Tissue-Specific Sample Dilution: An Important Parameter to Optimise Prior to Untargeted LC-MS Metabolomics

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Tissues for Sample Preparation Optimisation
2.2. Chemicals
2.3. Metabolite Extraction
2.4. Serial Dilution Experiment for Reconstitution Volume Determination
2.5. Ultra-Performance Liquid Chromatography (UPLC)-Mass Spectrometry Analysis of Lipids
2.6. Liquid Chromatography (LC)-Mass Spectrometry Analysis of Polar Metabolites
2.7. Data Processing
3. Result and Discussion
3.1. Workflow Summary and General Considerations
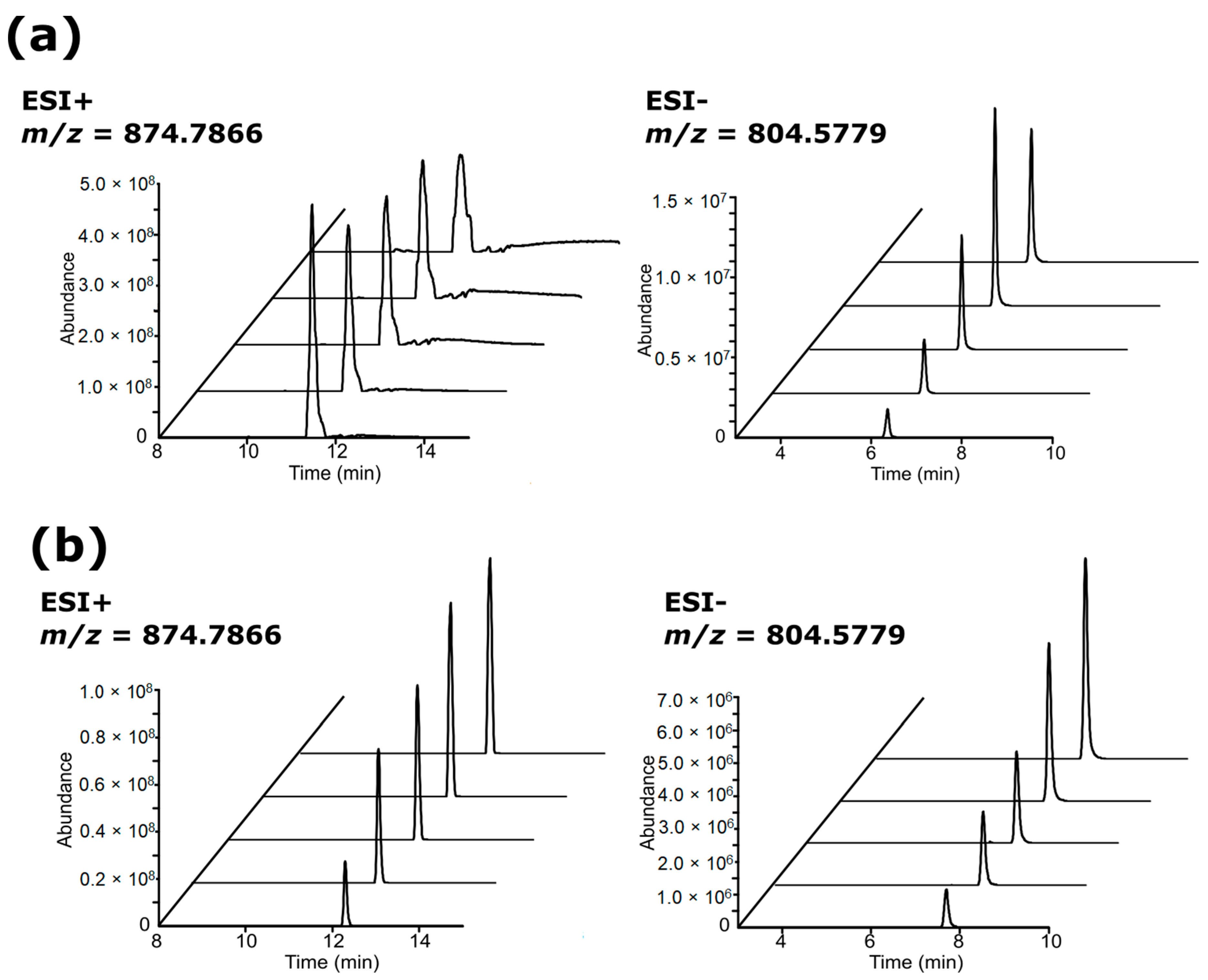
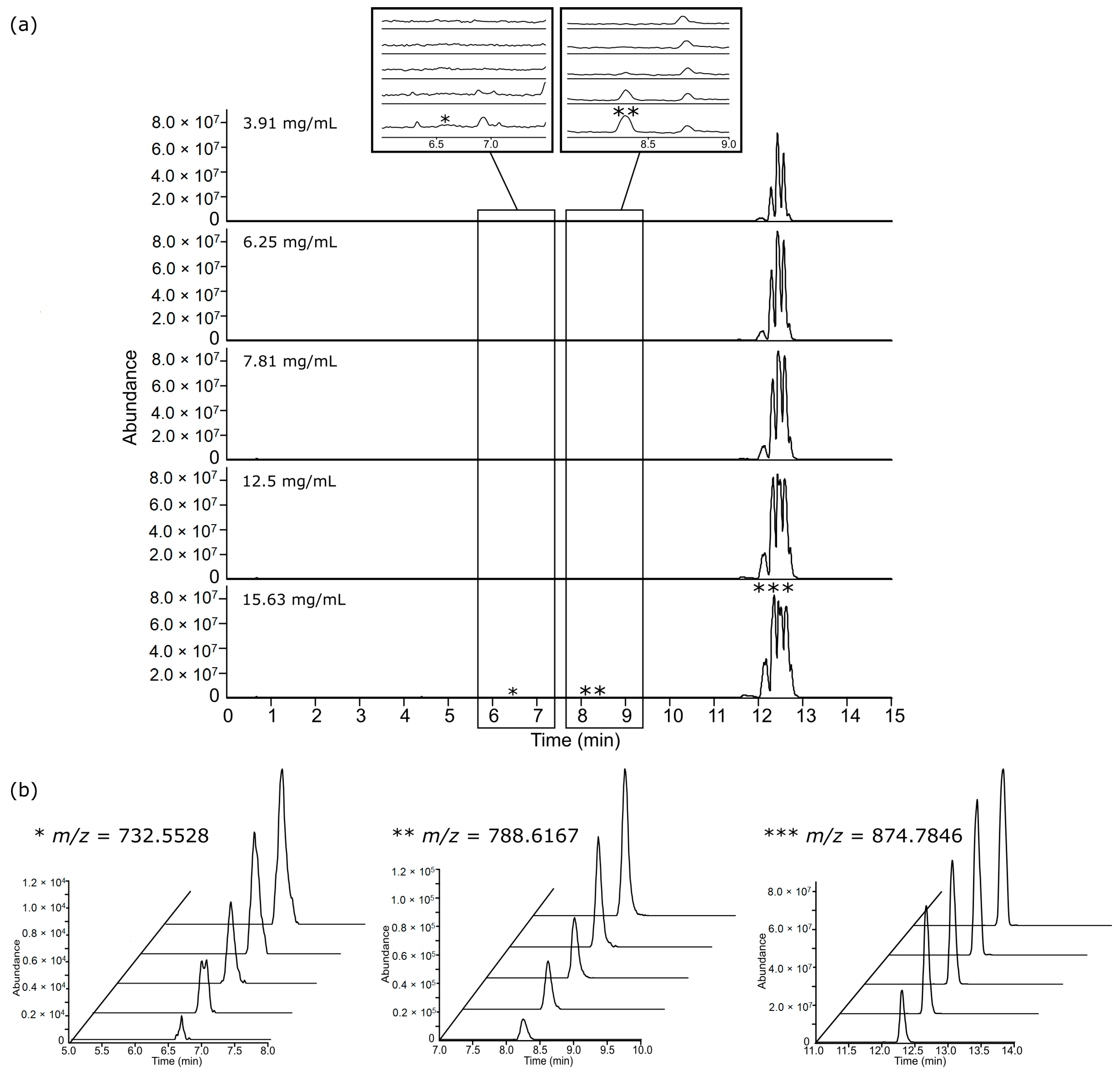
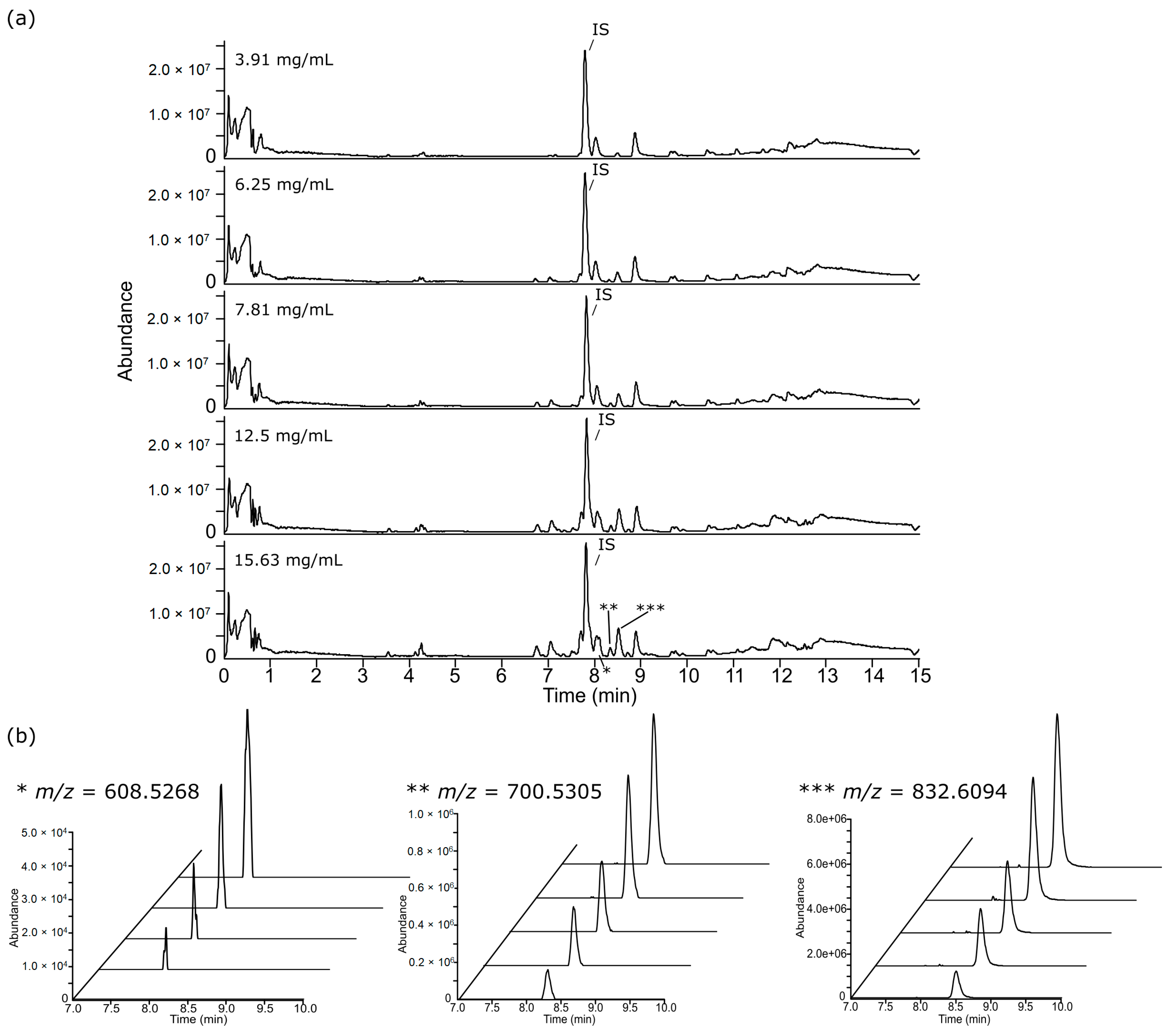
3.2. Chromatographic Examination
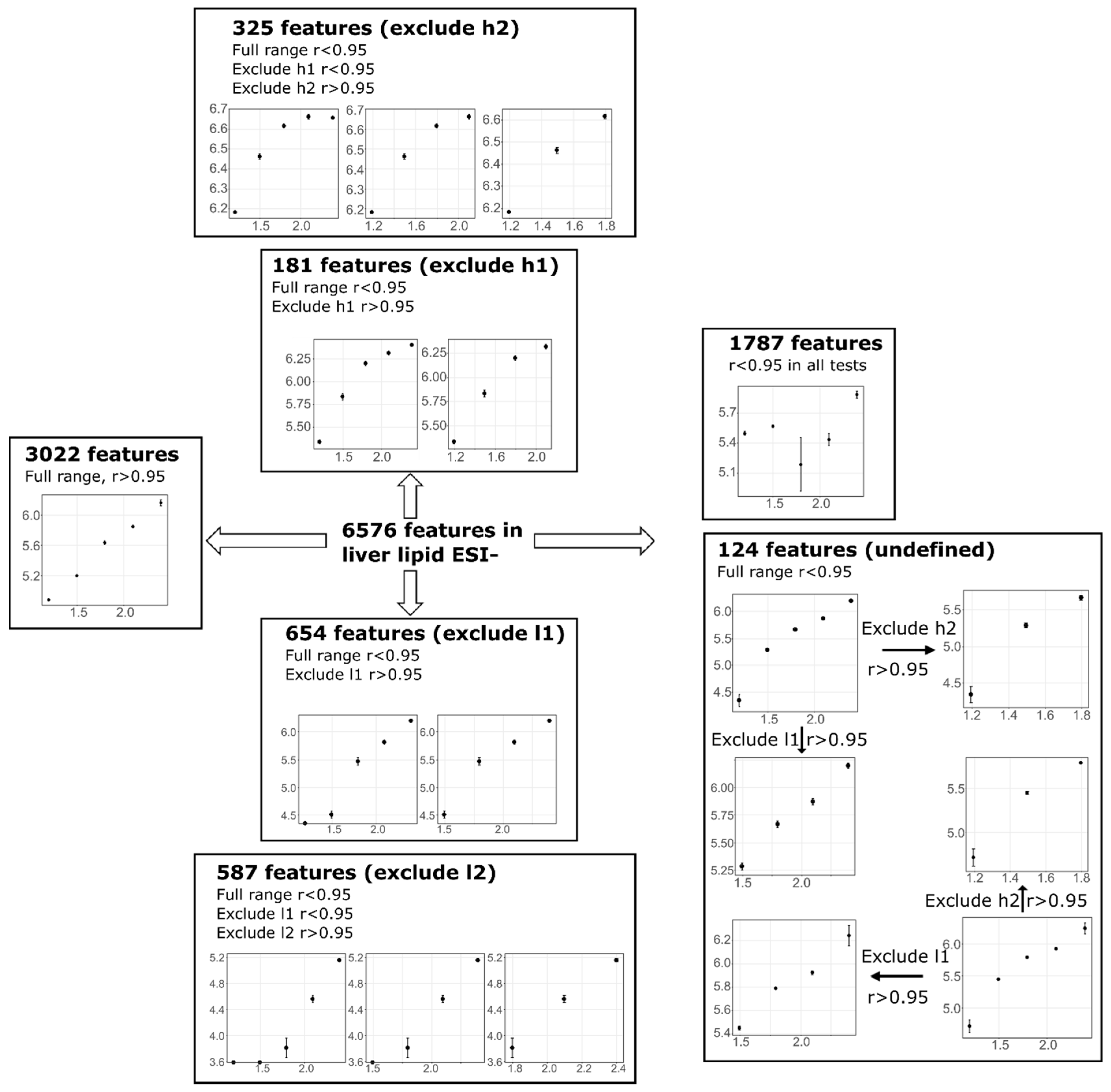
3.3. Feature Summary and Reproducibility
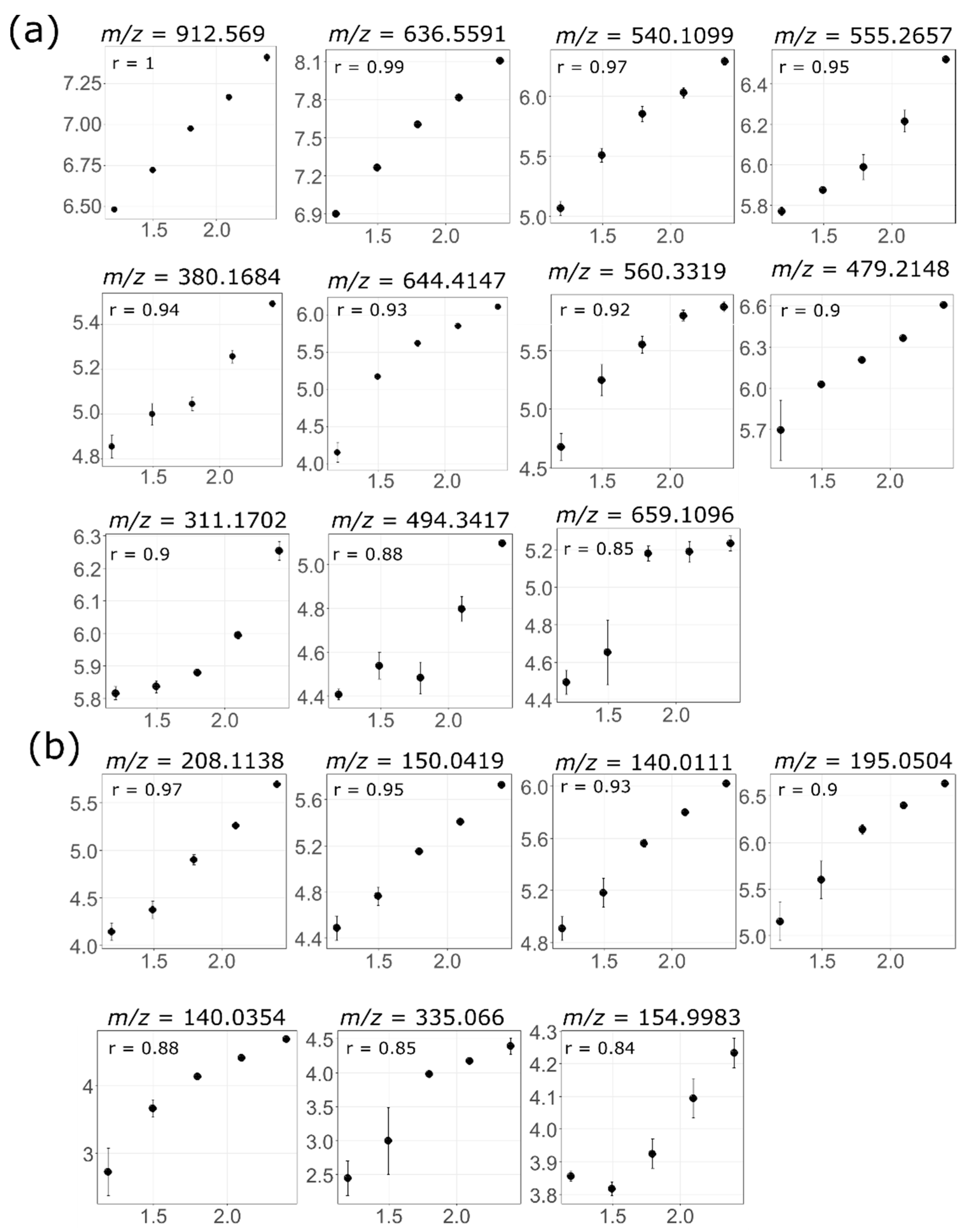
3.4. Feature Linearity Testing for Selection of Suitable Injection Concentration
3.5. Strength and Limitation of This Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Sadanala, K.C.; Kim, E.-K. A metabolomic approach to understanding the metabolic link between obesity and diabetes. Mol. Cells 2015, 38, 587. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Virtue, A.; Wang, H.; Yang, X.-F. Metabolomic analyses for atherosclerosis, diabetes, and obesity. Biomark. Res. 2013, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Orešič, M. Metabolomics, a novel tool for studies of nutrition, metabolism and lipid dysfunction. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Koulman, A.; Griffin, J.L. Towards metabolic biomarkers of insulin resistance and type 2 diabetes: Progress from the metabolome. Lancet Diabetes Endocrinol. 2014, 2, 65–75. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. In Functional Genomics; Springer: Berlin, Germany, 2002; pp. 155–171. [Google Scholar]
- Büscher, J.M.; Czernik, D.; Ewald, J.C.; Sauer, U.; Zamboni, N. Cross-platform comparison of methods for quantitative metabolomics of primary metabolism. Anal. Chem. 2009, 81, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.W.; Han, X. Lipidomics at the interface of structure and function in systems biology. Chem. Biol. 2011, 18, 284–291. [Google Scholar] [CrossRef]
- Rochat, B. From targeted quantification to untargeted metabolomics: Why LC-high-resolution-MS will become a key instrument in clinical labs. TrAC Trends Anal. Chem. 2016, 84, 151–164. [Google Scholar] [CrossRef]
- Naz, S.; dos Santos, D.C.M.; García, A.; Barbas, C. Analytical protocols based on LC–MS, GC–MS and CE–MS for nontargeted metabolomics of biological tissues. Bioanalysis 2014, 6, 1657–1677. [Google Scholar] [CrossRef]
- Reis, A.; Rudnitskaya, A.; Blackburn, G.J.; Fauzi, N.M.; Pitt, A.R.; Spickett, C.M. A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J. Lipid Res. 2013, 54, 1812–1824. [Google Scholar] [CrossRef]
- Vorkas, P.A.; Isaac, G.; Anwar, M.A.; Davies, A.H.; Want, E.J.; Nicholson, J.K.; Holmes, E. Untargeted UPLC-MS profiling pipeline to expand tissue metabolome coverage: Application to cardiovascular disease. Anal. Chem. 2015, 87, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Alves, A.C.; Ebbels, T.M.; Nicholson, J.K.; Want, E.J. Optimization and evaluation of metabolite extraction protocols for untargeted metabolic profiling of liver samples by UPLC-MS. Anal. Chem. 2010, 82, 7779–7786. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.D.; Dane, A.D.; Harms, A.; Strassburg, K.; Seifar, R.M.; Verdijk, L.B.; Kersten, S.; Berger, R.; Hankemeier, T.; Vreeken, R.J. Global profiling of the muscle metabolome: Method optimization, validation and application to determine exercise-induced metabolic effects. Metabolomics 2015, 11, 271–285. [Google Scholar] [CrossRef]
- Anwar, M.A.; Vorkas, P.A.; Li, J.V.; Shalhoub, J.; Want, E.J.; Davies, A.H.; Holmes, E. Optimization of metabolite extraction of human vein tissue for ultra performance liquid chromatography-mass spectrometry and nuclear magnetic resonance-based untargeted metabolic profiling. Analyst 2015, 140, 7586–7597. [Google Scholar] [CrossRef] [PubMed]
- Sargent, M. Guide to Achieving Reliable Quantitative LC-MS Measurements; RSC Analytical Methods Committee; LGC: Middlesex, UK, 2013. [Google Scholar]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC–MS. Nat. Protoc. 2010, 5, 1005. [Google Scholar] [CrossRef]
- Danne-Rasche, N.; Coman, C.; Ahrends, R. Nano-LC/NSI MS refines lipidomics by enhancing lipid coverage, measurement sensitivity, and linear dynamic range. Anal. Chem. 2018, 90, 8093–8101. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, L. Metabolomics of small numbers of cells: Metabolomic profiling of 100, 1000, and 10000 human breast cancer cells. Anal. Chem. 2017, 89, 11664–11671. [Google Scholar] [CrossRef]
- Cajka, T.; Smilowitz, J.T.; Fiehn, O. Validating quantitative untargeted lipidomics across nine liquid chromatography–High-resolution mass spectrometry platforms. Anal. Chem. 2017, 89, 12360–12368. [Google Scholar] [CrossRef]
- Taylor, P.J. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography–electrospray-tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef]
- Croixmarie, V.; Umbdenstock, T.; Cloarec, O.; Moreau, A.l.; Pascussi, J.-M.; Boursier-Neyret, C.; Walther, B. Integrated comparison of drug-related and drug-induced ultra performance liquid chromatography/mass spectrometry metabonomic profiles using human hepatocyte cultures. Anal. Chem. 2009, 81, 6061–6069. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Molnár, I.; Snyder, J.H.; Shen, G.-a.; Qi, X. Discrimination and quantification of true biological signals in metabolomics analysis based on liquid chromatography-mass spectrometry. Mol. Plant 2016, 9, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; Hollywood, K.A.; Jüllig, M.; Curtis, M.A.; Waldvogel, H.J.; Faull, R.L.; Unwin, R.D. Graded perturbations of metabolism in multiple regions of human brain in Alzheimer’s disease: Snapshot of a pervasive metabolic disorder. Biochimica Biophysica Acta (BBA)—Mol. Basis Dis. 2016, 1862, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, L.M.; Young, W.; Fraser, K.; Tannock, G.W.; Lee, J.; Roy, N.C. Digestive-resistant carbohydrates affect lipid metabolism in rats. Metabolomics 2016, 12, 79. [Google Scholar] [CrossRef]
- Fraser, K.; Harrison, S.J.; Lane, G.A.; Otter, D.E.; Hemar, Y.; Quek, S.-Y.; Rasmussen, S. Non-targeted analysis of tea by hydrophilic interaction liquid chromatography and high resolution mass spectrometry. Food Chem. 2012, 134, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography—Mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef]
- Burla, B.; Arita, M.; Arita, M.; Bendt, A.K.; Cazenave-Gassiot, A.; Dennis, E.A.; Ekroos, K.; Han, X.; Ikeda, K.; Liebisch, G. MS-based lipidomics of human blood plasma: A community-initiated position paper to develop accepted guidelines. J. Lipid Res. 2018, 59, 2001–2017. [Google Scholar] [CrossRef]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef]
- Waybright, T.J.; Van, Q.N.; Muschik, G.M.; Conrads, T.P.; Veenstra, T.D.; Issaq, H.J. LC-MS in metabonomics: Optimization of experimental conditions for the analysis of metabolites in human urine. J. Liq. Chromatogr. Relat. Technol. 2006, 29, 2475–2497. [Google Scholar] [CrossRef]
- Ortega, N.; Romero, M.-P.; Macià, A.; Reguant, J.; Anglès, N.; Morelló, J.-R.; Motilva, M.-J. Comparative study of UPLC–MS/MS and HPLC–MS/MS to determine procyanidins and alkaloids in cocoa samples. J. Food Compos. Anal. 2010, 23, 298–305. [Google Scholar] [CrossRef]
- Nordström, A.; O’Maille, G.; Qin, C.; Siuzdak, G. Nonlinear data alignment for UPLC−MS and HPLC−MS based metabolomics: Quantitative analysis of endogenous and exogenous metabolites in human serum. Anal. Chem. 2006, 78, 3289–3295. [Google Scholar] [CrossRef] [PubMed]
- Jankevics, A.; Merlo, M.E.; de Vries, M.; Vonk, R.J.; Takano, E.; Breitling, R. Separating the wheat from the chaff: A prioritisation pipeline for the analysis of metabolomics datasets. Metabolomics 2012, 8, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Römisch-Margl, W.; Prehn, C.; Bogumil, R.; Röhring, C.; Suhre, K.; Adamski, J. Procedure for tissue sample preparation and metabolite extraction for high-throughput targeted metabolomics. Metabolomics 2012, 8, 133–142. [Google Scholar] [CrossRef]
- Berg, M.; Vanaerschot, M.; Jankevics, A.; Cuypers, B.; Breitling, R.; Dujardin, J.-C. LC-MS metabolomics from study design to data-analysis–Using a versatile pathogen as a test case. Comput. Struct. Biotechnol. J. 2013, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Haid, M.; Muschet, C.; Wahl, S.; Römisch-Margl, W.; Prehn, C.; Möller, G.; Adamski, J. Long-term stability of human plasma metabolites during storage at −80 °C. J. Proteom. Res. 2017, 17, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Jové, M.; Moreno-Navarrete, J.M.; Pamplona, R.; Ricart, W.; Portero-Otín, M.; Fernández-Real, J.M. Human omental and subcutaneous adipose tissue exhibit specific lipidomic signatures. FASEB J. 2014, 28, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Furey, A.; Moriarty, M.; Bane, V.; Kinsella, B.; Lehane, M. Ion suppression; a critical review on causes, evaluation, prevention and applications. Talanta 2013, 115, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Majors, R. Sample Preparation Fundamentals for Chromatography; Agilent Technologies: Mississauga, ON, Canada, 2013. [Google Scholar]
- Sandra, K.; dos Santos Pereira, A.; Vanhoenacker, G.; David, F.; Sandra, P. Comprehensive blood plasma lipidomics by liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Pati, S.; Nie, B.; Arnold, R.D.; Cummings, B.S. Extraction, chromatographic and mass spectrometric methods for lipid analysis. Biomed. Chromatogr. 2016, 30, 695–709. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid | Reconstitution Volume (µL) | Concentration (mg/mL) |
| Liver | 100 | 250 |
| 200 | 125 | |
| 400 | 62.5 | |
| 800 | 31.25 | |
| 1600 | 15.63 | |
| Adipose | 1600 | 15.63 |
| 2000 | 12.5 | |
| 3200 | 7.81 | |
| 4000 | 6.25 | |
| 6400 | 3.91 | |
| Polar Metabolite | Reconstitution Volume (µL) | Concentration (mg/mL) |
| Liver | 50 | 250 |
| 100 | 125 | |
| 200 | 62.5 | |
| 400 | 31.25 | |
| 800 | 15.63 | |
| Adipose | 50 | 250 |
| 100 | 125 | |
| 200 | 62.5 | |
| 400 | 31.25 | |
| 800 | 15.63 |
| - | Concentration (mg/mL) | ESI (+) | ESI (−) | ||||
|---|---|---|---|---|---|---|---|
| #Non-Blank Features (tstat > 1, p < 0.05) | #Non-Blank Features with RSD < 30 | % | #Non-Blank Features (tstat > 1, p < 0.05) | #Non-Blank Features With RSD < 30 | % | ||
| Adipose lipids | 3.91 | 856 | 814 | 95.1 | 441 | 420 | 95.2 |
| 6.25 | 921 | 895 | 97.2 | 628 | 605 | 96.3 | |
| 7.81 | 939 | 914 | 97.3 | 751 | 710 | 94.5 | |
| 12.5 | 955 | 934 | 97.8 | 972 | 935 | 96.2 | |
| 15.63 | 985 | 942 | 95.6 | 1160 | 1127 | 97.2 | |
| Liver lipids | 15.63 | 1339 | 1189 | 88.8 | 2984 | 2789 | 93.5 |
| 31.25 | 1784 | 1510 | 84.6 | 4038 | 3820 | 94.6 | |
| 62.5 | 2634 | 2418 | 91.8 | 5122 | 4824 | 94.2 | |
| 125 | 3098 | 3047 | 98.4 | 5952 | 5659 | 95.1 | |
| 250 | 3210 | 3110 | 96.9 | 6576 | 6186 | 94.1 | |
| Adipose HILIC | 15.63 | 20 | 17 | 85.0 | 30 | 29 | 96.7 |
| 31.25 | 25 | 21 | 84.0 | 57 | 55 | 96.5 | |
| 62.5 | 77 | 72 | 93.5 | 72 | 62 | 86.1 | |
| 125 | 209 | 203 | 97.1 | 114 | 101 | 88.6 | |
| 250 | 239 | 217 | 90.8 | 176 | 154 | 87.5 | |
| Liver HILIC | 15.63 | 53 | 47 | 88.7 | 39 | 11 | 28.2 |
| 31.25 | 82 | 74 | 90.2 | 83 | 38 | 45.8 | |
| 62.5 | 435 | 413 | 94.9 | 236 | 221 | 93.6 | |
| 125 | 480 | 443 | 92.3 | 304 | 276 | 90.8 | |
| 250 | 553 | 496 | 89.7 | 380 | 349 | 91.8 | |
| Lipids | |||||||||
| Mode | Tissue | #Feature | r > 0.95 | Exclude l1 | Exclude l2 | Exclude h1 | Exclude h2 | Remain r < 0.95 | Undefined |
| ESI+ | Adipose | 985 | 428 | 23 | 5 | 56 | 188 | 235 | 50 |
| Liver | 3210 | 2119 | 422 | 299 | 20 | 25 | 314 | 11 | |
| ESI- | Adipose | 1160 | 304 | 121 | 42 | 25 | 47 | 576 | 45 |
| Liver | 6576 | 3022 | 654 | 587 | 181 | 325 | 1683 | 124 | |
| HILIC | |||||||||
| Mode | Tissue | #Feature | r > 0.9 | Exclude l1 | Exclude l2 | Exclude h1 | Exclude h2 | Remain r < 0.9 | Undefined |
| ESI+ | Adipose | 239 | 94 | 6 | 14 | 1 | 2 | 122 | 0 |
| Liver | 553 | 349 | 16 | 38 | 9 | 7 | 134 | 0 | |
| ESI- | Adipose | 176 | 70 | 26 | 12 | 0 | 1 | 67 | 0 |
| Liver | 380 | 136 | 39 | 60 | 2 | 1 | 142 | 0 | |
| Concentration | Non-Blank Features | r > 0.95 (Full) | r > 0.95 (Exclude l1) | r > 0.95 (Exclude l2) | r > 0.95 (Exclude h1) | r > 0.95 (Exclude h2) | Total Linear Features | ||
|---|---|---|---|---|---|---|---|---|---|
| Adipose lipids | ESI+ | 3.91 | 856 | 415 | - | - | 56 | 183 | 654 |
| 6.25 | 921 | 426 | 22 | - | 56 | 187 | 691 | ||
| 7.81 | 939 | 428 | 22 | 5 | 56 | 187 | 698 | ||
| 12.5 | 955 | 428 | 22 | 5 | 56 | - | 511 | ||
| 15.63 | 985 | 428 | 23 | 5 | - | - | 456 | ||
| ESI− | 3.91 | 441 | 264 | - | - | 25 | 31 | 320 | |
| 6.25 | 628 | 290 | 95 | - | 25 | 35 | 445 | ||
| 7.81 | 751 | 296 | 107 | 35 | 25 | 35 | 498 | ||
| 12.5 | 972 | 302 | 119 | 41 | 25 | - | 487 | ||
| 15.63 | 1160 | 304 | 121 | 42 | - | - | 467 | ||
| Liver lipids | ESI+ | 15.63 | 1339 | 1285 | - | - | 13 | 14 | 1312 |
| 31.25 | 1784 | 1570 | 145 | - | 15 | 16 | 1746 | ||
| 62.5 | 2634 | 1939 | 344 | 198 | 18 | 21 | 2520 | ||
| 125 | 3098 | 2083 | 414 | 295 | 19 | - | 2811 | ||
| 250 | 3210 | 2119 | 422 | 299 | - | - | 2840 | ||
| ESI− | 15.63 | 2984 | 2178 | - | - | 111 | 173 | 2462 | |
| 31.25 | 4038 | 2517 | 441 | - | 138 | 203 | 3299 | ||
| 62.5 | 5122 | 2817 | 558 | 420 | 174 | 235 | 4204 | ||
| 125 | 5952 | 2955 | 635 | 556 | 180 | - | 4326 | ||
| 250 | 6576 | 3022 | 654 | 587 | - | - | 4263 | ||
| Adipose HILIC | ESI+ | 15.63 | 20 | 4 | - | - | 0 | 1 | 5 |
| 31.25 | 25 | 5 | 0 | - | 0 | 1 | 6 | ||
| 62.5 | 77 | 29 | 0 | 7 | 1 | 1 | 38 | ||
| 125 | 209 | 89 | 4 | 14 | 1 | - | 108 | ||
| 250 | 239 | 94 | 6 | 14 | - | - | 114 | ||
| ESI− | 15.63 | 30 | 27 | - | - | 0 | 0 | 27 | |
| 31.25 | 57 | 42 | 12 | - | 0 | 0 | 54 | ||
| 62.5 | 72 | 49 | 17 | 3 | 0 | 0 | 69 | ||
| 125 | 114 | 59 | 23 | 8 | 0 | - | 90 | ||
| 250 | 176 | 70 | 26 | 12 | - | - | 108 | ||
| Liver HILIC | ESI+ | 15.63 | 53 | 29 | - | - | 1 | 3 | 33 |
| 31.25 | 82 | 42 | 2 | - | 3 | 5 | 52 | ||
| 62.5 | 435 | 311 | 11 | 26 | 9 | 6 | 363 | ||
| 125 | 480 | 331 | 14 | 30 | 9 | - | 384 | ||
| 250 | 553 | 349 | 16 | 38 | - | - | 403 | ||
| ESI− | 15.63 | 39 | 34 | - | - | 0 | 0 | 34 | |
| 31.25 | 83 | 60 | 5 | - | 2 | 0 | 67 | ||
| 62.5 | 236 | 118 | 22 | 44 | 2 | 0 | 186 | ||
| 125 | 304 | 126 | 35 | 51 | 2 | - | 214 | ||
| 250 | 380 | 136 | 39 | 60 | - | - | 235 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.E.; Kruger, M.C.; Cooper, G.J.S.; Poppitt, S.D.; Fraser, K. Tissue-Specific Sample Dilution: An Important Parameter to Optimise Prior to Untargeted LC-MS Metabolomics. Metabolites 2019, 9, 124. https://doi.org/10.3390/metabo9070124
Wu ZE, Kruger MC, Cooper GJS, Poppitt SD, Fraser K. Tissue-Specific Sample Dilution: An Important Parameter to Optimise Prior to Untargeted LC-MS Metabolomics. Metabolites. 2019; 9(7):124. https://doi.org/10.3390/metabo9070124
Chicago/Turabian StyleWu, Zhanxuan E., Marlena C. Kruger, Garth J.S. Cooper, Sally D. Poppitt, and Karl Fraser. 2019. "Tissue-Specific Sample Dilution: An Important Parameter to Optimise Prior to Untargeted LC-MS Metabolomics" Metabolites 9, no. 7: 124. https://doi.org/10.3390/metabo9070124
APA StyleWu, Z. E., Kruger, M. C., Cooper, G. J. S., Poppitt, S. D., & Fraser, K. (2019). Tissue-Specific Sample Dilution: An Important Parameter to Optimise Prior to Untargeted LC-MS Metabolomics. Metabolites, 9(7), 124. https://doi.org/10.3390/metabo9070124