4) | |
| PGS#, 15 μM | 5.3 ± 0.6 | 5.0 ± 0.6 | 4.1 ± 0.4 |
| PGS#, 40 μM | 10.4 ± 1.0 | 9.8 ± 0.9 | 8.9 ± 0.8 |
| ATVS lactone*, 8 μM | 2.8 ± 0.2/0.47 ± 0.08 | 2.7 ± 0.25/0.48 ± 0.08 | 2.7 ± 0.16/0.52 ± 0.06 |
| ATVS lactone*, 18 μM | 3.2 ± 0.2/1.1 ± 0.1 | 3.2 ± 0.2/0.97 ± 0.12 | 3.1 ± 0.18/0.87 ± 0.13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, L.; Denisov, I.G.; Grinkova, Y.V.; Sligar, S.G.; Fæste, C.K. Biotransformation of the Mycotoxin Enniatin B1 by CYP P450 3A4 and Potential for Drug-Drug Interactions. Metabolites 2019, 9, 158. https://doi.org/10.3390/metabo9080158
Ivanova L, Denisov IG, Grinkova YV, Sligar SG, Fæste CK. Biotransformation of the Mycotoxin Enniatin B1 by CYP P450 3A4 and Potential for Drug-Drug Interactions. Metabolites. 2019; 9(8):158. https://doi.org/10.3390/metabo9080158
Chicago/Turabian StyleIvanova, Lada, Ilia G. Denisov, Yelena V. Grinkova, Stephen G. Sligar, and Christiane K. Fæste. 2019. "Biotransformation of the Mycotoxin Enniatin B1 by CYP P450 3A4 and Potential for Drug-Drug Interactions" Metabolites 9, no. 8: 158. https://doi.org/10.3390/metabo9080158
APA StyleIvanova, L., Denisov, I. G., Grinkova, Y. V., Sligar, S. G., & Fæste, C. K. (2019). Biotransformation of the Mycotoxin Enniatin B1 by CYP P450 3A4 and Potential for Drug-Drug Interactions. Metabolites, 9(8), 158. https://doi.org/10.3390/metabo9080158